
Therapeutic Strategies to Enhance Tumor Antigenicity: Making the Tumor Detectable by the Immune System



{kind=link}
Abstract
:1. Introduction
2. The Basis of an Efficient Immune Response in Cancer
3. Cancer Mechanisms of Immune Escape
4. Current Immunotherapy Approaches
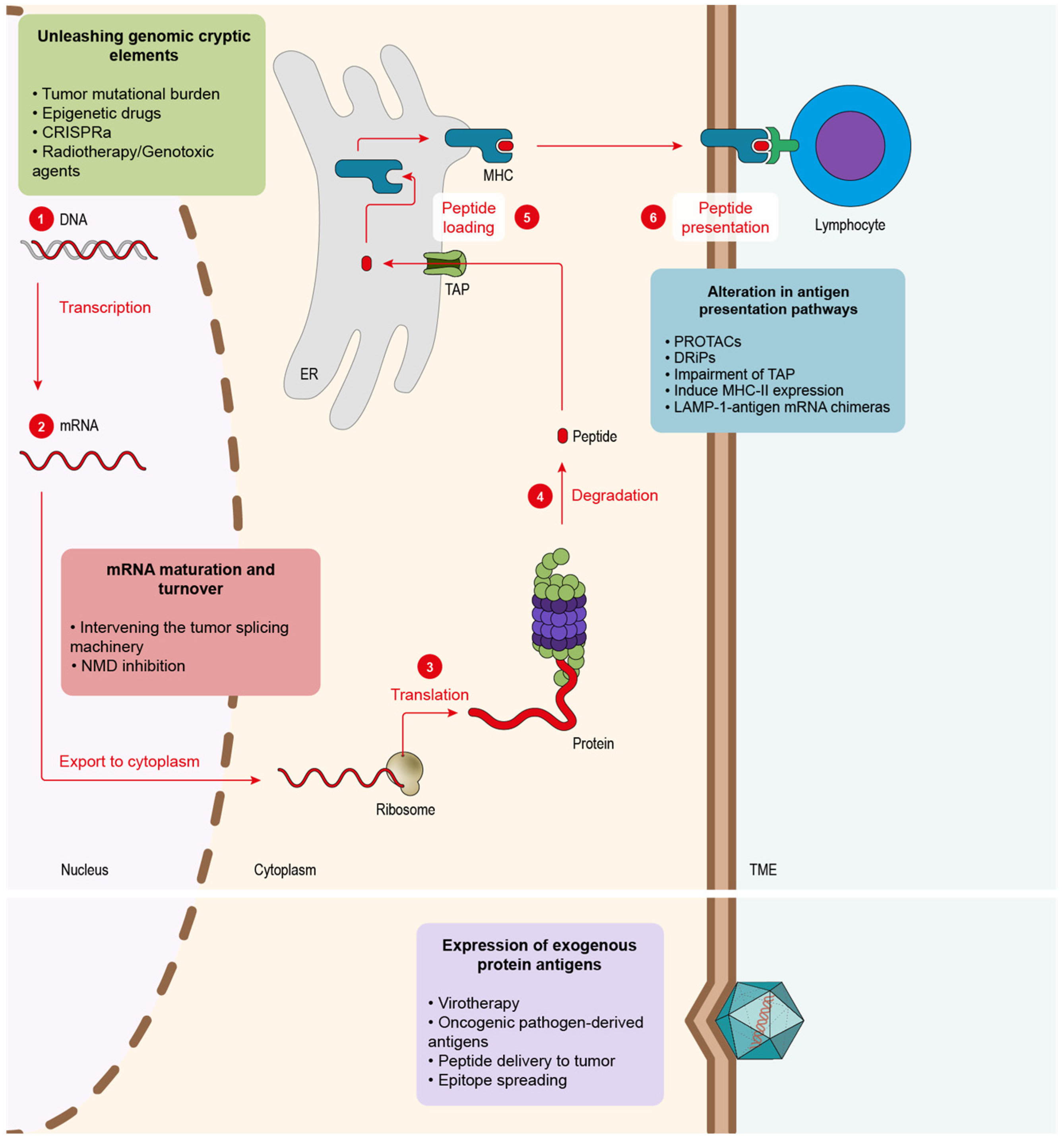
5. Main Approaches to Enhance Tumor Antigenicity
5.1. Alteration in Antigen Presentation Pathways to Elicit Tumor Antigens
5.2. Alteration of mRNA Maturation and Turnover to Foster New Tumor Antigens
5.3. Unleashing Genomic Cryptic Elements
5.4. Expression of Exogenous Protein Antigens
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; Coggeshall, M.; et al. Global, Regional, and National Life Expectancy, All-Cause Mortality, and Cause-Specific Mortality for 249 Causes of Death, 1980–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive Cellular Therapies: The Current Landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Korman, A.J.; Garrett-Thomson, S.C.; Lonberg, N. The Foundations of Immune Checkpoint Blockade and the Ipilimumab Approval Decennial. Nat. Rev. Drug Discov. 2021, 21, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.H. Efforts to Explain Spontaneous Regression of Cancer. J. Surg. Oncol. 1981, 17, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Oiseth, S.J.; Aziz, M.S. Cancer Immunotherapy: A Brief Review of the History, Possibilities, and Challenges Ahead. J. Cancer Metastasis Treat. 2017, 3, 250. [Google Scholar] [CrossRef]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10, 2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.F.A.P.; Mitchell, G.F.; Weiss, N.S. Cellular Basis of the Immunological Defects in Thymectomized Mice. Nature 1967, 214, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice: I. Morphology, Quantitation, Tissue Distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” Killer Cells in the Mouse. I. Cytotoxic Cells with Specificity for Mouse Moloney Leukemia Cells. Specificity and Distribution According to Genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Malissen, B.; Grégoire, C.; Malissen, M.; Roncagalli, R. Integrative Biology of T Cell Activation. Nat. Immunol. 2014, 15, 790–797. [Google Scholar] [CrossRef]
- Zarnitsyna, V.I.; Evavold, B.D.; Schoettle, L.N.; Blattman, J.N.; Antia, R. Estimating the Diversity, Completeness, and Cross-Reactivity of the T Cell Repertoire. Front. Immunol. 2013, 4, 485. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.; Alt, F.W. Unraveling V(D)J Recombination: Insights into Gene Regulation. Cell 2004, 116, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and Negative Selection of the T Cell Repertoire: What Thymocytes See (and Don’t See). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, H.; Matsumoto, T. Mechanisms of Tolerance Induction by Dendritic Cells in Vivo. Front. Immunol. 2018, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.K.; Weiss, A. Insights into the Initiation of TCR Signaling. Nat. Immunol. 2014, 15, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; Ledbetter, J.A.; Gillespie, M.M.; Lindsten, T.; Thompson, C.B. T-Cell Proliferation Involving the CD28 Pathway Is Associated with Cyclosporine-Resistant Interleukin 2 Gene Expression. Mol. Cell. Biol. 1987, 7, 4472–4481. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, P.; Cohn, M. A Theory of Self-Nonself Discrimination. Science 1970, 169, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.L.; Jenkins, M.K.; Schwartz, R.H. Clonal Expansion versus Functional Clonal Inactivation: A Costimulatory Signalling Pathway Determines the Outcome of T Cell Antigen Receptor Occupancy. Annu. Rev. Immunol. 1989, 7, 445–480. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H.; Mueller, D.L.; Jenkins, M.K.; Quill, H. T-Cell Clonal Anergy. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 605–610. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory Cytokines as a Third Signal for T Cell Activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Curtsinger, J.M.; Schmidt, C.S.; Mondino, A.; Lins, D.C.; Kedl, R.M.; Jenkins, M.K.; Mescher, M.F. Inflammatory Cytokines Provide a Third Signal for Activation of Naive CD4+ and CD8+ T Cells. J. Immunol. 1999, 162, 3256–3262. [Google Scholar] [PubMed]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodland, D.L.; Kohlmeier, J.E. Migration, Maintenance and Recall of Memory T Cells in Peripheral Tissues. Nat. Rev. Immunol. 2009, 9, 153–161. [Google Scholar] [CrossRef]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident Memory T Cells, Critical Components in Tumor Immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.A.; Teixeiro, E. TCR Signaling in T Cell Memory. Front. Immunol. 2015, 6, 617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of Immune Evasion by Tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-β Directly Targets Cytotoxic T Cell Functions during Tumor Evasion of Immune Surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.C.; Balko, J.M. Mechanisms of MHC-I Downregulation and Role in Immunotherapy Response. Front. Immunol. 2022, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Korkolopoulou, P.; Kaklamanis, L.; Pezzella, F.; Harris, A.L.; Gatter, K.C. Loss of Antigen-Presenting Molecules (MHC Class I and TAP-1) in Lung Cancer. Br. J. Cancer 1996, 73, 148–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in Patients with Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, C.S.; Doi, T.; Jang, R.W.-J.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.-P.; et al. KEYNOTE-059 Cohort 1: Efficacy and Safety of Pembrolizumab (Pembro) Monotherapy in Patients with Previously Treated Advanced Gastric Cancer. J. Clin. Oncol. 2017, 35, 4003. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.N.; Lanier, B.J.; et al. Eradication of B-Lineage Cells and Regression of Lymphoma in a Patient Treated with Autologous T Cells Genetically Engineered to Recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and Persistence of Adoptively Transferred Autologous CD19-Targeted T Cells in Patients with Relapsed or Chemotherapy Refractory B-Cell Leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with Irradiated Tumor Cells Engineered to Secrete Murine Granulocyte-Macrophage Colony-Stimulating Factor Stimulates Potent, Specific, and Long-Lasting Anti-Tumor Immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [Green Version]
- Soiffer, R.J.; Kooshesh, K.A.; Ho, V. Whole Tumor Cell Vaccines Engineered to Secrete GM-CSF (GVAX). ImmunoMedicine 2021, 1, e1025. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-Deletion-Derived Tumour-Specific Neoantigens and the Immunogenic Phenotype: A Pan-Cancer Analysis. Lancet Oncol 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Blass, E.; Ott, P.A. Advances in the Development of Personalized Neoantigen-Based Therapeutic Cancer Vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Fang, H.; Wu, Y.; Narzisi, G.; O’Rawe, J.A.; Barrón, L.T.J.; Rosenbaum, J.; Ronemus, M.; Iossifov, I.; Schatz, M.C.; Lyon, G.J. Reducing INDEL Calling Errors in Whole Genome and Exome Sequencing Data. Genome Med. 2014, 6, 89. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.P.; Petersen, B.L.; Johansen, I.E.; Niu, Y.; Yang, Z.; Chamberlain, C.A.; Met, Ö.; Wandall, H.H.; Frödin, M. INDEL Detection, the “Achilles Heel” of Precise Genome Editing: A Survey of Methods for Accurate Profiling of Gene Editing Induced Indels. Nucleic Acids Res. 2021, 48, 11958–11981. [Google Scholar] [CrossRef]
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697. [Google Scholar] [CrossRef]
- Massafra, V.; Tundo, S.; Dietzig, A.; Ducret, A.; Jost, C.; Klein, C.; Kontermann, R.E.; Knoetgen, H.; Steegmaier, M.; Romagnani, A.; et al. Proteolysis-Targeting Chimeras Enhance T Cell Bispecific Antibody-Driven T Cell Activation and Effector Function through Increased MHC Class I Antigen Presentation in Cancer Cells. J. Immunol. 2021, 207, 493–504. [Google Scholar] [CrossRef]
- Del Val, M.; Antón, L.C.; Ramos, M.; Muñoz-Abad, V.; Campos-Sánchez, E. Endogenous TAP-Independent MHC-I Antigen Presentation: Not Just the ER Lumen. Curr. Opin. Immunol. 2020, 64, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Van Hall, T.; Wolpert, E.Z.; Van Veelen, P.; Laban, S.; Van Der Veer, M.; Roseboom, M.; Bres, S.; Grufman, P.; De Ru, A.; Meiring, H.; et al. Selective Cytotoxic T-Lymphocyte Targeting of Tumor Immune Escape Variants. Nat. Med. 2006, 12, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.C.; Van Hall, T. Importance of TAP-Independent Processing Pathways. Mol. Immunol. 2013, 55, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Garrido, G.; Schrand, B.; Rabasa, A.; Levay, A.; D’Eramo, F.; Berezhnoy, A.; Modi, S.; Gefen, T.; Marijt, K.; Doorduijn, E.; et al. Tumor-Targeted Silencing of the Peptide Transporter {TAP} Induces Potent Antitumor Immunity. Nat. Commun. 2019, 10, 3773. [Google Scholar] [CrossRef] [Green Version]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T Cell Help in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-Specific MHC-II Expression Represents a Tumour-Autonomous Phenotype and Predicts Response to Anti-PD-1/PD-L1 Therapy. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Andres, F.; Yufeng, L.; Dongquan, C.; William, E.G.; Katherine, L.U.; Natalie, D.M.; Erinn, D.K.; Todd, C.B.; Christos, V.; Donald, J.B.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Mortara, L.; Castellani, P.; Meazza, R.; Tosi, G.; De Lerma Barbaro, A.; Procopio, F.A.; Comes, A.; Zardi, L.; Ferrini, S.; Accolla, R.S. CIITA-Induced MHC Class II Expression in Mammary Adenocarcinoma Leads to a Th1 Polarization of the Tumor Microenvironment, Tumor Rejection, and Specific Antitumor Memory. Clin. Cancer Res. 2006, 12, 3435–3443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.K.; Boczkowski, D.; Morse, M.; Cumming, R.I.; Lyerly, H.K.; Gilboa, E. Induction of Primary Carcinoembryonic Antigen (CEA)-Specific Cytotoxic T Lymphocytes in Vitro Using Human Dendritic Cells Transfected with RNA. Nat. Biotechnol. 1998, 16, 364–369. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Antón, L.C.; Bennink, J.R. Defective Ribosomal Products (DRiPs): A Major Source of Antigenic Peptides for MHC Class I Molecules? J. Immunol. 1996, 157, 1823–1826. [Google Scholar]
- Anton, L.C.; Yewdell, J.W. Translating {DRiPs}: {MHC} Class {I} Immunosurveillance of Pathogens and Tumors. J. Leukoc. Biol. 2014, 95, 551–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, N.P.; Smith, S.A.; Wong, Y.C.; Tan, C.T.; Dudek, N.L.; Flesch, I.E.A.; Lin, L.C.W.; Tscharke, D.C.; Purcell, A.W. Kinetics of Antigen Expression and Epitope Presentation during Virus Infection. PLoS Pathog. 2013, 9, e1003129. [Google Scholar] [CrossRef] [Green Version]
- Esquivel, F.; Yewdell, J.; Bennink, J. RMA/S Cells Present Endogenously Synthesized Cytosolic Proteins to Class i-Restricted Cytotoxic T Lymphocytes. J. Exp. Med. 1992, 175, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Kishton, R.J.; Angel, M.; Conn, C.S.; Dalla-Venezia, N.; Marcel, V.; Vincent, A.; Catez, F.; Ferré, S.; Ayadi, L.; et al. Ribosomal Proteins Regulate MHC Class I Peptide Generation for Immunosurveillance. Mol. Cell 2019, 73, 1162–1173.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, C.J. SF3B1 Mutations in Chronic Lymphocytic Leukemia. Blood 2013, 121, 4627–4634. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Xu, L.; Suzuki, T.; Yoshikawa, T.; Sakamoto, H.; Uemura, H.; Yoshizawa, A.C.; Suzuki, Y.; Nakatsura, T.; Ishihama, Y.; et al. Aberrant Splicing Isoforms Detected by Full-Length Transcriptome Sequencing as Transcripts of Potential Neoantigens in Non-Small Cell Lung Cancer. Genome Biol. 2021, 22, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.X.; De Neef, E.; Thomas, J.D.; Sabio, E.; Rousseau, B.; Gigoux, M.; Knorr, D.A.; Greenbaum, B.; Elhanati, Y.; Hogg, S.J.; et al. Pharmacologic Modulation of RNA Splicing Enhances Anti-Tumor Immunity. Cell 2021, 184, 4032–4047. [Google Scholar] [CrossRef]
- North, K.; Benbarche, S.; Liu, B.; Pangallo, J.; Chen, S.; Stahl, M.; Bewersdorf, J.P.; Stanley, R.F.; Erickson, C.; Cho, H.; et al. Synthetic Introns Enable Splicing Factor Mutation-Dependent Targeting of Cancer Cells. Nat. Biotechnol. 2022, 40, 1103–1113. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Lim, E.L.; Xu, H.; Liu, P.; Al-Bakir, M.; Wong, Y.N.S.; Rowan, A.; Funt, S.A.; Merghoub, T.; et al. Escape from Nonsense-Mediated Decay Associates with Anti-Tumor Immunogenicity. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Supek, F.; Lehner, B. The Rules and Impact of Nonsense-Mediated MRNA Decay in Human Cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [Green Version]
- Pastor, F.; Kolonias, D.; Giangrande, P.H.; Gilboa, E. Induction of Tumour Immunity by Targeted Inhibition of Nonsense-Mediated MRNA Decay. Nature 2010, 465, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xie, X.; Yeganeh, P.N.; Lee, D.J.; Valle-Garcia, D.; Meza-Sosa, K.F.; Junqueira, C.; Su, J.; Luo, H.R.; Hide, W.; et al. Immunotherapy for Breast Cancer Using EpCAM Aptamer Tumor-Targeted Gene Knockdown. Proc. Natl. Acad. Sci. USA 2021, 118, e2022830118. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia-Crivelli, D.; Villanueva, H.; Menon, A.P.; Zheleva, A.; Moreno, B.; Villalba-Esparza, M.; Pastor, F. A Pan-Tumor-SiRNA Aptamer Chimera to Block Nonsense-Mediated MRNA Decay (NMD) Inflames and Suppresses Tumor Progression. Mol. Ther. Nucleic Acids 2022. [Google Scholar] [CrossRef]
- Bokhari, A.; Jonchere, V.; Lagrange, A.; Bertrand, R.; Svrcek, M.; Marisa, L.; Buhard, O.; Greene, M.; Demidova, A.; Jia, J.; et al. Targeting Nonsense-Mediated {mRNA} Decay in Colorectal Cancers with Microsatellite Instability. Oncogenesis 2018, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haen, S.P.; Löffler, M.W.; Rammensee, H.G.; Brossart, P. Towards New Horizons: Characterization, Classification and Implications of the Tumour Antigenic Repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Törngren, T.; Kvist, A.; et al. Mutational and Putative Neoantigen Load Predict Clinical Benefit of Adoptive T Cell Therapy in Melanoma. Nat. Commun. 2017, 8, 1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Sharma, S.; Advani, D.; Khosla, A.; Kumar, P.; Ambasta, R.K. Unboxing the Molecular Modalities of Mutagens in Cancer. Environ. Sci. Pollut. Res. 2021, 1–49. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- De Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive Elements May Comprise over Two-Thirds of the Human Genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.V.; Vernaz, G.; Putman, A.L.K.; Miska, E.A. Taming Transposable Elements in Vertebrates: From Epigenetic Silencing to Domestication. Trends Genet. 2022, 38, 529–553. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuvanagiri, M.; Lewis, J.; Putzker, K.; Becker, J.P.; Leicht, S.; Krijgsveld, J.; Batra, R.; Turnwald, B.; Jovanovic, B.; Hauer, C.; et al. 5-Azacytidine Inhibits Nonsense-Mediated Decay in a MYC-Dependent Fashion. EMBO Mol. Med. 2014, 6, 1593–1609. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Rei, M.; Woodhouse, I.; Ferris, K.; Kirschner, S.; Chandran, A.; Gileadi, U.; Chen, J.-L.; Pereira Pinho, M.; Ariosa-Morejon, Y.; et al. Decitabine Increases Neoantigen and Cancer Testis Antigen Expression to Enhance T-Cell–Mediated Toxicity against Glioblastoma. Neuro. Oncol. 2022, noac107. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Gruber, E.; Maher, B.; Waltham, M.; Sabouri-Thompson, Z.; Jong, I.; Luong, Q.; Levy, S.; Kumar, B.; Brasacchio, D.; et al. Integrated Clinical and Genomic Evaluation of Guadecitabine (SGI-110) in Peripheral T-Cell Lymphoma. Leukemia 2022, 36, 1654–1665. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Chow, R.D.; Bai, Z.; Zhu, L.; Errami, Y.; Dai, X.; Dong, M.B.; Ye, L.; Zhang, X.; Renauer, P.A.; et al. Multiplexed Activation of Endogenous Genes by CRISPRa Elicits Potent Antitumor Immunity. Nat. Immunol. 2019, 20, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, C.; Formenti, S.C.; Demaria, S.; Lhuillier, C.; Rudqvist, N.; Yamazaki, T.; Zhang, T.; Charpentier, M.; Galluzzi, L.; Dephoure, N.; et al. Radiotherapy-Exposed CD8+ and CD4+ Neoantigens Enhance Tumor Control. J. Clin. Invest. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Hietala, A.; Joutsen, J.; Vaarala, S.; Säily, M. A Rare and Complete Response to Combination Therapy with Radiation and Nivolumab in a Patient with Metastatic Urothelial Cancer. BMJ Case Rep. 2022, 15, e246653. [Google Scholar] [CrossRef]
- Giri, A.K.; Aittokallio, T. DNMT Inhibitors Increase Methylation in the Cancer Genome. Front. Pharmacol. 2019, 10, 385. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic Virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Santos Apolonio, J.; Lima de Souza Gonçalves, V.; Cordeiro Santos, M.L.; Silva Luz, M.; Silva Souza, J.V.; Rocha Pinheiro, S.L.; de Souza, W.R.; Sande Loureiro, M.; de Melo, F.F. Oncolytic Virus Therapy in Cancer: A Current Review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Lucas, A.; McFadden, G. Secreted Immunomodulatory Viral Proteins as Novel Biotherapeutics. J. Immunol. 2004, 173, 4765–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laspidea, V.; Fueyo, J.; Alonso, M.M.; Laspidea, V.; Labiano, S.; Marrodán, L.; Ausejo-mauleon, I.; De Nava, D.; Herrador-cañete, G. Exploiting 4-1BB Immune Checkpoint to Enhance the Efficacy of Oncolytic Virotherapy for Diffuse Intrinsic Pontine Gliomas. JCI Insight 2022, 7, e154812. [Google Scholar] [CrossRef]
- Tian, L.; Liu, T.; Jiang, S.; Cao, Y.; Kang, K.; Su, H.; Ren, G.; Wang, Z.; Xiao, W.; Li, D. Oncolytic Newcastle Disease Virus Expressing the Co-Stimulator OX40L as Immunopotentiator for Colorectal Cancer Therapy. Gene Ther. 2021, 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Newman, J.H.; Chesson, C.B.; Herzog, N.L.; Bommareddy, P.K.; Aspromonte, S.M.; Pepe, R.; Estupinian, R.; Aboelatta, M.M.; Buddhadev, S.; Tarabichi, S.; et al. Intratumoral Injection of the Seasonal Flu Shot Converts Immunologically Cold Tumors to Hot and Serves as an Immunotherapy for Cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 1119–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tempera, I.; Lieberman, P.M. Oncogenic Viruses as Entropic Drivers of Cancer Evolution. Front. Virol. 2021, 1, 28. [Google Scholar] [CrossRef]
- Cook, K.W.; Durrant, L.G.; Brentville, V.A. Current Strategies to Enhance Anti-Tumour Immunity. Biomedicines 2018, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.J.; Bu, W.; Nguyen, L.A.; Batchelor, J.D.; Kim, J.H.; Pittaluga, S.; Fuller, J.R.; Nguyen, H.; Chou, T.H.; Cohen, J.I.; et al. A Bivalent Epstein-Barr Virus Vaccine Induces Neutralizing Antibodies That Block Infection and Confer Immunity in Humanized Mice. Sci. Transl. Med. 2022, 14, eabf3685. [Google Scholar] [CrossRef]
- Ilca, T.; Boyle, L.H. The Ins and Outs of TAPBPR. Curr. Opin. Immunol. 2020, 64, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Tudor Ilca, F.; Neerincx, A.; Wills, M.R.; De La Roche, M.; Boyle, L.H. Utilizing TAPBPR to Promote Exogenous Peptide Loading onto Cell Surface MHC I Molecules. Proc. Natl. Acad. Sci. USA 2018, 115, E9353–E9361. [Google Scholar] [CrossRef] [Green Version]
- Kavunja, H.W.; Lang, S.; Sungsuwan, S.; Yin, Z.; Huang, X. Delivery of Foreign Cytotoxic T Lymphocyte Epitopes to Tumor Tissues for Effective Antitumor Immunotherapy against Pre-Established Solid Tumors in Mice. Cancer Immunol. Immunother. 2017, 66, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Vanderlugt, C.L.; Miller, S.D. Epitope Spreading in Immune-Mediated Diseases: Implications for Immunotherapy. Nat. Rev. Immunol. 2002, 2, 85–95. [Google Scholar] [CrossRef]
- Brossart, P. The Role of Antigen Spreading in the Efficacy of Immunotherapies. Clin. Cancer Res. 2020, 26, 4442–4447. [Google Scholar] [CrossRef]
- El-Shami, K.; Tirosh, B.; Bar-Haîm, E.; Carmon, L.; Vadai, E.; Fridkin, M.; Feldman, M.; Eisenbach, L. MHC Class I-Restricted Epitope Spreading in the Context of Tumor Rejection Following Vaccination with a Single Immunodominant CTL Epitope. Eur. J. Immunol. 1999, 29, 3295–3301. [Google Scholar] [CrossRef]
- Wierecky, J.; Müller, M.R.; Wirths, S.; Halder-Oehler, E.; Dörfel, D.; Schmidt, S.M.; Häntschel, M.; Brugger, W.; Schröder, S.; Horger, M.S.; et al. Immunologic and Clinical Responses after Vaccinations with Peptide-Pulsed Dendritic Cells in Metastatic Renal Cancer Patients. Cancer Res. 2006, 66, 5910–5918. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meraviglia-Crivelli, D.; Zheleva, A.; Barainka, M.; Moreno, B.; Villanueva, H.; Pastor, F. Therapeutic Strategies to Enhance Tumor Antigenicity: Making the Tumor Detectable by the Immune System. Biomedicines 2022, 10, 1842. https://doi.org/10.3390/biomedicines10081842
Meraviglia-Crivelli D, Zheleva A, Barainka M, Moreno B, Villanueva H, Pastor F. Therapeutic Strategies to Enhance Tumor Antigenicity: Making the Tumor Detectable by the Immune System. Biomedicines. 2022; 10(8):1842. https://doi.org/10.3390/biomedicines10081842
Chicago/Turabian StyleMeraviglia-Crivelli, Daniel, Angelina Zheleva, Martin Barainka, Beatriz Moreno, Helena Villanueva, and Fernando Pastor. 2022. "Therapeutic Strategies to Enhance Tumor Antigenicity: Making the Tumor Detectable by the Immune System" Biomedicines 10, no. 8: 1842. https://doi.org/10.3390/biomedicines10081842
APA StyleMeraviglia-Crivelli, D., Zheleva, A., Barainka, M., Moreno, B., Villanueva, H., & Pastor, F. (2022). Therapeutic Strategies to Enhance Tumor Antigenicity: Making the Tumor Detectable by the Immune System. Biomedicines, 10(8), 1842. https://doi.org/10.3390/biomedicines10081842