

Transferrin-Modified Triptolide Liposome Targeting Enhances Anti-Hepatocellular Carcinoma Effects
 ,
,
Abstract
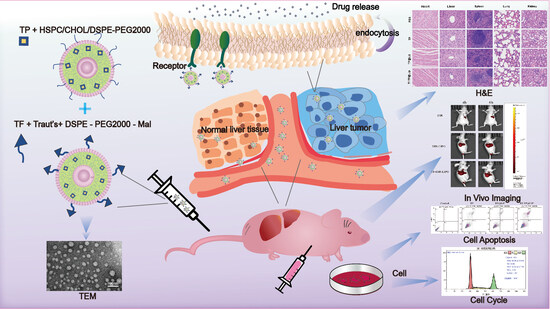
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Animals
2.4. Preparation of Triptolide Liposomes (TP@LIP) and Triptolide Liposomes Modified by Transferrin (TF-TP@LIP)
2.5. Characterization of Liposomes
2.6. In Vitro Drug Release from LIPs
2.7. Hemolytic Examination of TF-TP@LIP
2.8. Cellular Uptake
2.9. In Vitro Cytotoxicity
2.10. Analysis of Apoptosis and the Cell Cycle by Flow Cytometry
2.11. Animal Tumor Models
2.12. Live Fluorescence Imaging
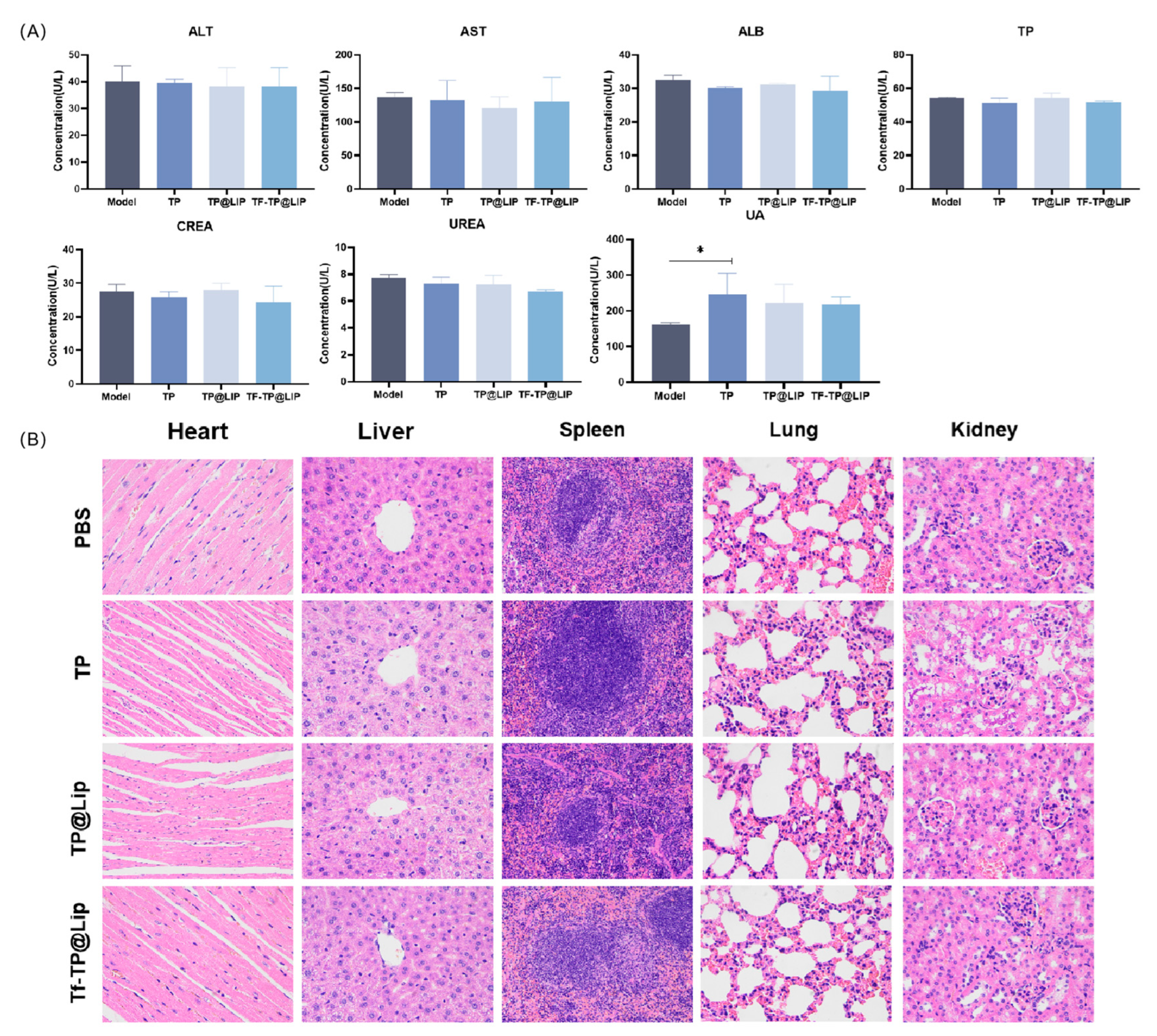
2.13. Safety Evaluation
2.14. Statistical Analysis
3. Results and Discussion
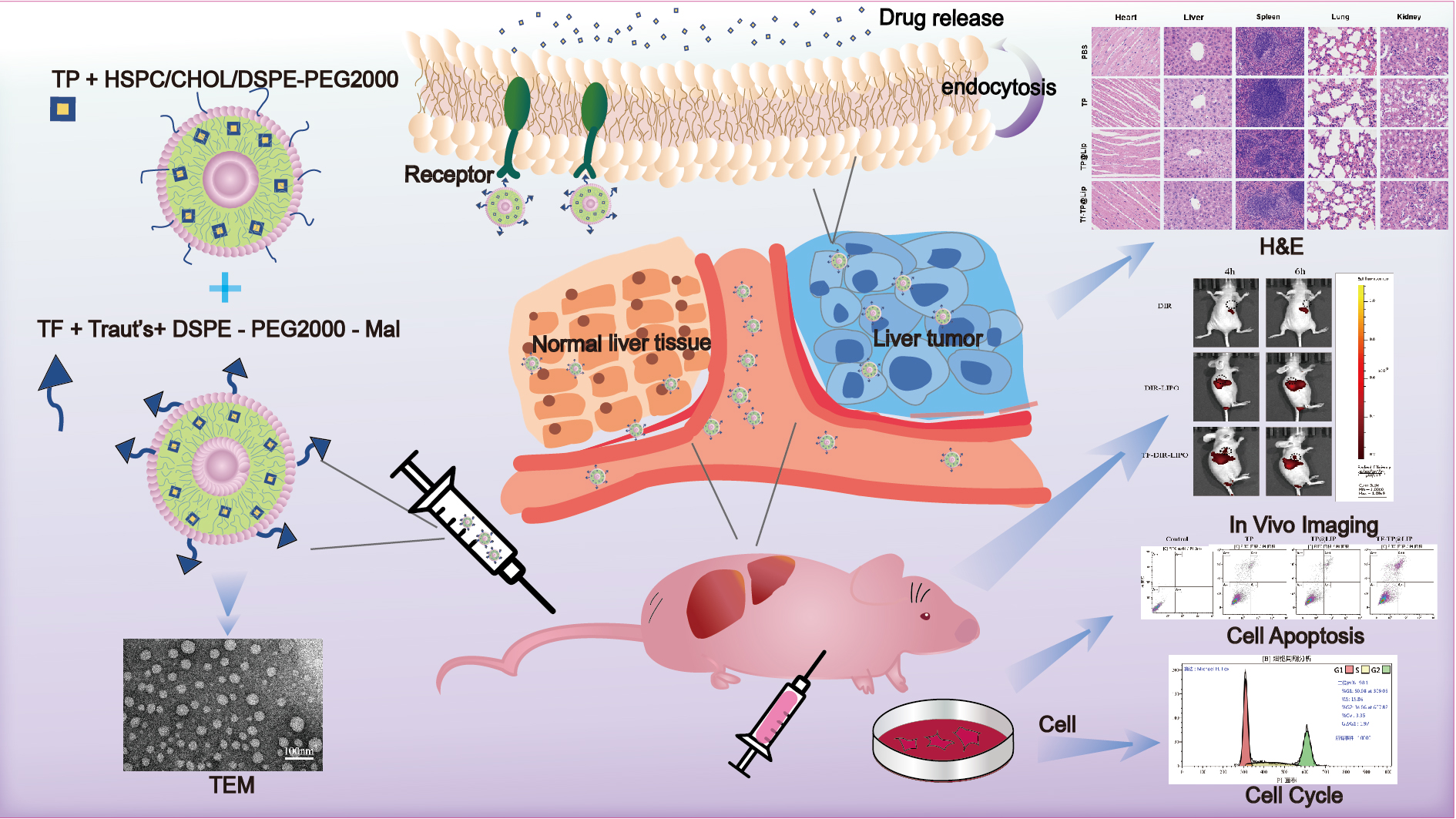
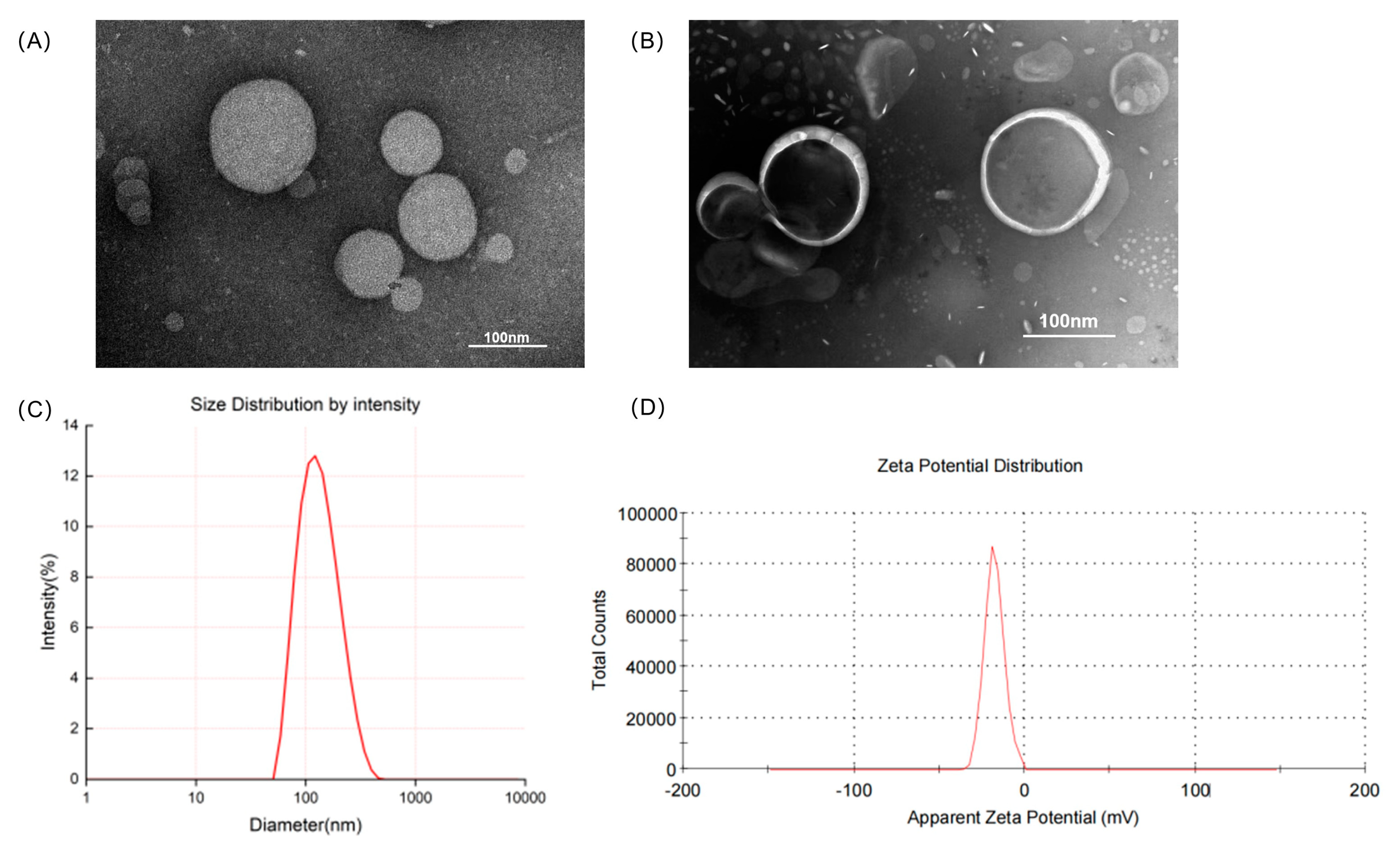
3.1. Characterization of the Liposomes
3.2. In Vitro Release of TF-TP@LIP
3.3. Hemolytic Activity of Liposomes
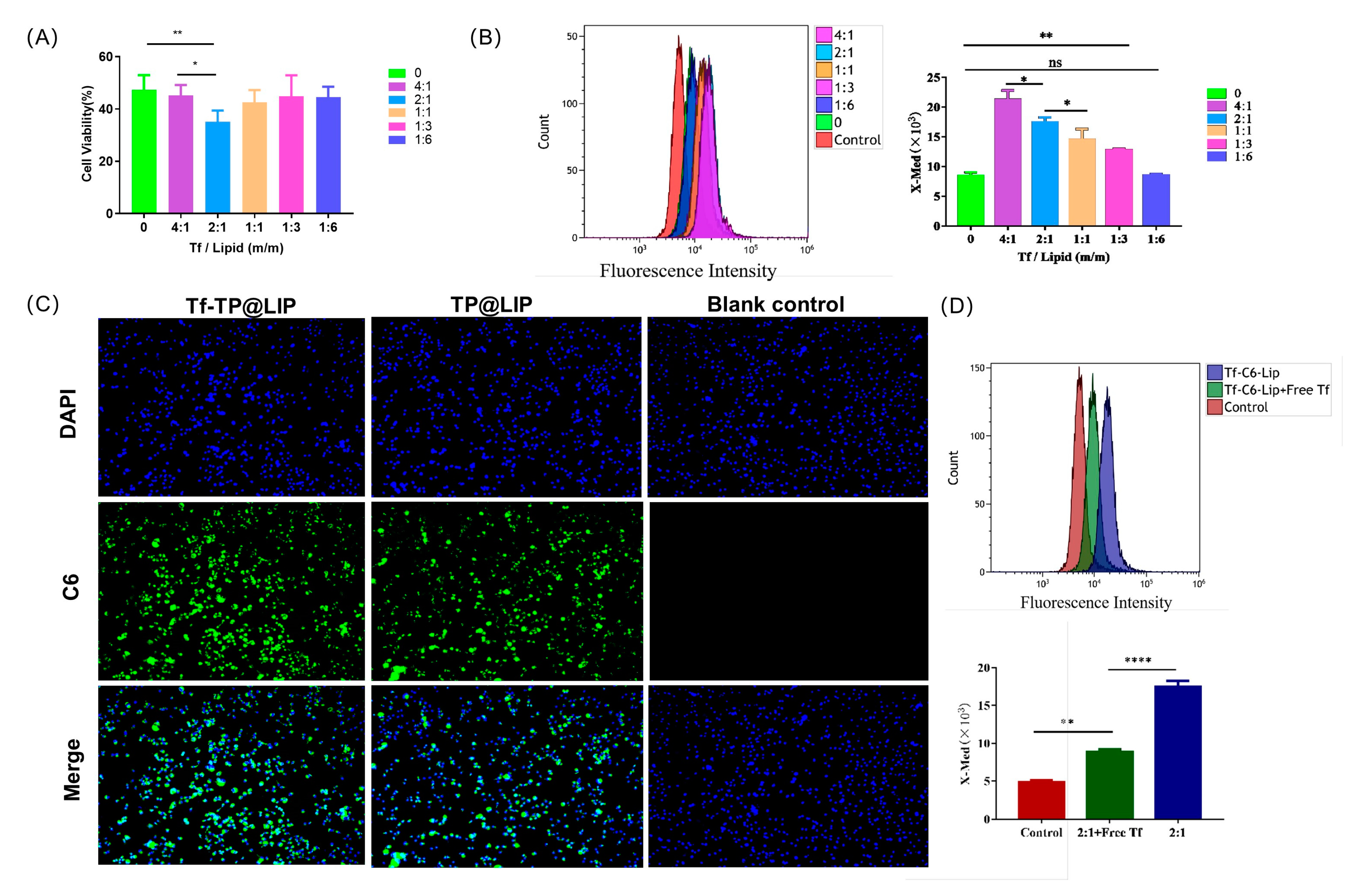
3.4. Cellular Uptake and Targeting Effects In Vitro
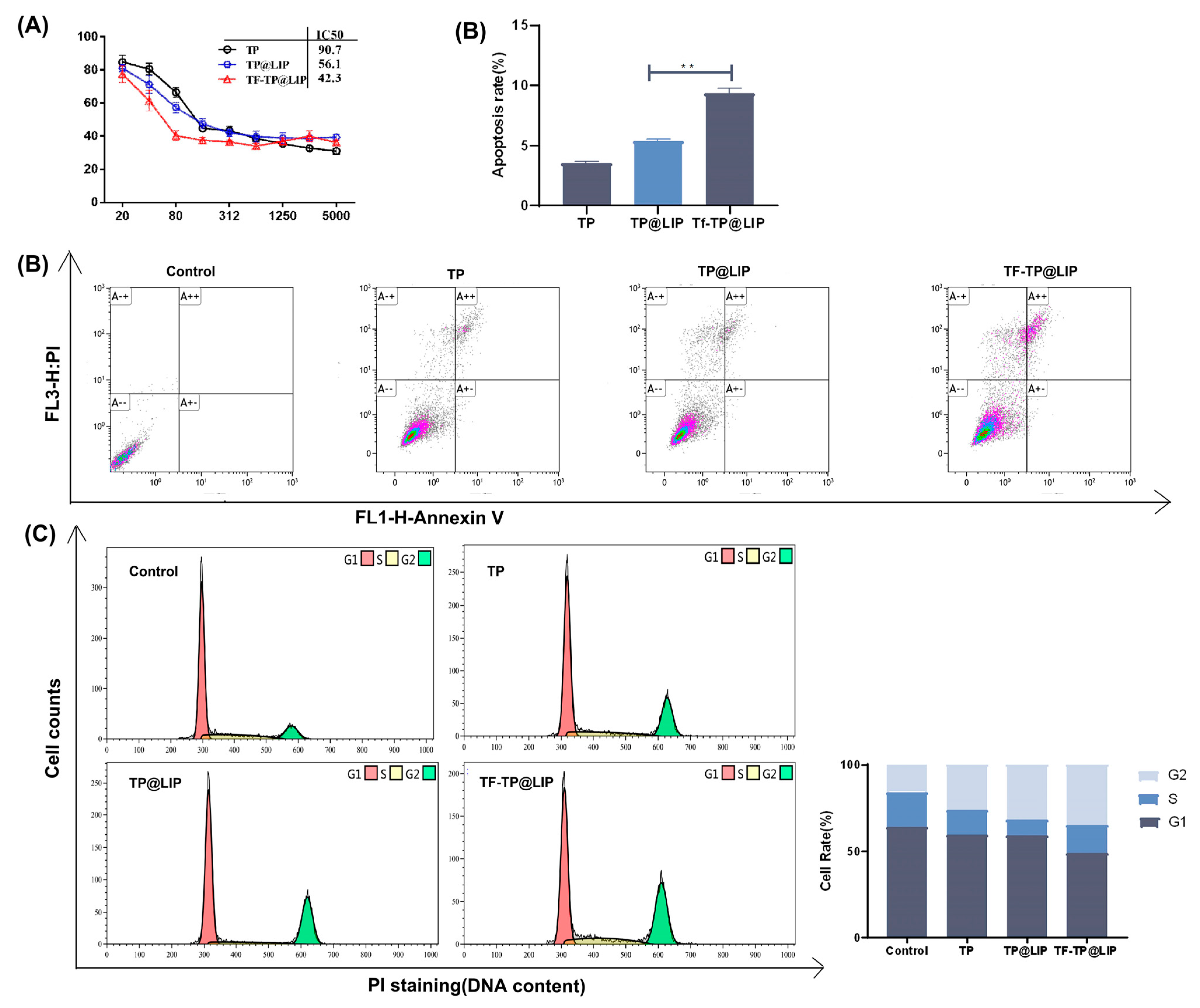
3.5. Anti-tumor Effects of TF-TP@LIP In Vitro
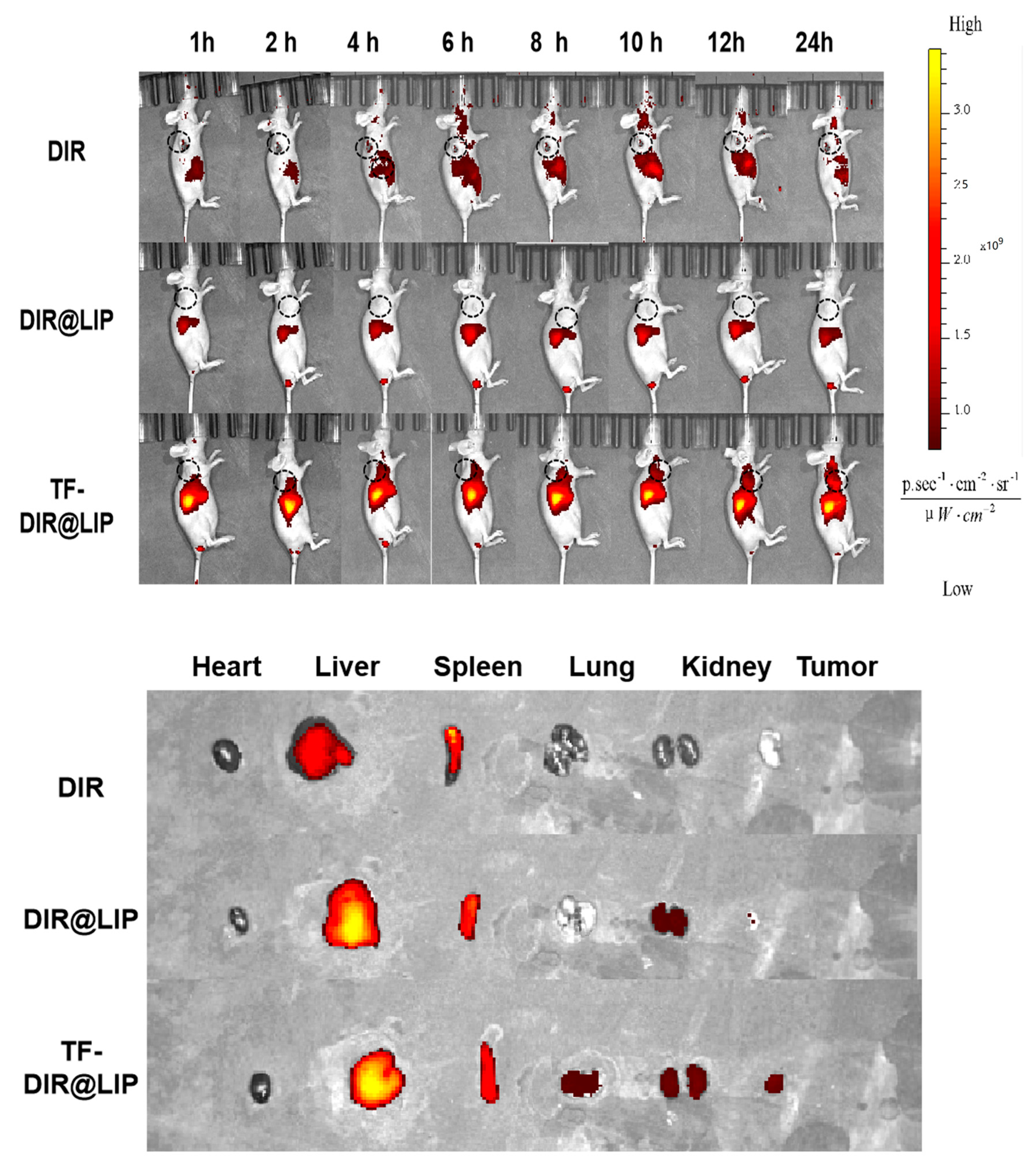
3.6. In Vivo Real-Time Imaging
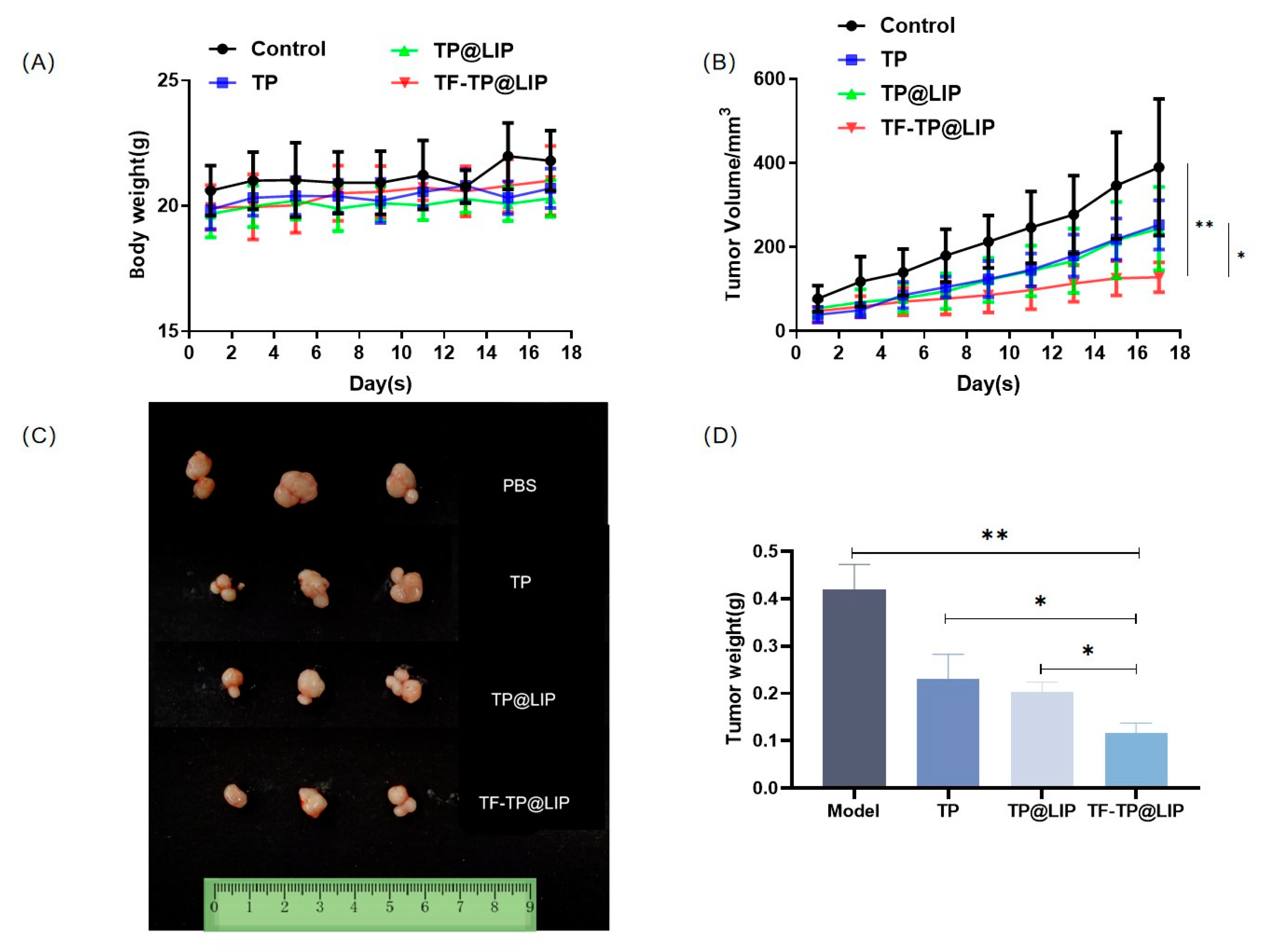
3.7. In Vivo Anti-Hepatoma Efficacy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ladju, R.B.; Ulhaq, Z.S.; Soraya, G.V. Nanotheranostics: A powerful next-generation solution to tackle hepatocellular carcinoma. World J. Gastroenterol. 2022, 28, 176–187. [Google Scholar] [CrossRef]
- Baffy, G. The impact of network medicine in gastroenterology and hepatology. Clin. Gastroenterol. Hepatol. 2013, 11, 1240–1244. [Google Scholar] [CrossRef]
- Maluccio, M.; Covey, A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J. Clin. 2012, 62, 394–399. [Google Scholar] [CrossRef]
- Zhao, C.; Wu, X.; Chen, J.; Qian, G. The therapeutic effect of IL-21 combined with IFN-γ inducing CD4(+)CXCR5(+)CD57(+)T cells differentiation on hepatocellular carcinoma. J. Adv. Res. 2022, 36, 89–99. [Google Scholar] [CrossRef]
- Li, Z.; Yang, G.; Han, L.; Wang, R.; Gong, C.; Yuan, Y. Sorafenib and triptolide loaded cancer cell-platelet hybrid membrane-camouflaged liquid crystalline lipid nanoparticles for the treatment of hepatocellular carcinoma. J. Nanobiotechnol. 2021, 19, 360. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Court, W.A.; Dailey, R.G., Jr.; Gilmore, C.J.; Bryan, R.F. Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J. Am. Chem. Soc. 1972, 94, 7194–7195. [Google Scholar] [CrossRef]
- Hou, W.; Liu, B.; Xu, H. Triptolide: Medicinal chemistry, chemical biology and clinical progress. Eur. J. Med. Chem. 2019, 176, 378–392. [Google Scholar] [CrossRef]
- Kim, S.T.; Kim, S.Y.; Lee, J.; Kim, K.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Park, J.O. Triptolide as a novel agent in pancreatic cancer: The validation using patient derived pancreatic tumor cell line. BMC Cancer 2018, 18, 1103. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Shen, Y.; Liao, M.M.; Mao, X.; Mi, G.J.; You, C.; Guo, Q.Y.; Li, W.J.; Wang, X.Y.; Lin, N.; et al. Galactosylated chitosan triptolide nanoparticles for overcoming hepatocellular carcinoma: Enhanced therapeutic efficacy, low toxicity, and validated network regulatory mechanisms. Nanomedicine 2019, 15, 86–97. [Google Scholar] [CrossRef]
- Meng, C.; Zhu, H.; Song, H.; Wang, Z.; Huang, G.; Li, D.; Ma, Z.; Ma, J.; Qin, Q.; Sun, X.; et al. Targets and molecular mechanisms of triptolide in cancer therapy. Chin. J. Cancer Res. 2014, 26, 622–626. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, Y.I. Triptolide induces apoptosis of PMA-treated THP-1 cells through activation of caspases, inhibition of NF-κB and activation of MAPKs. Int. J. Oncol. 2013, 43, 1169–1175. [Google Scholar] [CrossRef]
- Liu, H.J.; Wang, M.; Hu, X.; Shi, S.; Xu, P. Enhanced Photothermal Therapy through the In Situ Activation of a Temperature and Redox Dual-Sensitive Nanoreservoir of Triptolide. Small 2020, 16, e2003398. [Google Scholar] [CrossRef]
- Pefanis, E.; Wang, J.; Rothschild, G.; Lim, J.; Kazadi, D.; Sun, J.; Federation, A.; Chao, J.; Elliott, O.; Liu, Z.P.; et al. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell 2015, 161, 774–789. [Google Scholar] [CrossRef]
- Qin, G.; Li, P.; Xue, Z. Triptolide induces protective autophagy and apoptosis in human cervical cancer cells by downregulating Akt/mTOR activation. Oncol. Lett. 2018, 16, 3929–3934. [Google Scholar] [CrossRef]
- Fu, Q.; Huang, X.; Shu, B.; Xue, M.; Zhang, P.; Wang, T.; Liu, L.; Jiang, Z.; Zhang, L. Inhibition of mitochondrial respiratory chain is involved in triptolide-induced liver injury. Fitoterapia 2011, 82, 1241–1248. [Google Scholar] [CrossRef]
- Cao, L.J.; Hou, Z.Y.; Li, H.D.; Zhang, B.K.; Fang, P.F.; Xiang, D.X.; Li, Z.H.; Gong, H.; Deng, Y.; Ma, Y.X.; et al. The Ethanol Extract of Licorice (Glycyrrhiza uralensis) Protects against Triptolide-Induced Oxidative Stress through Activation of Nrf2. Evid. Based Complement. Altern. Med. 2017, 2017, 2752389. [Google Scholar] [CrossRef]
- Wang, S.R.; Chen, X.; Ling, S.; Ni, R.Z.; Guo, H.; Xu, J.W. MicroRNA expression, targeting, release dynamics and early-warning biomarkers in acute cardiotoxicity induced by triptolide in rats. Biomed. Pharmacother. 2019, 111, 1467–1477. [Google Scholar] [CrossRef]
- Ma, B.; Qi, H.; Li, J.; Xu, H.; Chi, B.; Zhu, J.; Yu, L.; An, G.; Zhang, Q. Triptolide disrupts fatty acids and peroxisome proliferator-activated receptor (PPAR) levels in male mice testes followed by testicular injury: A GC-MS based metabolomics study. Toxicology 2015, 336, 84–95. [Google Scholar] [CrossRef]
- Sun, R.; Dai, J.; Ling, M.; Yu, L.; Yu, Z.; Tang, L. Delivery of triptolide: A combination of traditional Chinese medicine and nanomedicine. J. Nanobiotechnol. 2022, 20, 194. [Google Scholar] [CrossRef]
- Yu, L.; Wang, Z.; Mo, Z.; Zou, B.; Yang, Y.; Sun, R.; Ma, W.; Yu, M.; Zhang, S.; Yu, Z. Synergetic delivery of triptolide and Ce6 with light-activatable liposomes for efficient hepatocellular carcinoma therapy. Acta Pharm. Sin. B 2021, 11, 2004–2015. [Google Scholar] [CrossRef]
- Fulton, M.D.; Najahi-Missaoui, W. Liposomes in Cancer Therapy: How Did We Start and Where Are We Now. Int. J. Mol. Sci. 2023, 24, 6615. [Google Scholar] [CrossRef]
- Wei, Y.; Gu, X.; Cheng, L.; Meng, F.; Storm, G.; Zhong, Z. Low-toxicity transferrin-guided polymersomal doxorubicin for potent chemotherapy of orthotopic hepatocellular carcinoma in vivo. Acta Biomater. 2019, 92, 196–204. [Google Scholar] [CrossRef]
- Kakudo, T.; Chaki, S.; Futaki, S.; Nakase, I.; Akaji, K.; Kawakami, T.; Maruyama, K.; Kamiya, H.; Harashima, H. Transferrin-modified liposomes equipped with a pH-sensitive fusogenic peptide: An artificial viral-like delivery system. Biochemistry 2004, 43, 5618–5628. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ishida, T.; Okada, Y.; Ise, S.; Harashima, H.; Kiwada, H. Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. Int. J. Pharm. 2007, 329, 94–102. [Google Scholar] [CrossRef]
- Daniels, T.R.; Bernabeu, E.; Rodríguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta 2012, 1820, 291–317. [Google Scholar] [CrossRef]
- Choi, C.H.; Alabi, C.A.; Webster, P.; Davis, M.E. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc. Natl. Acad. Sci. USA 2010, 107, 1235–1240. [Google Scholar] [CrossRef]
- Koshkaryev, A.; Piroyan, A.; Torchilin, V.P. Increased apoptosis in cancer cells in vitro and in vivo by ceramides in transferrin-modified liposomes. Cancer Biol. Ther. 2012, 13, 50–60. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng. 2006, 8, 343–375. [Google Scholar] [CrossRef]
- Li, X.; Ding, L.; Xu, Y.; Wang, Y.; Ping, Q. Targeted delivery of doxorubicin using stealth liposomes modified with transferrin. Int. J. Pharm. 2009, 373, 116–123. [Google Scholar] [CrossRef]
- Sakpakdeejaroen, I.; Somani, S.; Laskar, P.; Mullin, M.; Dufès, C. Regression of Melanoma Following Intravenous Injection of Plumbagin Entrapped in Transferrin-Conjugated, Lipid-Polymer Hybrid Nanoparticles. Int. J. Nanomed. 2021, 16, 2615–2631. [Google Scholar] [CrossRef]
- Gowda, B.H.J.; Ahmed, M.G.; Alshehri, S.A.; Wahab, S.; Vora, L.K.; Singh Thakur, R.R.; Kesharwani, P. The cubosome-based nanoplatforms in cancer therapy: Seeking new paradigms for cancer theranostics. Environ. Res. 2023, 273, 116894. [Google Scholar] [CrossRef]
- Wang, K.; Shang, F.; Chen, D.; Cao, T.; Wang, X.; Jiao, J.; He, S.; Liang, X. Protein liposomes-mediated targeted acetylcholinesterase gene delivery for effective liver cancer therapy. J. Nanobiotechnol. 2021, 19, 31. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, B.; Su, X.; Qiao, P.; Di, L.; Zhao, X. Preparation of transferrin modified triptolide liposome and in vitro evaluation. Chin. Tradit. Herb. Drugs 2022, 53, 687–695. (In Chinese) [Google Scholar]
- Lin, Y.; Wan, Y.; Du, X.; Li, J.; Wei, J.; Li, T.; Li, C.; Liu, Z.; Zhou, M.; Zhong, Z. TAT-modified serum albumin nanoparticles for sustained-release of tetramethylpyrazine and improved targeting to spinal cord injury. J. Nanobiotechnol. 2021, 19, 28. [Google Scholar] [CrossRef]
- Maritim, S.; Boulas, P.; Lin, Y. Comprehensive analysis of liposome formulation parameters and their influence on encapsulation, stability and drug release in glibenclamide liposomes. Int. J. Pharm. 2021, 592, 120051. [Google Scholar] [CrossRef]
- Driscoll, D.F. Globule-size distribution in injectable 20% lipid emulsions: Compliance with USP requirements. Am. J. Health Syst. Pharm. 2007, 64, 2032–2036. [Google Scholar] [CrossRef]
- Kuai, R.; Yuan, W.; Qin, Y.; Chen, H.; Tang, J.; Yuan, M.; Zhang, Z.; He, Q. Efficient delivery of payload into tumor cells in a controlled manner by TAT and thiolytic cleavable PEG co-modified liposomes. Mol. Pharm. 2010, 7, 1816–1826. [Google Scholar] [CrossRef]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef]
- Zheng, Y.; Kong, F.; Liu, S.; Liu, X.; Pei, D.; Miao, X. Membrane protein-chimeric liposome-mediated delivery of triptolide for targeted hepatocellular carcinoma therapy. Drug Deliv. 2021, 28, 2033–2043. [Google Scholar] [CrossRef]
- Sakpakdeejaroen, I.; Somani, S.; Laskar, P.; Mullin, M.; Dufès, C. Transferrin-bearing liposomes entrapping plumbagin for targeted cancer therapy. J. Interdiscip. Nanomed. 2019, 4, 54–71. [Google Scholar] [CrossRef]
- Liu, X.; Dong, S.; Dong, M.; Li, Y.; Sun, Z.; Zhang, X.; Wang, Y.; Teng, L.; Wang, D. Transferrin-conjugated liposomes loaded with carnosic acid inhibit liver cancer growth by inducing mitochondria-mediated apoptosis. Int. J. Pharm. 2021, 607, 121034. [Google Scholar] [CrossRef]
- Sutar, Y.B.; Mali, J.K.; Telvekar, V.N.; Rajmani, R.S.; Singh, A. Transferrin conjugates of antitubercular drug isoniazid: Synthesis and in vitro efficacy. Eur. J. Med. Chem. 2019, 183, 111713. [Google Scholar] [CrossRef]
- Weissleder, R.; Pittet, M.J. Imaging in the era of molecular oncology. Nature 2008, 452, 580–589. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Tube | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 2% Red Blood Cells Suspension (μL) | 500 | 500 | 500 | 500 | 500 |
| Saline solution (μL) | 450 | 400 | 300 | 500 | |
| Distilled water (μL) | 500 | ||||
| Sample (μL) | 50 | 100 | 200 |
| Liposomes | Size (nm) | PDI | Zeta Potential (mV) | EE% | DL% |
|---|---|---|---|---|---|
| TF-Blank@LIP | 125.02 ± 2.20 | 0.19 ± 0.05 | −23.90 ± 2.30 | ||
| TP@LIP | 121.10 ± 3.40 | 0.15 ± 0.03 | −17.50 ± 0.20 | 88.23 ± 0.14 | 10.26 ± 0.02 |
| TF-TP@LIP | 130.33 ± 1.89 | 0.20 ± 0.04 | −23.20 ± 0.90 | 85.33 ± 0.41 | 9.96 ± 0.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Yang, Y.; Su, X.; Xie, Y.; Liang, Y.; Zhou, T.; Wu, Y.; Di, L. Transferrin-Modified Triptolide Liposome Targeting Enhances Anti-Hepatocellular Carcinoma Effects. Biomedicines 2023, 11, 2869. https://doi.org/10.3390/biomedicines11102869
Zhao X, Yang Y, Su X, Xie Y, Liang Y, Zhou T, Wu Y, Di L. Transferrin-Modified Triptolide Liposome Targeting Enhances Anti-Hepatocellular Carcinoma Effects. Biomedicines. 2023; 11(10):2869. https://doi.org/10.3390/biomedicines11102869
Chicago/Turabian StyleZhao, Xiaoli, Yifan Yang, Xuerong Su, Ying Xie, Yiyao Liang, Tong Zhou, Yangqian Wu, and Liuqing Di. 2023. "Transferrin-Modified Triptolide Liposome Targeting Enhances Anti-Hepatocellular Carcinoma Effects" Biomedicines 11, no. 10: 2869. https://doi.org/10.3390/biomedicines11102869
APA StyleZhao, X., Yang, Y., Su, X., Xie, Y., Liang, Y., Zhou, T., Wu, Y., & Di, L. (2023). Transferrin-Modified Triptolide Liposome Targeting Enhances Anti-Hepatocellular Carcinoma Effects. Biomedicines, 11(10), 2869. https://doi.org/10.3390/biomedicines11102869