
Monoclonal Antibodies for Bacterial Pathogens: Mechanisms of Action and Engineering Approaches for Enhanced Effector Functions


Abstract
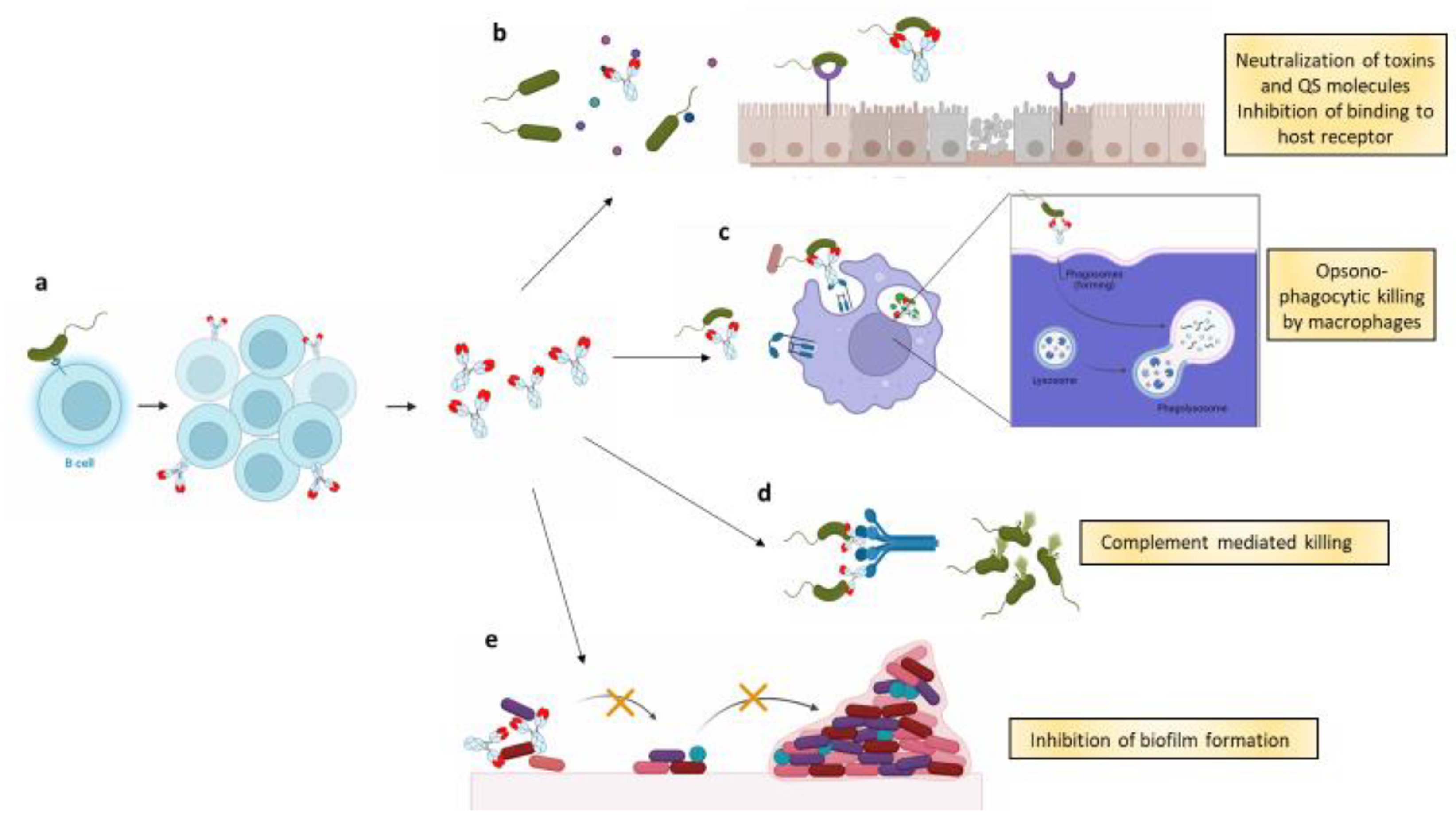
:1. Introduction
2. Mechanisms of Action of mAbs against Pathogenic Bacteria
3. Factors Influencing mAb Effector Functions
4. mAb Fc Engineering
4.1. Fc Engineering for Enhanced Receptor Engagement
4.2. Fc Engineering for Increasing Valency
4.3. Fc Glyco-Engineering
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, A.; Oxenius, A. Single B Cell Technologies for Monoclonal Antibody Discovery. Trends Immunol. 2021, 42, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Luciani, M.; Iannetti, L. Monoclonal Antibodies and Bacterial Virulence. Virulence 2017, 8, 635–636. [Google Scholar] [CrossRef] [PubMed]
- Speziale, P.; Pietrocola, G. Monoclonal Antibodies Targeting Surface-Exposed and Secreted Proteins from Staphylococci. Vaccines 2021, 9, 459. [Google Scholar] [CrossRef]
- Ojima-Kato, T.; Morishita, S.; Uchida, Y.; Nagai, S.; Kojima, T.; Nakano, H. Rapid Generation of Monoclonal Antibodies from Single B Cells by Ecobody Technology. Antibodies 2018, 7, 38. [Google Scholar] [CrossRef]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2018, 8, 1751. [Google Scholar] [CrossRef]
- Mullard, A. FDA Approves 100th Monoclonal Antibody Product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef]
- Castelli, M.S.; McGonigle, P.; Hornby, P.J. The Pharmacology and Therapeutic Applications of Monoclonal Antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Man, A.; Luo, H.; Levitskaya, S.V.; Macapagal, N.; Newell, K.J. Optimization of a Platform Process Operating Space for a Monoclonal Antibody Susceptible to Reversible and Irreversible Aggregation Using a Solution Stability Screening Approach. J. Chromatogr. A 2019, 1597, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The Therapeutic Monoclonal Antibody Market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.L.; Suscovich, T.J.; Fortune, S.M.; Alter, G. Beyond Binding: Antibody Effector Functions in Infectious Diseases. Nat. Rev. Immunol. 2018, 18, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed]
- van Erp, E.A.; Luytjes, W.; Ferwerda, G.; van Kasteren, P.B. Fc-Mediated Antibody Effector Functions During Respiratory Syncytial Virus Infection and Disease. Front. Immunol. 2019, 10, 548. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Chow, S.-K.; Abboud, N.; Casadevall, A.; Ravetch, J.V. Human IgG Fc Domain Engineering Enhances Antitoxin Neutralizing Antibody Activity. J. Clin. Investig. 2014, 124, 725–729. [Google Scholar] [CrossRef]
- Kang, T.H.; Jung, S.T. Boosting Therapeutic Potency of Antibodies by Taming Fc Domain Functions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Mimoto, F.; Kuramochi, T.; Katada, H.; Igawa, T.; Hattori, K. Fc Engineering to Improve the Function of Therapeutic Antibodies. Curr. Pharm. Biotechnol. 2016, 17, 1298–1314. [Google Scholar] [CrossRef]
- Berger, M.; Shankar, V.; Vafai, A. Therapeutic Applications of Monoclonal Antibodies. Am. J. Med. Sci. 2002, 324, 14–30. [Google Scholar] [CrossRef]
- Zurawski, D.V.; McLendon, M.K. Monoclonal Antibodies as an Antibacterial Approach Against Bacterial Pathogens. Antibiotics 2020, 9, 155. [Google Scholar] [CrossRef]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic Resistance in Microbes: History, Mechanisms, Therapeutic Strategies and Future Prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef]
- Tsai, C.-W.; Morris, S. Approval of Raxibacumab for the Treatment of Inhalation Anthrax Under the US Food and Drug Administration “Animal Rule”. Front. Microbiol. 2015, 6, 1320. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, S. Raxibacumab. MAbs 2009, 1, 531–538. [Google Scholar] [CrossRef]
- Yamamoto, B.J.; Shadiack, A.M.; Carpenter, S.; Sanford, D.; Henning, L.N.; Gonzales, N.; O’Connor, E.; Casey, L.S.; Serbina, N.V. Obiltoxaximab Prevents Disseminated Bacillus Anthracis Infection and Improves Survival during Pre- and Postexposure Prophylaxis in Animal Models of Inhalational Anthrax. Antimicrob. Agents Chemother. 2016, 60, 5796–5805. [Google Scholar] [CrossRef]
- Greig, S.L. Obiltoxaximab: First Global Approval. Drugs 2016, 76, 823–830. [Google Scholar] [CrossRef]
- Markham, A. Bezlotoxumab: First Global Approval. Drugs 2016, 76, 1793–1798. [Google Scholar] [CrossRef]
- Lee, Y.; Lim, W.I.; Bloom, C.I.; Moore, S.; Chung, E.; Marzella, N. Ezlotoxumab (Zinplava) for Clostridium Difficile Infection: The First Monoclonal Antibody Approved to Pre-Vent the Recurrence of a Bacterial Infection. Pharm. Ther. 2017, 42, 735–738. [Google Scholar]
- Brennan, F.R.; Cavagnaro, J.; McKeever, K.; Ryan, P.C.; Schutten, M.M.; Vahle, J.; Weinbauer, G.F.; Marrer-Berger, E.; Black, L.E. Safety Testing of Monoclonal Antibodies in Non-Human Primates: Case Studies Highlighting Their Impact on Human Risk Assessment. MAbs 2018, 10, 1–17. [Google Scholar] [CrossRef]
- Pal, S.; Tifrea, D.F.; de la Maza, L.M. Characterization of the Horizontal and Vertical Sexual Transmission of Chlamydia Genital Infections in a New Mouse Model. Infect. Immun. 2019, 87, e00834-18. [Google Scholar] [CrossRef]
- Rice, P.A.; Shafer, W.M.; Ram, S.; Jerse, A.E. Neisseria Gonorrhoeae: Drug Resistance, Mouse Models, and Vaccine Development. Annu. Rev. Microbiol. 2017, 71, 665–686. [Google Scholar] [CrossRef]
- Gulati, S.; Beurskens, F.J.; de Kreuk, B.-J.; Roza, M.; Zheng, B.; DeOliveira, R.B.; Shaughnessy, J.; Nowak, N.A.; Taylor, R.P.; Botto, M.; et al. Complement Alone Drives Efficacy of a Chimeric Antigonococcal Monoclonal Antibody. PLoS Biol. 2019, 17, e3000323. [Google Scholar] [CrossRef]
- Kaufmann, G.F.; Park, J.; Mee, J.M.; Ulevitch, R.J.; Janda, K.D. The Quorum Quenching Antibody RS2-1G9 Protects Macrophages from the Cytotoxic Effects of the Pseudomonas Aeruginosa Quorum Sensing Signalling Molecule N-3-Oxo-Dodecanoyl-Homoserine Lactone. Mol. Immunol. 2008, 45, 2710–2714. [Google Scholar] [CrossRef]
- Diago-Navarro, E.; Calatayud-Baselga, I.; Sun, D.; Khairallah, C.; Mann, I.; Ulacia-Hernando, A.; Sheridan, B.; Shi, M.; Fries, B.C. Antibody-Based Immunotherapy To Treat and Prevent Infection with Hypervirulent Klebsiella Pneumoniae. Clin. Vaccine Immunol. 2017, 24, e00456-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.H. Lipopolysaccharide: Basic Biochemistry, Intracellular Signaling, and Physiological Impacts in the Gut. Intest. Res. 2014, 12, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Thiem, S.; Steinert, M.; Purvis, D.; Lugmayr, V.; Treutlein, U.; Plobner, L.; Leiser, R.-M.; Hust, M.; Dübel, S. Human Anti-Lipopolysaccharid (LPS) Antibodies against Legionella with High Species Specificity. Hum. Antibodies 2019, 26, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.S.; Pelletier, M.; Cheng, L.; Pennini, M.E.; Bonnell, J.; Cvitkovic, R.; Chang, C.; Xiao, X.; Cameroni, E.; Corti, D.; et al. Anti-LPS Antibodies Protect against Klebsiella Pneumoniae by Empowering Neutrophil-Mediated Clearance without Neutralizing TLR4. JCI Insight 2017, 2, e92774. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.K.; Adams, F.G.; Brown, M.H. Diversity and Function of Capsular Polysaccharide in Acinetobacter Baumannii. Front. Microbiol. 2019, 9, 3301. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M.; Phan, H.M.; Watson, D.A.; Baughn, R.E. Antibody to Capsular Polysaccharide of Streptococcus Pneumoniae at the Time of Hospital Admission for Pneumococcal Pneumonia. J. Infect. Dis. 2000, 182, 158–167. [Google Scholar] [CrossRef]
- Avila-Calderón, E.D.; Ruiz-Palma, M.d.S.; Aguilera-Arreola, M.G.; Velázquez-Guadarrama, N.; Ruiz, E.A.; Gomez-Lunar, Z.; Witonsky, S.; Contreras-Rodríguez, A. Outer Membrane Vesicles of Gram-Negative Bacteria: An Outlook on Biogenesis. Front. Microbiol. 2021, 12, 557902. [Google Scholar] [CrossRef]
- van der Pol, L.; Stork, M.; van der Ley, P. Outer Membrane Vesicles as Platform Vaccine Technology. Biotechnol. J. 2015, 10, 1689–1706. [Google Scholar] [CrossRef]
- Pizarro-Cerdá, J.; Cossart, P. Bacterial Adhesion and Entry into Host Cells. Cell 2006, 124, 715–727. [Google Scholar] [CrossRef]
- Yougbare, I.; McTague, A.; He, L.; Choy, C.H.; Su, J.; Gajewska, B.; Azizi, A. Anti-FIM and Anti-FHA Antibodies Inhibit Bordetella Pertussis Growth and Reduce Epithelial Cell Inflammation Through Bacterial Aggregation. Front. Immunol. 2020, 11, 605273. [Google Scholar] [CrossRef]
- Leininger, E.; Probst, P.G.; Brennan, M.J.; Kenimer, J.G. Inhibition of Bordetella Pertussis Filamentous Hemagglutinin-Mediated Cell Adherence with Monoclonal Antibodies. FEMS Microbiol. Lett. 1993, 106, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Langen, H.; Naito, M.; Pieters, J. A Coat Protein on Phagosomes Involved in the Intracellular Survival of Mycobacteria. Cell 1999, 97, 435–447. [Google Scholar] [CrossRef]
- Kumar, S.K.; Singh, P.; Sinha, S. Naturally Produced Opsonizing Antibodies Restrict the Survival of Mycobacterium Tuberculosis in Human Macrophages by Augmenting Phagosome Maturation. Open Biol. 2015, 5, 150171. [Google Scholar] [CrossRef]
- Escobar, A.; Rodas, P.I.; Acuña-Castillo, C. Macrophage–Neisseria Gonorrhoeae Interactions: A Better Understanding of Pathogen Mechanisms of Immunomodulation. Front. Immunol. 2018, 9, 3044. [Google Scholar] [CrossRef] [PubMed]
- Kurbatfinski, N.; Goodman, S.D.; Bakaletz, L.O. A Humanized Monoclonal Antibody Potentiates Killing of Diverse Biofilm-Forming Respiratory Tract Pathogens by Antibiotics. Antimicrob. Agents Chemother. 2022, 66, e01877-21. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival Mechanisms of Clinically Relevant Microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus Biofilm: An Emerging Battleground in Microbial Communities. Antimicrob. Resist. Infect. Control. 2019, 8, 76. [Google Scholar] [CrossRef]
- Danese, P.N. Antibiofilm Approaches. Chem. Biol. 2002, 9, 873–880. [Google Scholar] [CrossRef]
- Sun, D.; Accavitti, M.A.; Bryers, J.D. Inhibition of Biofilm Formation by Monoclonal Antibodies against Staphylococcus Epidermidis RP62A Accumulation-Associated Protein. Clin. Vaccine Immunol. 2005, 12, 93–100. [Google Scholar] [CrossRef]
- de Vor, L.; van Dijk, B.; van Kessel, K.; Kavanaugh, J.S.; de Haas, C.; Aerts, P.C.; Viveen, M.C.; Boel, E.C.; Fluit, A.C.; Kwiecinski, J.M.; et al. Human Monoclonal Antibodies against Staphylococcus Aureus Surface Antigens Recognize In Vitro and In Vivo Biofilm. eLife 2022, 11, e67301. [Google Scholar] [CrossRef]
- Xiong, Y.Q.; Estellés, A.; Li, L.; Abdelhady, W.; Gonzales, R.; Bayer, A.S.; Tenorio, E.; Leighton, A.; Ryser, S.; Kauvar, L.M. A Human Biofilmdisrupting Monoclonal Antibody Potentiates Antibiotic Efficacy in Rodent Models of Both Staphylococcus Aureus and Acinetobacter Baumannii Infections. Antimicrob. Agents Chemother. 2017, 61, e00904-17. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez de Aldecoa, A.L.; Zafra, O.; González-Pastor, J.E. Mechanisms and Regulation of Extracellular DNA Release and Its Biological Roles in Microbial Communities. Front. Microbiol. 2017, 8, 1390. [Google Scholar] [CrossRef]
- Miller, M.B.; Bassler, B.L. Quorum Sensing in Bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef]
- Prescott, R.D.; Decho, A.W. Flexibility and Adaptability of Quorum Sensing in Nature. Trends Microbiol. 2020, 28, 436–444. [Google Scholar] [CrossRef]
- DiGiandomenico, A.; Keller, A.E.; Gao, C.; Rainey, G.J.; Warrener, P.; Camara, M.M.; Bonnell, J.; Fleming, R.; Bezabeh, B.; Dimasi, N.; et al. A Multifunctional Bispecific Antibody Protects against Pseudomonas Aeruginosa. Sci. Transl. Med. 2014, 6, 262ra155. [Google Scholar] [CrossRef]
- Tkaczyk, C.; Kasturirangan, S.; Minola, A.; Jones-Nelson, O.; Gunter, V.; Shi, Y.Y.; Rosenthal, K.; Aleti, V.; Semenova, E.; Warrener, P.; et al. Multimechanistic Monoclonal Antibodies (MAbs) Targeting Staphylococcus Aureus Alpha-Toxin and Clumping Factor A: Activity and Efficacy Comparisons of a MAb Combination and an Engineered Bispecific Antibody Approach. Antimicrob. Agents Chemother. 2017, 61, e00629-17. [Google Scholar] [CrossRef]
- Cleary, K.L.S.; Chan, H.T.C.; James, S.; Glennie, M.J.; Cragg, M.S. Antibody Distance from the Cell Membrane Regulates Antibody Effector Mechanisms. J. Immunol. 2017, 198, 3999–4011. [Google Scholar] [CrossRef]
- Shim, H. One Target, Different Effects: A Comparison of Distinct Therapeutic Antibodies against the Same Targets. Exp. Mol. Med. 2011, 43, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Bakalar, M.H.; Joffe, A.M.; Schmid, E.M.; Son, S.; Podolski, M.; Fletcher, D.A. Size-Dependent Segregation Controls Macrophage Phagocytosis of Antibody-Opsonized Targets. Cell 2018, 174, 131–142.e13. [Google Scholar] [CrossRef]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered Antibody Fc Variants with Enhanced Effector Function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef]
- Sips, M.; Krykbaeva, M.; Diefenbach, T.J.; Ghebremichael, M.; Bowman, B.A.; Dugast, A.-S.; Boesch, A.W.; Streeck, H.; Kwon, D.S.; Ackerman, M.E.; et al. Fc Receptor-Mediated Phagocytosis in Tissues as a Potent Mechanism for Preventive and Therapeutic HIV Vaccine Strategies. Mucosal Immunol. 2016, 9, 1584–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, G.L.; Chen, H.; Karki, S.; Lazar, G.A. Engineered Fc Variant Antibodies with Enhanced Ability to Recruit Complement and Mediate Effector Functions. MAbs 2010, 2, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax Toxin Protective Antigen Is Activated by a Cell Surface Protease with the Sequence Specificity and Catalytic Properties of Furin. Proc. Natl. Acad. Sci. USA 1992, 89, 10277–10281. [Google Scholar] [CrossRef]
- de Jong, R.N.; Beurskens, F.J.; Verploegen, S.; Strumane, K.; van Kampen, M.D.; Voorhorst, M.; Horstman, W.; Engelberts, P.J.; Oostindie, S.C.; Wang, G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. [Google Scholar] [CrossRef]
- Zwarthoff, S.A.; Widmer, K.; Kuipers, A.; Strasser, J.; Ruyken, M.; Aerts, P.C.; de Haas, C.J.C.; Ugurlar, D.; den Boer, M.A.; Vidarsson, G.; et al. C1q Binding to Surface-Bound IgG Is Stabilized by C1r 2 s 2 Proteases. Proc. Natl. Acad. Sci. USA 2021, 118, e2102787118. [Google Scholar] [CrossRef]
- Berends, E.T.M.; Dekkers, J.F.; Nijland, R.; Kuipers, A.; Soppe, J.A.; van Strijp, J.A.G.; Rooijakkers, S.H.M. Distinct Localization of the Complement C5b-9 Complex on Gram-Positive Bacteria. Cell. Microbiol. 2013, 15, 1955–1968. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Pelletier, M.; Cvitkovic, R.; Bonnell, J.; Chang, C.-Y.; Koksal, A.C.; O’Connor, E.; Gao, X.; Yu, X.-Q.; et al. Enhancement of Antibody Functions through Fc Multiplications. MAbs 2017, 9, 393–403. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative Molecular Formats and Therapeutic Applications for Bispecific Antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Liu, H.; Saxena, A.; Sidhu, S.S.; Wu, D. Fc Engineering for Developing Therapeutic Bispecific Antibodies and Novel Scaffolds. Front. Immunol. 2017, 8, 38. [Google Scholar] [CrossRef]
- Zheng, K.; Yarmarkovich, M.; Bantog, C.; Bayer, R.; Patapoff, T.W. Influence of Glycosylation Pattern on the Molecular Properties of Monoclonal Antibodies. MAbs 2014, 6, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The Impact of Glycosylation on the Biological Function and Structure of Human Immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef]
- Wang, T.T.; Ravetch, J.V. Functional Diversification of IgGs through Fc Glycosylation. J. Clin. Investig. 2019, 129, 3492–3498. [Google Scholar] [CrossRef]
- Ferrara, C.; Grau, S.; Jäger, C.; Sondermann, P.; Brünker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique Carbohydrate–Carbohydrate Interactions Are Required for High Affinity Binding between FcγRIII and Antibodies Lacking Core Fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef]
- Ackerman, M.E.; Crispin, M.; Yu, X.; Baruah, K.; Boesch, A.W.; Harvey, D.J.; Dugast, A.-S.; Heizen, E.L.; Ercan, A.; Choi, I.; et al. Natural Variation in Fc Glycosylation of HIV-Specific Antibodies Impacts Antiviral Activity. J. Clin. Investig. 2013, 123, 2183–2192. [Google Scholar] [CrossRef]
- Irvine, E.B.; Alter, G. Understanding the Role of Antibody Glycosylation through the Lens of Severe Viral and Bacterial Diseases. Glycobiology 2020, 30, 241–253. [Google Scholar] [CrossRef]
- Lu, L.L.; Chung, A.W.; Rosebrock, T.R.; Ghebremichael, M.; Yu, W.H.; Grace, P.S.; Schoen, M.K.; Tafesse, F.; Martin, C.; Leung, V.; et al. A Functional Role for Antibodies in Tuberculosis. Cell 2016, 167, 433–443.e14. [Google Scholar] [CrossRef]
- Chen, X.; Shi, M.; Tong, X.; Kim, H.K.; Wang, L.-X.; Schneewind, O.; Missiakas, D. Glycosylation-Dependent Opsonophagocytic Activity of Staphylococcal Protein A Antibodies. Proc. Natl. Acad. Sci. USA 2020, 117, 22992–23000. [Google Scholar] [CrossRef]


{kind=link}
| Fc Engineering | Mechanism | Reference |
|---|---|---|
| Fc engineering for enhanced effector engagement | Selective engagement of particular classes of human FcγRs | [15] |
| Fc mutation leading to hexamerization upon antigen binding. This leads to greater engagement of Fc to C1q and C3b | [57,58] | |
| Fc portion multiplication, leading to incremental binding to FcgR, FcRn, and C1q through incremented avidity effect | [60] | |
| Fc engineering for increased valency | Antibody carries two different Fabs, each specific for one antigen | [47] |
| Fc glycol-engineering | Addition of oligosaccharides to a conserved Asn297 N-glycosylation site in the heavy chain Fc region | [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vacca, F.; Sala, C.; Rappuoli, R. Monoclonal Antibodies for Bacterial Pathogens: Mechanisms of Action and Engineering Approaches for Enhanced Effector Functions. Biomedicines 2022, 10, 2126. https://doi.org/10.3390/biomedicines10092126
Vacca F, Sala C, Rappuoli R. Monoclonal Antibodies for Bacterial Pathogens: Mechanisms of Action and Engineering Approaches for Enhanced Effector Functions. Biomedicines. 2022; 10(9):2126. https://doi.org/10.3390/biomedicines10092126
Chicago/Turabian StyleVacca, Fabiola, Claudia Sala, and Rino Rappuoli. 2022. "Monoclonal Antibodies for Bacterial Pathogens: Mechanisms of Action and Engineering Approaches for Enhanced Effector Functions" Biomedicines 10, no. 9: 2126. https://doi.org/10.3390/biomedicines10092126
APA StyleVacca, F., Sala, C., & Rappuoli, R. (2022). Monoclonal Antibodies for Bacterial Pathogens: Mechanisms of Action and Engineering Approaches for Enhanced Effector Functions. Biomedicines, 10(9), 2126. https://doi.org/10.3390/biomedicines10092126