
A Human Stem Cell-Derived Neurosensory–Epithelial Circuitry on a Chip to Model Herpes Simplex Virus Reactivation

 ,
,  , , , ,
, , , ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
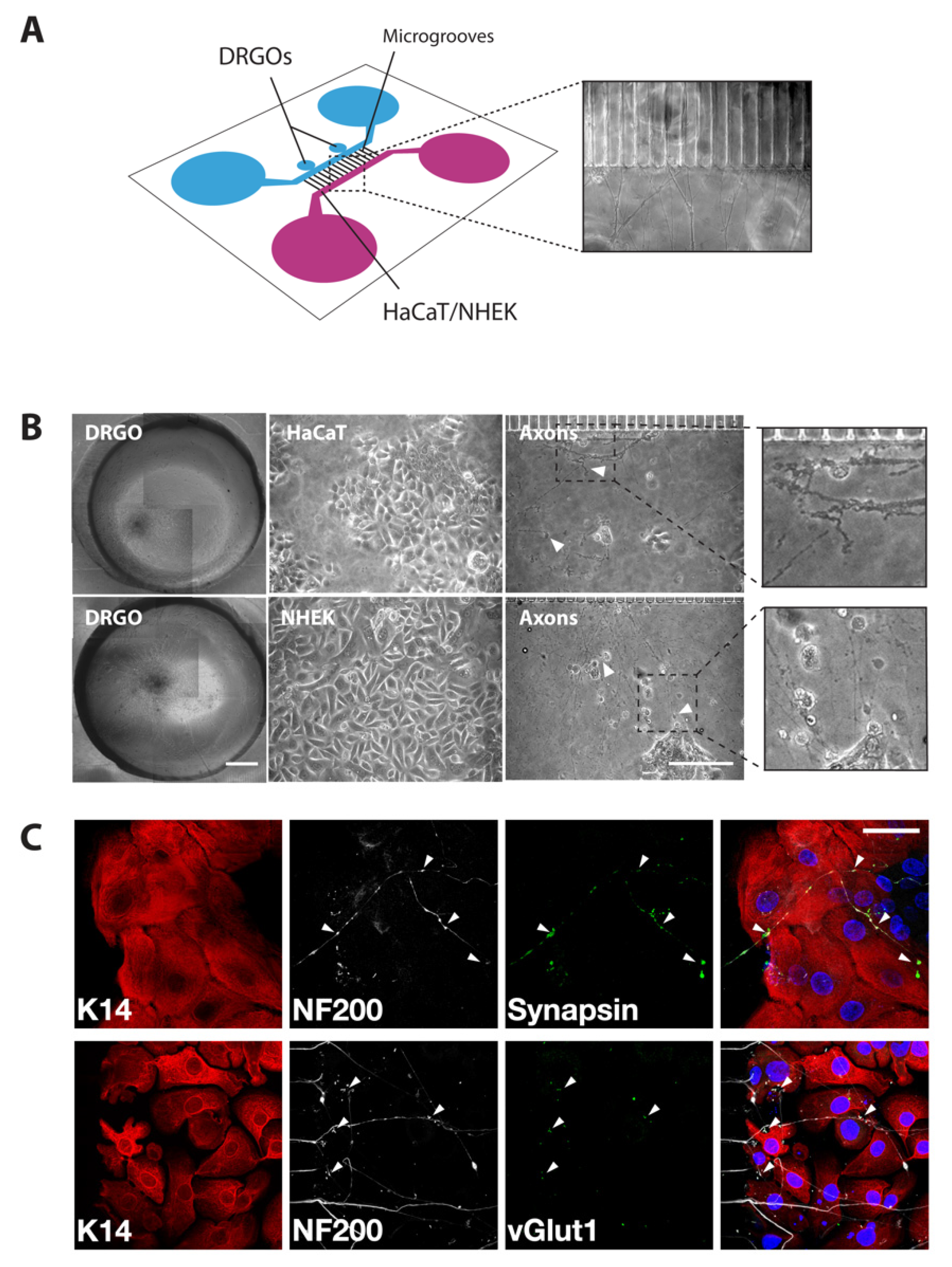
3.1. Setting the Neuronal–Epithelial Coculture Model in a Patterned Microfluidic Chip Culture System
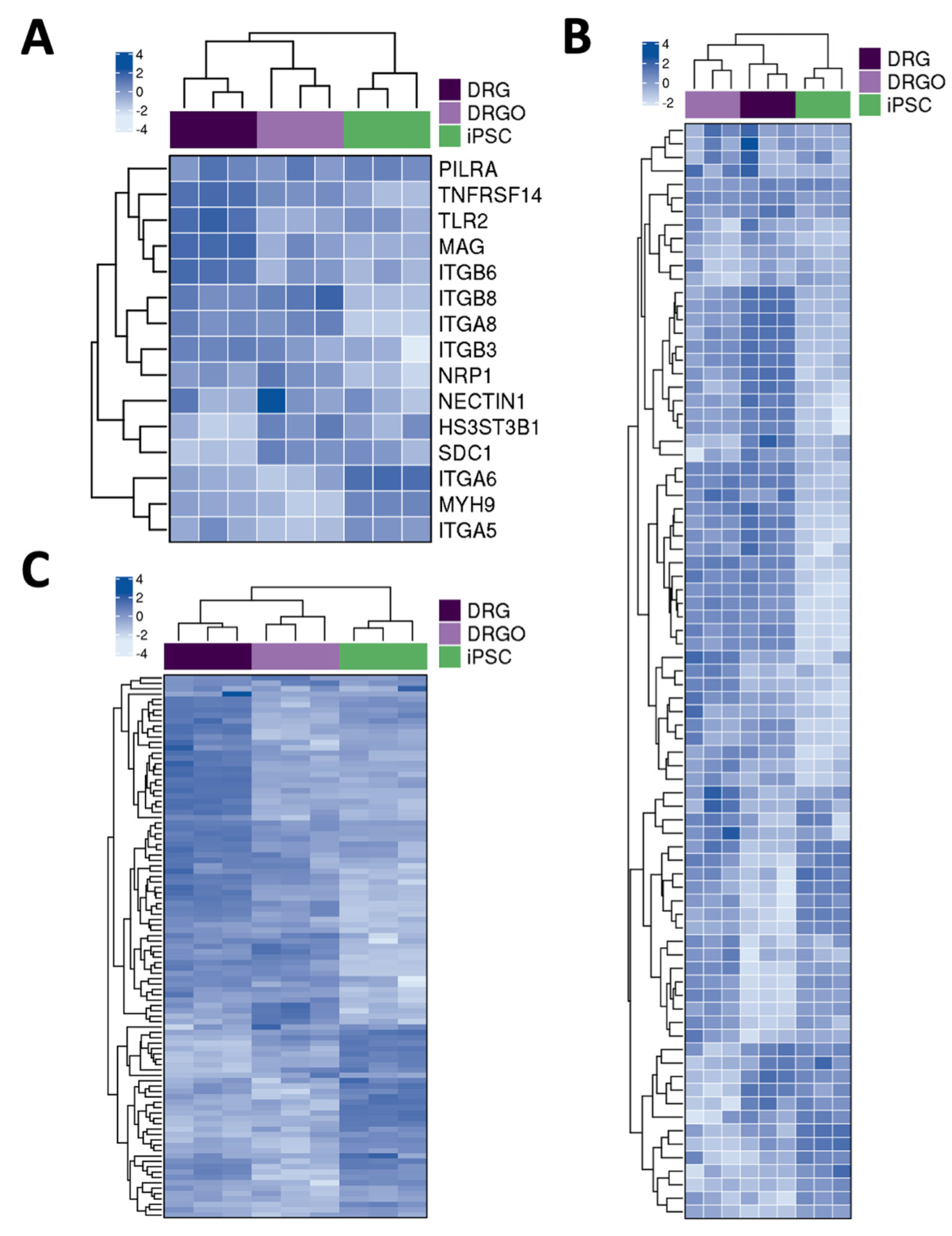
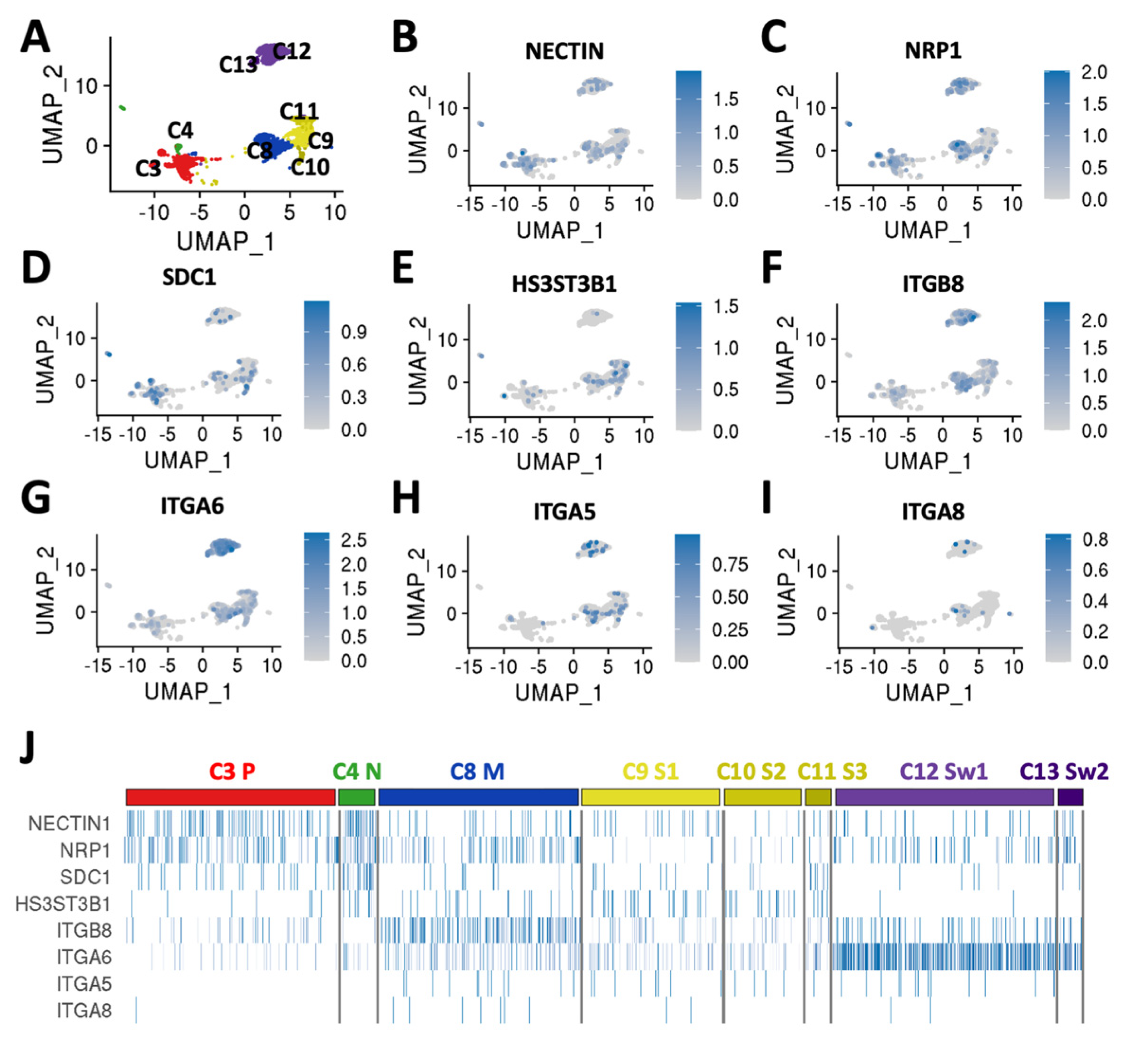
3.2. DRGO Gene Expression Competence to HSV-1 Infection
3.3. Validation of the Neuronal–Epithelial Microfluidic Platform for Viral Infectivity Studies
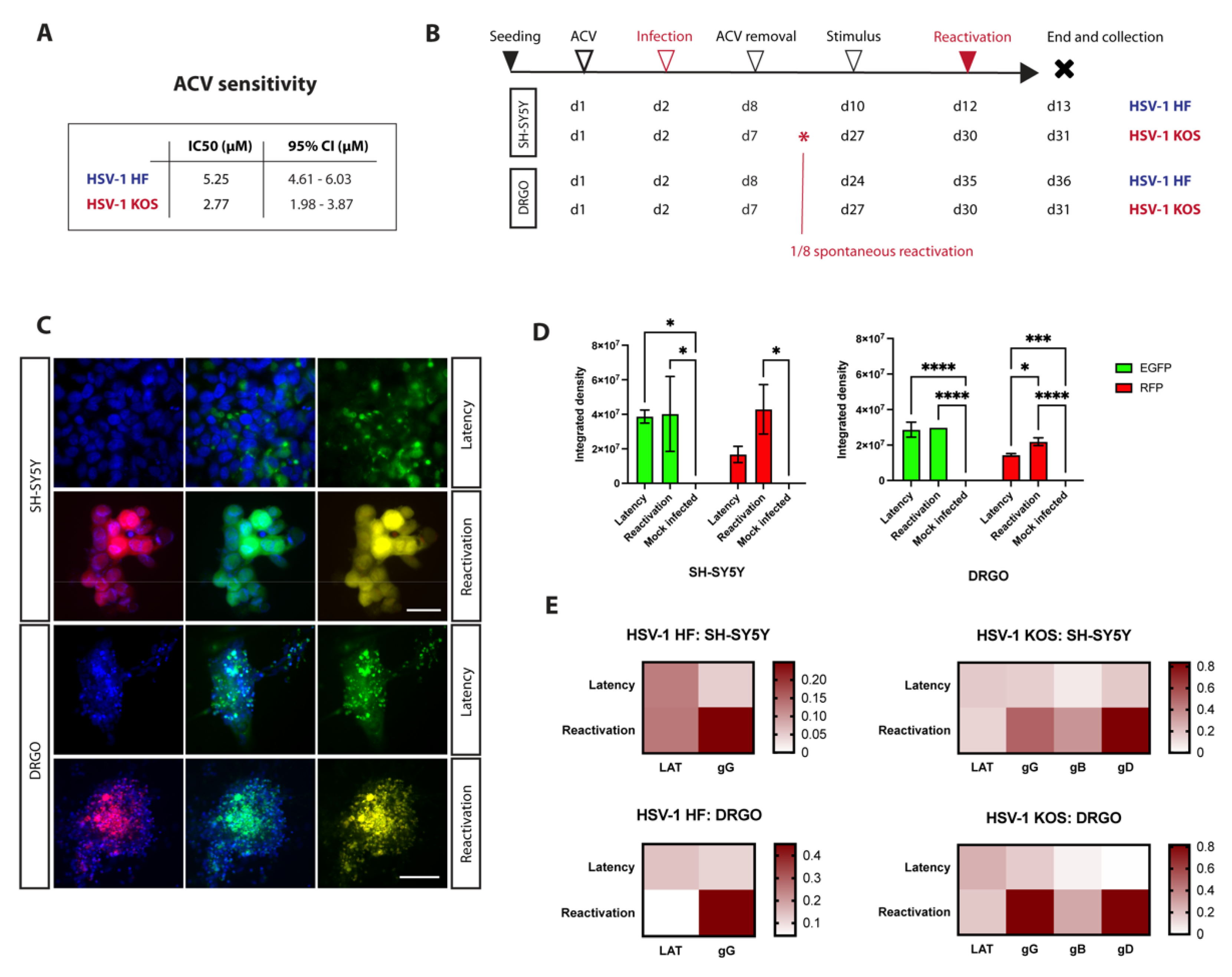
3.4. Successful Establishment of HSV-1 Latency in DRGO Neurons
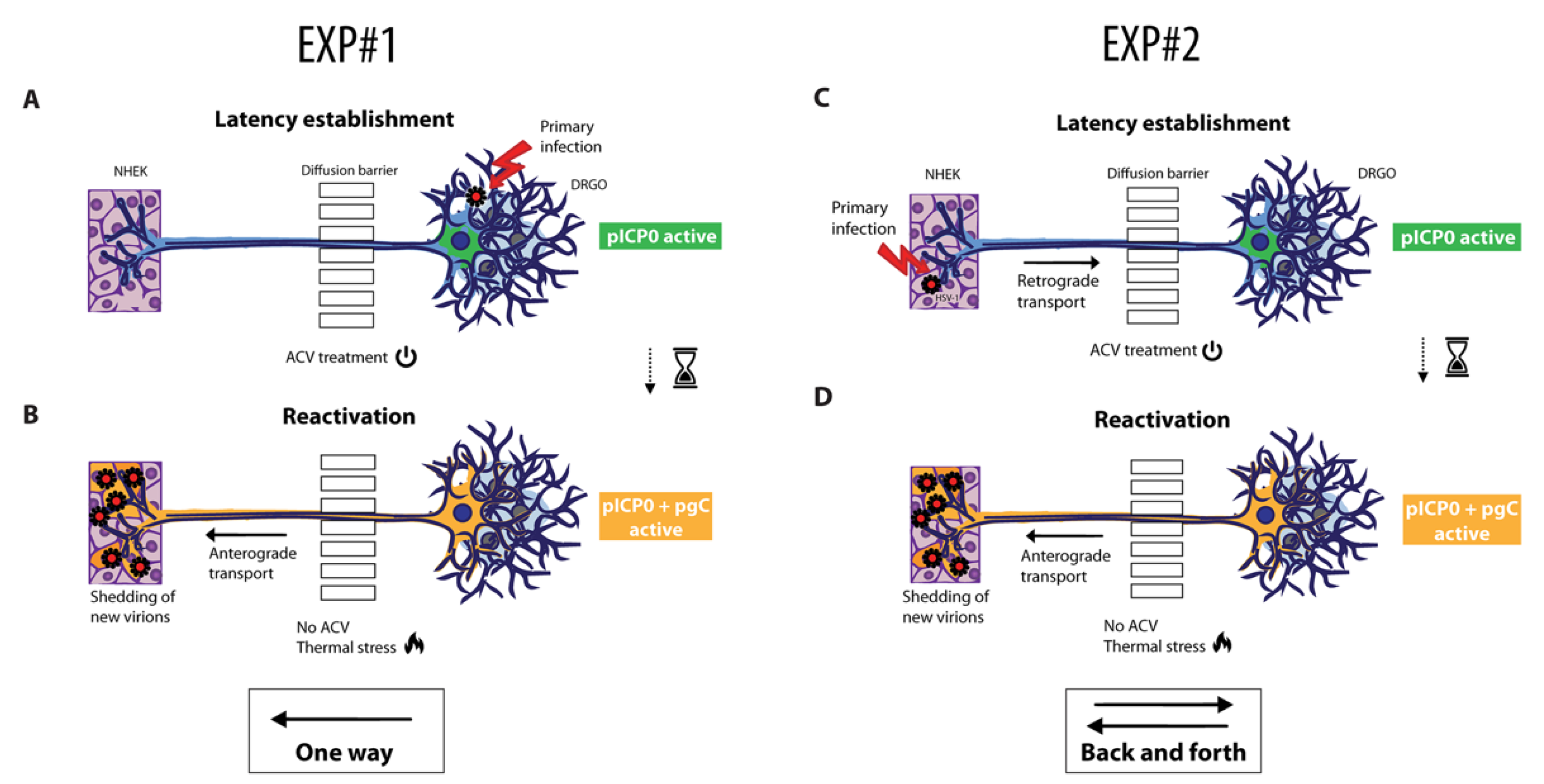
3.5. Experimental Settings for Latency and Reactivation in Microfluidic Culture System
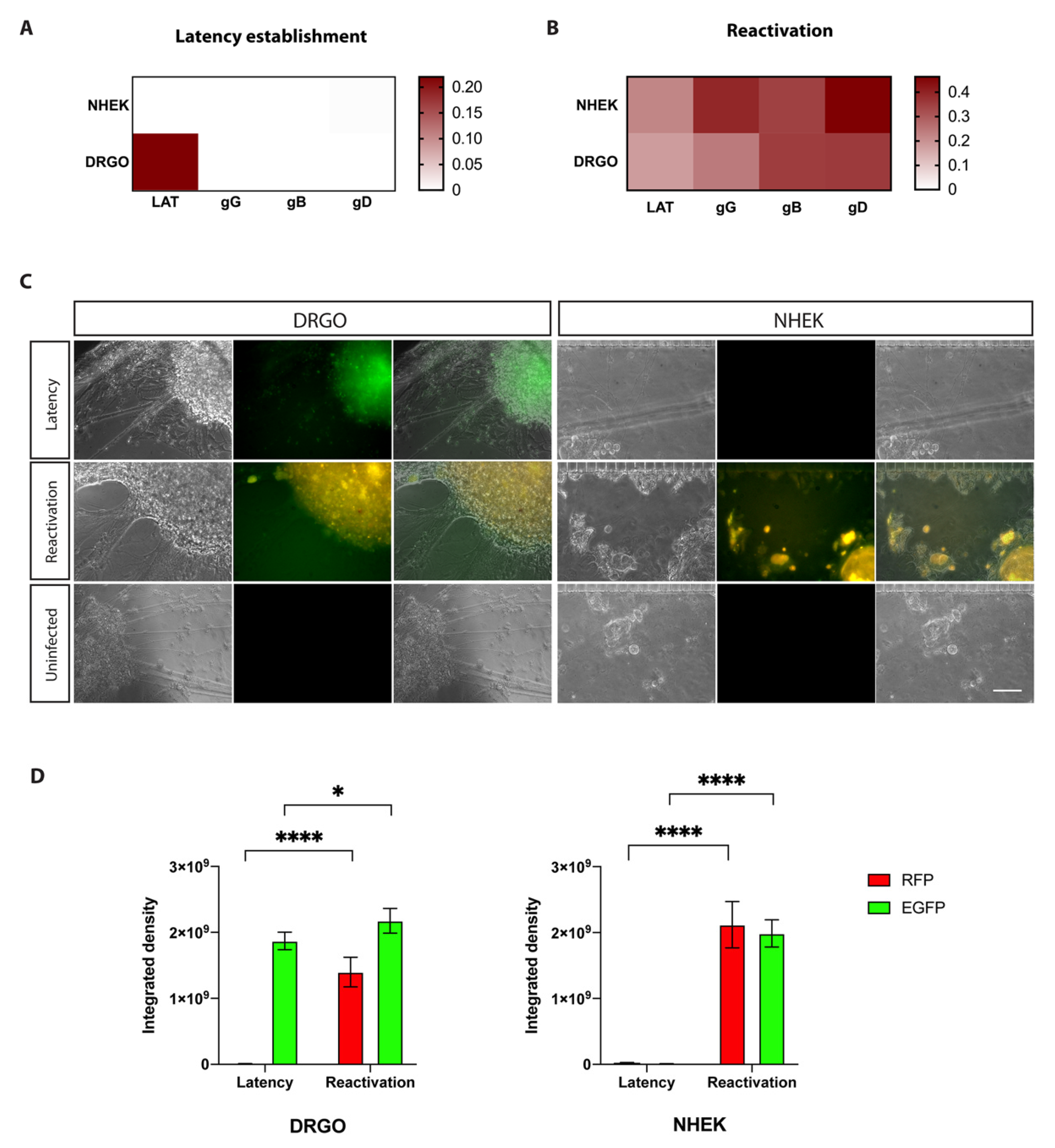
3.5.1. Anterograde Virus Spread in Neuronal–Epithelial Culture System
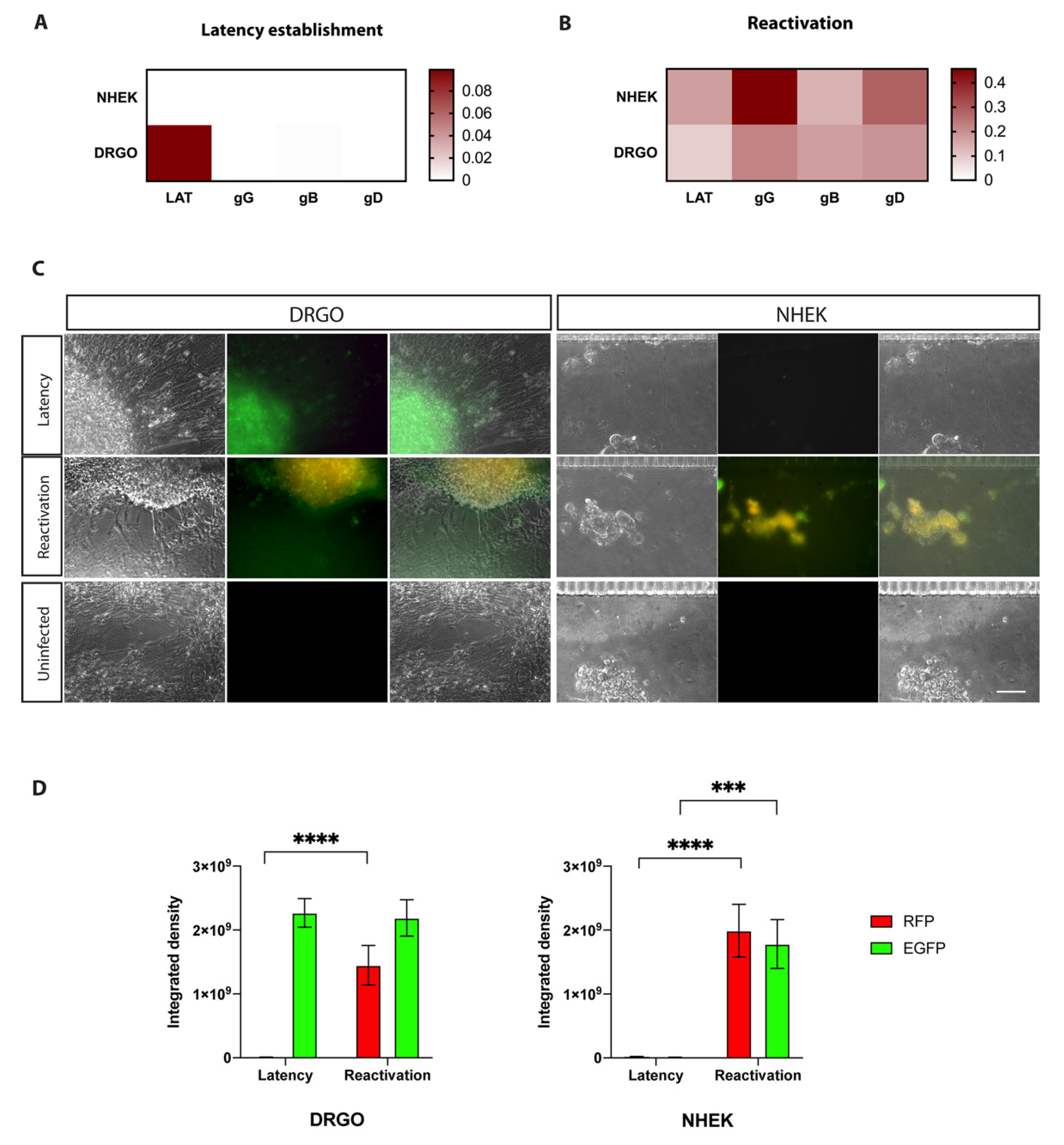
3.5.2. Validation of the Retrograde–Anterograde Virus Spread
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leger, A.J.S.; Koelle, D.M.; Kinchington, P.R.; Verjans, G.M.G.M. Local Immune Control of Latent Herpes Simplex Virus Type 1 in Ganglia of Mice and Man. Front. Immunol. 2021, 12, 723809. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Reyes, A.; Farías, M.A.; Riedel, C.A.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Crosstalk Between Epithelial Cells, Neurons and Immune Mediators in HSV-1 Skin Infection. Front. Immunol. 2021, 12, 662234. [Google Scholar] [CrossRef] [PubMed]
- BenMohamed, L.; Osorio, N.; Khan, A.A.; Srivastava, R.; Huang, L.; Krochmal, J.J.; Garcia, J.M.; Simpson, J.L.; Wechsler, S.L. Prior Corneal Scarification and Injection of Immune Serum Are Not Required Before Ocular HSV-1 Infection for UV-B-Induced Virus Reactivation and Recurrent Herpetic Corneal Disease in Latently Infected Mice. Curr. Eye Res. 2016, 41, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Berges, B.K.; Tanner, A. Modelling of Human Herpesvirus Infections in Humanized Mice. J. Gen. Virol. 2014, 95, 2106–2117. [Google Scholar] [CrossRef] [PubMed]
- Chiuppesi, F.; Vannucci, L.; Luca, A.D.; Lai, M.; Matteoli, B.; Freer, G.; Manservigi, R.; Ceccherini-Nelli, L.; Maggi, F.; Bendinelli, M.; et al. A Lentiviral Vector-Based, Herpes Simplex Virus 1 (HSV-1) Glycoprotein B Vaccine Affords Cross-Protection against HSV-1 and HSV-2 Genital Infections. J. Virol. 2012, 86, 6563–6574. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.-W.; Ling, P.; Tung, Y.-Y.; Hsu, S.-M.; Chen, S.-H. In Vivo Reactivation of Latent Herpes Simplex Virus 1 in Mice Can Occur in the Brain before Occurring in the Trigeminal Ganglion. J. Virol. 2014, 88, 11264–11270. [Google Scholar] [CrossRef]
- Xu, K.; Liu, X.-N.; Zhang, H.-B.; An, N.; Wang, Y.; Zhang, Z.-C.; Wang, Y.-N. Replication-Defective HSV-1 Effectively Targets Trigeminal Ganglion and Inhibits Viral Pathopoiesis by Mediating Interferon Gamma Expression in SH-SY5Y Cells. J. Mol. Neurosci. 2014, 53, 78–86. [Google Scholar] [CrossRef]
- Edwards, T.G.; Bloom, D.C. Lund Human Mesencephalic (LUHMES) Neuronal Cell Line Supports Herpes Simplex Virus 1 Latency In Vitro. J. Virol. 2019, 93, e02210-18. [Google Scholar] [CrossRef]
- Pourchet, A.; Modrek, A.; Placantonakis, D.; Mohr, I.; Wilson, A. Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons. Pathogens 2017, 6, 24. [Google Scholar] [CrossRef]
- Thellman, N.M.; Botting, C.; Madaj, Z.; Triezenberg, S.J. An Immortalized Human Dorsal Root Ganglion Cell Line Provides a Novel Context To Study Herpes Simplex Virus 1 Latency and Reactivation. J. Virol. 2017, 91, e00080-17. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Zhou, W.; Scott-McKean, J.J.; Emmerling, K.L.; Cai, G.; Krah, D.L.; Costa, A.C.; Freed, C.R.; Levin, M.J. Human Sensory Neurons Derived from Induced Pluripotent Stem Cells Support Varicella-Zoster Virus Infection. PLoS ONE 2012, 7, e53010. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, L.; Bloom, D.C.; Naciri, J.N.; Smith, A.; Edwards, T.G.; McClain, L.; Callio, J.A.; Jessup, M.; Wood, J.; Chowdari, K.; et al. Modeling HSV-1 Infections in Human CNS Neuronal Cells Using Two-Dimensional and Three- Dimensional Cultures Derived from Induced Pluripotent Stem Cells. J. Virol. 2019, 93, 1–56. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-P.; Qiu, Y.; Zhang, B.; Chen, G.; Chen, Q.; Wang, M.; Mo, F.; Xu, J.; Wu, J.; Zhang, R.-R.; et al. Zika Virus Infection Induces RNAi-Mediated Antiviral Immunity in Human Neural Progenitors and Brain Organoids. Cell Res. 2019, 29, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Sloutskin, A.; Kinchington, P.R.; Goldstein, R.S. Productive vs Non-Productive Infection by Cell-Free Varicella Zoster Virus of Human Neurons Derived from Embryonic Stem Cells Is Dependent upon Infectious Viral Dose. Virology 2013, 443, 285–293. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Davis, A.; Miranda-Saksena, M.; Fernandez, M.A.; Kelly, B.J.; Jones, C.A.; LaVail, J.H.; Xue, J.; Lai, J.; Cunningham, A.L. The Basic Domain of Herpes Simplex Virus 1 PUS9 Recruits Kinesin-1 To Facilitate Egress from Neurons. J. Virol. 2016, 90, 2102–2111. [Google Scholar] [CrossRef]
- Howard, P.W.; Wright, C.C.; Howard, T.; Johnson, D.C. Herpes Simplex Virus GE/GI Extracellular Domains Promote Axonal Transport and Spread from Neurons to Epithelial Cells. J. Virol. 2014, 88, 11178–11186. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Boadle, R.A.; Diefenbach, R.J.; Cunningham, A.L. Dual Role of Herpes Simplex Virus 1 PUS9 in Virus Anterograde Axonal Transport and Final Assembly in Growth Cones in Distal Axons. J. Virol. 2015, 90, 2653–2663. [Google Scholar] [CrossRef]
- Iannielli, A.; Ugolini, G.S.; Cordiglieri, C.; Bido, S.; Rubio, A.; Colasante, G.; Valtorta, M.; Cabassi, T.; Rasponi, M.; Broccoli, V. Reconstitution of the Human Nigro-Striatal Pathway on-a-Chip Reveals OPA1-Dependent Mitochondrial Defects and Loss of Dopaminergic Synapses. Cell Rep. 2019, 29, 4646–4656.e4. [Google Scholar] [CrossRef]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An In Vitro Model of Latency and Reactivation of Varicella Zoster Virus in Human Stem Cell-Derived Neurons. PLoS Pathog. 2015, 11, e1004885. [Google Scholar] [CrossRef]
- Mazzara, P.G.; Muggeo, S.; Luoni, M.; Massimino, L.; Zaghi, M.; Valverde, P.T.-T.; Brusco, S.; Marzi, M.J.; Palma, C.; Colasante, G.; et al. Frataxin Gene Editing Rescues Friedreich’s Ataxia Pathology in Dorsal Root Ganglia Organoid-Derived Sensory Neurons. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal Keratinization in a Spontaneously Immortalized Aneuploid Human Keratinocyte Cell Line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Klammer, A.M.; Naciri, J.N.; Yeung, J.; Demers, M.; Milosevic, J.; Kinchington, P.R.; Bloom, D.C.; Nimgaonkar, V.L.; D’Aiuto, L. Patterns of Herpes Simplex Virus 1 Infection in Neural Progenitor Cells. J. Virol. 2020, 94, e00994-20. [Google Scholar] [CrossRef] [PubMed]
- Decman, V.; Kinchington, P.R.; Harvey, S.A.K.; Hendricks, R.L. Gamma Interferon Can Block Herpes Simplex Virus Type 1 Reactivation from Latency, Even in the Presence of Late Gene Expression. J. Virol. 2005, 79, 10339–10347. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Knickelbein, J.E.; Ferko, C.; Hendricks, R.L.; Kinchington, P.R. Development and Pathogenic Evaluation of Recombinant Herpes Simplex Virus Type 1 Expressing Two Fluorescent Reporter Genes from Different Lytic Promoters. Virology 2008, 378, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Schang, L.M.; Phillips, J.; Schaffer, P.A. Requirement for Cellular Cyclin-Dependent Kinases in Herpes Simplex Virus Replication and Transcription. J. Virol. 1998, 72, 5626–5637. [Google Scholar] [CrossRef]
- Halford, W.P.; Gebhardt, B.M.; Carr, D.J. Mechanisms of Herpes Simplex Virus Type 1 Reactivation. J. Virol. 1996, 70, 5051–5060. [Google Scholar] [CrossRef]
- Lynas, C.; Laycock, K.A.; Cook, S.D.; Hill, T.J.; Blyth, W.A.; Maitland, N.J. Detection of Herpes Simplex Virus Type 1 Gene Expression in Latently and Productively Infected Mouse Ganglia Using the Polymerase Chain Reaction. J. Gen. Virol. 1989, 70, 2345–2355. [Google Scholar] [CrossRef]
- LeBlanc, R.A.; Pesnicak, L.; Godleski, M.; Straus, S.E. The Comparative Effects of Famciclovir and Valacyclovir on Herpes Simplex Virus Type 1 Infection, Latency, and Reactivation in Mice. J. Infect. Dis. 1999, 180, 594–599. [Google Scholar] [CrossRef]
- Grams, T.R.; Edwards, T.G.; Bloom, D.C. Herpes Simplex Virus 1 Strains 17 Syn + and KOS(M) Differ Greatly in Their Ability To Reactivate from Human Neurons In Vitro. J. Virol. 2020, 94, e00796-20. [Google Scholar] [CrossRef]
- Hill, J.M.; Field, M.A.R.; Haruta, Y. Strain Specificity of Spontaneous and Adrenergically Induced HSV-1 Ocular Reactivation in Latently Infected Rabbits. Curr. Eye Res. 2009, 6, 91–97. [Google Scholar] [CrossRef]
- Balliet, J.W.; Kushnir, A.S.; Schaffer, P.A. Construction and Characterization of a Herpes Simplex Virus Type I Recombinant Expressing Green Fluorescent Protein: Acute Phase Replication and Reactivation in Mice. Virology 2007, 361, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Person, S. Incorporation of the Green Fluorescent Protein into the Herpes Simplex Virus Type 1 Capsid. J. Virol. 1998, 72, 7563–7568. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Schmidt, E.E.; Halford, W.P. ICP0 Dismantles Microtubule Networks in Herpes Simplex Virus-Infected Cells. PLoS ONE 2010, 5, e10975. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J.; Antoine, T.E.; Farooq, A.V.; Valyi-Nagy, T.; Shukla, D. An Investigative Peptide–Acyclovir Combination to Control Herpes Simplex Virus Type 1 Ocular Infection. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6373–6379. [Google Scholar] [CrossRef]
- Sari, T.K.; Gianopulos, K.A.; Weed, D.J.; Schneider, S.M.; Pritchard, S.M.; Nicola, A.V. Herpes Simplex Virus Glycoprotein C Regulates Low-PH Entry. mSphere 2020, 5, e00826-19. [Google Scholar] [CrossRef]
- Taylor, A.M.; Blurton-Jones, M.; Rhee, S.W.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. A Microfluidic Culture Platform for CNS Ax-onal Injury, Regeneration and Transport. Nat. Methods 2005, 2, 599–605. [Google Scholar] [CrossRef]
- Gebhardt, B.M.; Halford, W.P. Evidence That Spontaneous Reactivation of Herpes Virus Does Not Occur in Mice. Virol. J. 2005, 2, 67. [Google Scholar] [CrossRef]
- Mott, K.R.; Chentoufi, A.A.; Carpenter, D.; BenMohamed, L.; Wechsler, S.L.; Ghiasi, H. The Role of a Glycoprotein K (GK) CD8+ T-Cell Epitope of Herpes Simplex Virus on Virus Replication and Pathogenicity. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2903–2912. [Google Scholar] [CrossRef]
- Zhang, X.; Castelli, F.A.; Zhu, X.; Wu, M.; Maillère, B.; BenMohamed, L. Gender-Dependent HLA-DR-Restricted Epitopes Identified from Herpes Simplex Virus Type 1 Glycoprotein D▿. Clin. Vaccine Immunol. 2008, 15, 1436–1449. [Google Scholar] [CrossRef]
- Chentoufi, A.A.; Zhang, X.; Lamberth, K.; Dasgupta, G.; Bettahi, I.; Nguyen, A.; Wu, M.; Zhu, X.; Mohebbi, A.; Buus, S.; et al. HLA-A*0201-Restricted CD8+ Cytotoxic T Lymphocyte Epitopes Identified from Herpes Simplex Virus Glycoprotein D. J. Immunol. 2008, 180, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Taneja, V.; David, C.S. HLA Class II Transgenic Mice as Models of Human Diseases. Immunol. Rev. 1999, 169, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Koelle, D.M.; Reymond, S.N.; Chen, H.; Kwok, W.W.; McClurkan, C.; Gyaltsong, T.; Petersdorf, E.W.; Rotkis, W.; Talley, A.R.; Harrison, D.A. Tegument-Specific, Virus-Reactive CD4 T Cells Localize to the Cornea in Herpes Simplex Virus Interstitial Keratitis in Humans. J. Virol. 2000, 74, 10930–10938. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, G.; BenMohamed, L. Of Mice and Not Humans: How Reliable Are Animal Models for Evaluation of Herpes CD8+-T Cell-Epitopes-Based Immunotherapeutic Vaccine Candidates? Vaccine 2011, 29, 5824–5836. [Google Scholar] [CrossRef] [PubMed]
- Belote, R.L.; Simon, S.M. Ca2+ Transients in Melanocyte Dendrites and Dendritic Spine-like Structures Evoked by Cell-to-Cell Signaling. J. Cell Biol. 2019, 219, e201902014. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Lee, L.Y.; Garber, D.A.; Schaffer, P.A.; Knipe, D.M.; Coen, D.M. Neither LAT nor Open Reading Frame P Mutations Increase Expression of Spliced or Intron-Containing ICP0 Transcripts in Mouse Ganglia Latently Infected with Herpes Simplex Virus. J. Virol. 2002, 76, 4764–4772. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.L.; Shieh, M.T.; Sawtell, N.M. Analysis of Herpes Simplex Virus ICP0 Promoter Function in Sensory Neurons during Acute Infection, Establishment of Latency, and Reactivation in Vivo. J. Virol. 2003, 77, 12319–12330. [Google Scholar] [CrossRef]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs Expressed by Herpes Simplex Virus 1 during Latent Infection Regulate Viral MRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Direction | Sequence | Amplicon (bp) | Ref. |
|---|---|---|---|---|
| BRN3A | forward | CGTACCACACGATGAACAGC | 123 | |
| reverse | AGGAGATGTGGTCCAGCAGA | |||
| NAV1.7 | forward | ACCTATCTCTGCTTCAAGTTGC | 90 | |
| reverse | TGGGCTGCTTGTCTACATTAAC | |||
| ACTB | forward | ACCCCAGCCATGTACGTT | 198 | |
| reverse | GGTGAGGATCTTCATGAGGTAG | |||
| ICP0-3′ | TCGACCAGGGCACCCTAGT | [26] | ||
| LAT | forward | GACAGCAAAAATCCCCTGAG | 192 | [27] |
| reverse | ACGAGGGAAAACAATAAGGG | |||
| gG | forward | CTGTTCTCGTTCCTCACTGCCT | 81 | [28] |
| reverse | CAAAAACGATAAGGTGTGGATGAC | |||
| gB | forward | CCAGTCGCCAGCACAAACTCG | 135 | |
| reverse | GCACACCACCGACCTCAAGTACAACC | |||
| gD | forward | CCTGTCCCATCCGAACGCAGC | 384 | |
| reverse | GCAGCAGGGTGCTCGTGTATGG | |||
| GADPH | forward | GAATCTACTGGCGTCTTCACC | 293 | |
| reverse | GTCATGAGCCCTTCCACGATGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzara, P.G.; Criscuolo, E.; Rasponi, M.; Massimino, L.; Muggeo, S.; Palma, C.; Castelli, M.; Clementi, M.; Burioni, R.; Mancini, N.; et al. A Human Stem Cell-Derived Neurosensory–Epithelial Circuitry on a Chip to Model Herpes Simplex Virus Reactivation. Biomedicines 2022, 10, 2068. https://doi.org/10.3390/biomedicines10092068
Mazzara PG, Criscuolo E, Rasponi M, Massimino L, Muggeo S, Palma C, Castelli M, Clementi M, Burioni R, Mancini N, et al. A Human Stem Cell-Derived Neurosensory–Epithelial Circuitry on a Chip to Model Herpes Simplex Virus Reactivation. Biomedicines. 2022; 10(9):2068. https://doi.org/10.3390/biomedicines10092068
Chicago/Turabian StyleMazzara, Pietro Giuseppe, Elena Criscuolo, Marco Rasponi, Luca Massimino, Sharon Muggeo, Cecilia Palma, Matteo Castelli, Massimo Clementi, Roberto Burioni, Nicasio Mancini, and et al. 2022. "A Human Stem Cell-Derived Neurosensory–Epithelial Circuitry on a Chip to Model Herpes Simplex Virus Reactivation" Biomedicines 10, no. 9: 2068. https://doi.org/10.3390/biomedicines10092068
APA StyleMazzara, P. G., Criscuolo, E., Rasponi, M., Massimino, L., Muggeo, S., Palma, C., Castelli, M., Clementi, M., Burioni, R., Mancini, N., Broccoli, V., & Clementi, N. (2022). A Human Stem Cell-Derived Neurosensory–Epithelial Circuitry on a Chip to Model Herpes Simplex Virus Reactivation. Biomedicines, 10(9), 2068. https://doi.org/10.3390/biomedicines10092068