
The Na/K-ATPase α1/Src Signaling Axis Regulates Mitochondrial Metabolic Function and Redox Signaling in Human iPSC-Derived Cardiomyocytes

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Induced Pluripotent Stem Cells (hiPSC): WT and A420P Mutant Lines
2.2. Differentiation of hiPSC into Cardiomyocytes (iCM)
2.3. Immunostaining and Fluorescence Microscopy
2.4. Video imaging
2.5. Electron Microscopy Imaging
2.6. RNA Purification, Reverse Transcription, and Real-Time qPCR
2.7. Ouabain-sensitive Na/K-ATPase Activity
2.8. Cell Lysis and Western Blotting
2.9. Seahorse Extracellular Flux Analysis
2.10. Protein Carbonylation
2.11. Detection of Reactive Oxygen Species (ROS)
2.12. Statistical Analysis
3. Results
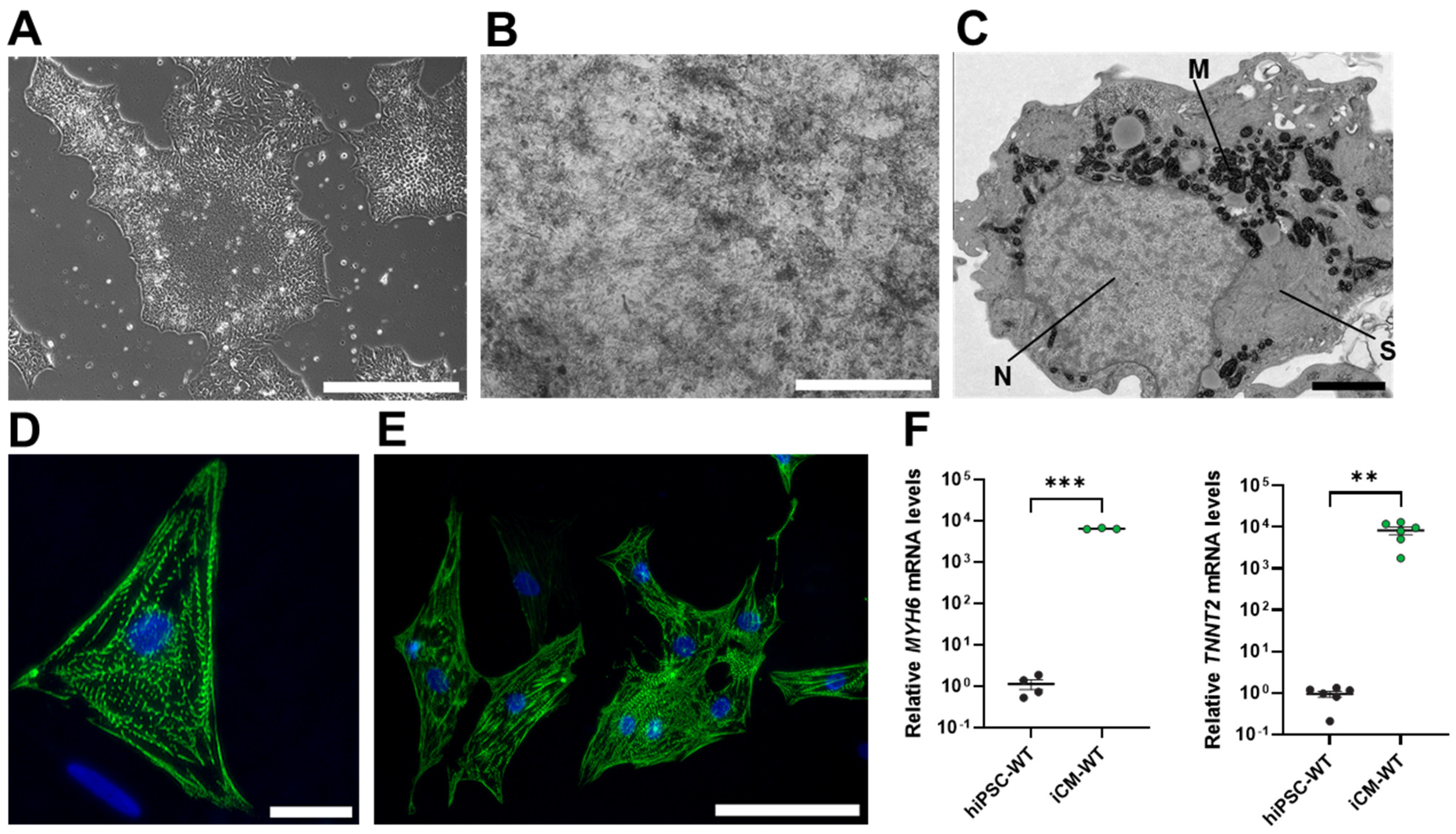
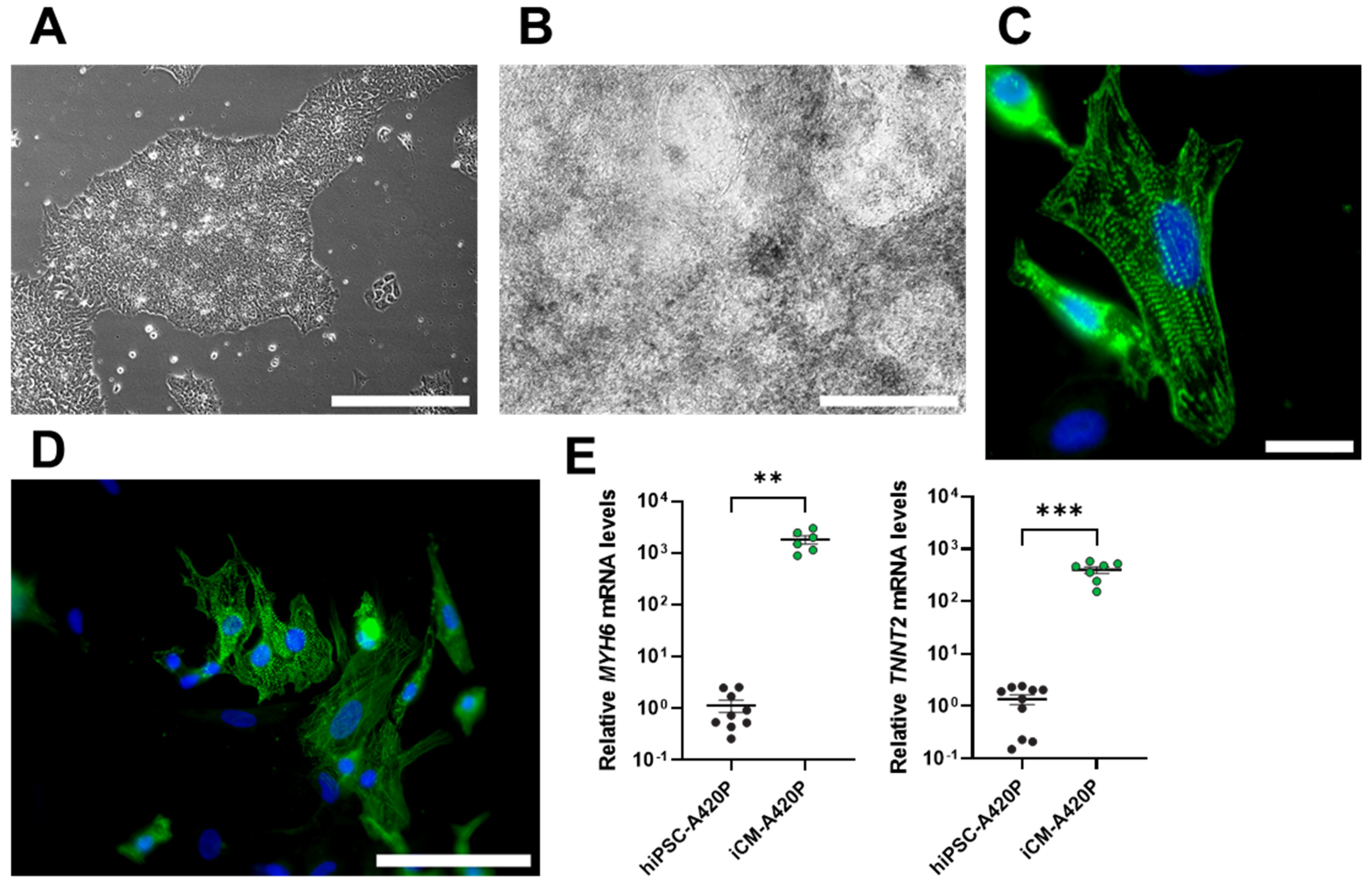
3.1. Characterization of WT and A420P hiPSC-Derived Cardiomyocytes (iCM)
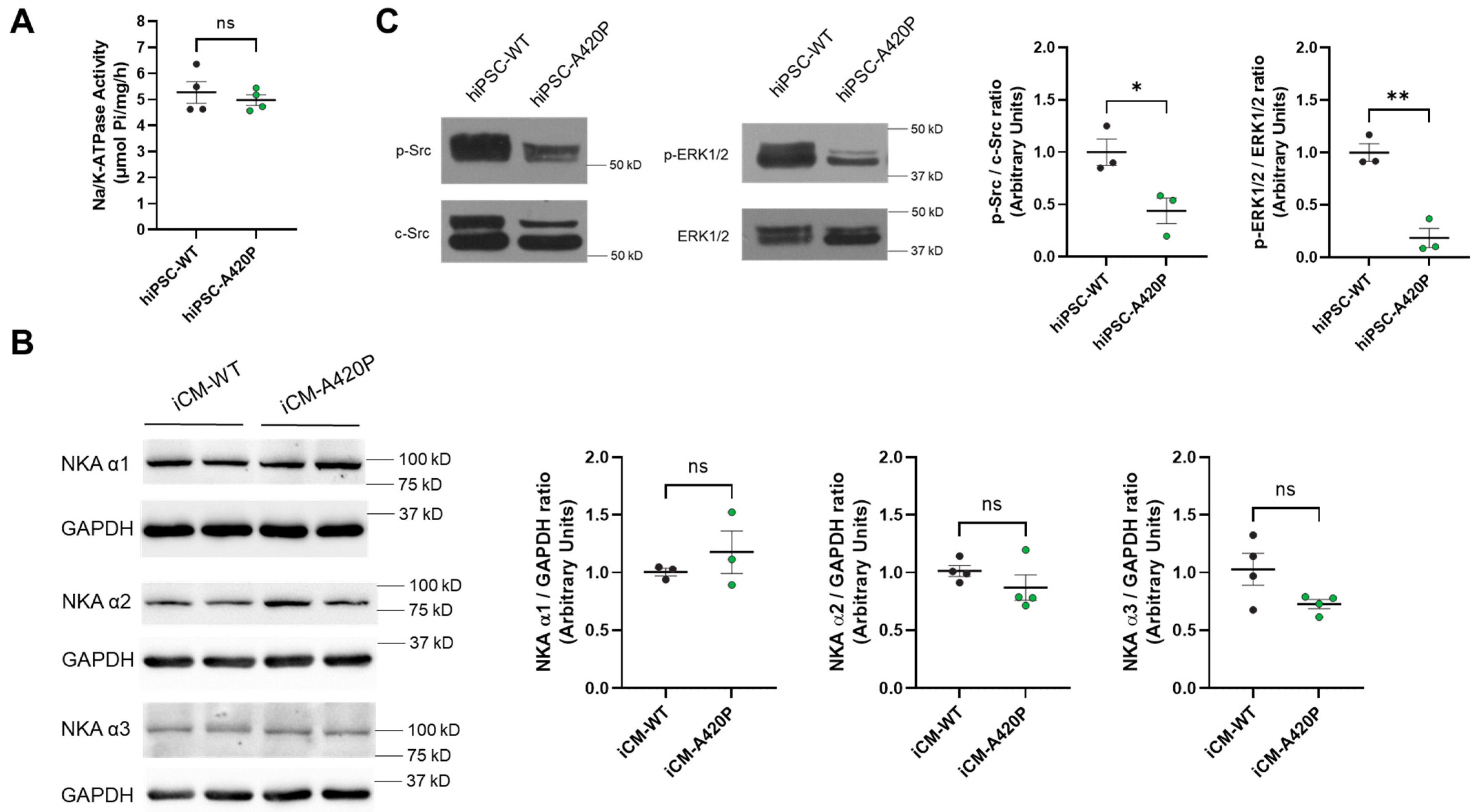
3.2. Characterization of NKA Expression, Na+/K+-ATPase Activity, and Basal NKA Signaling Axis in A420P hiPSC and iCM
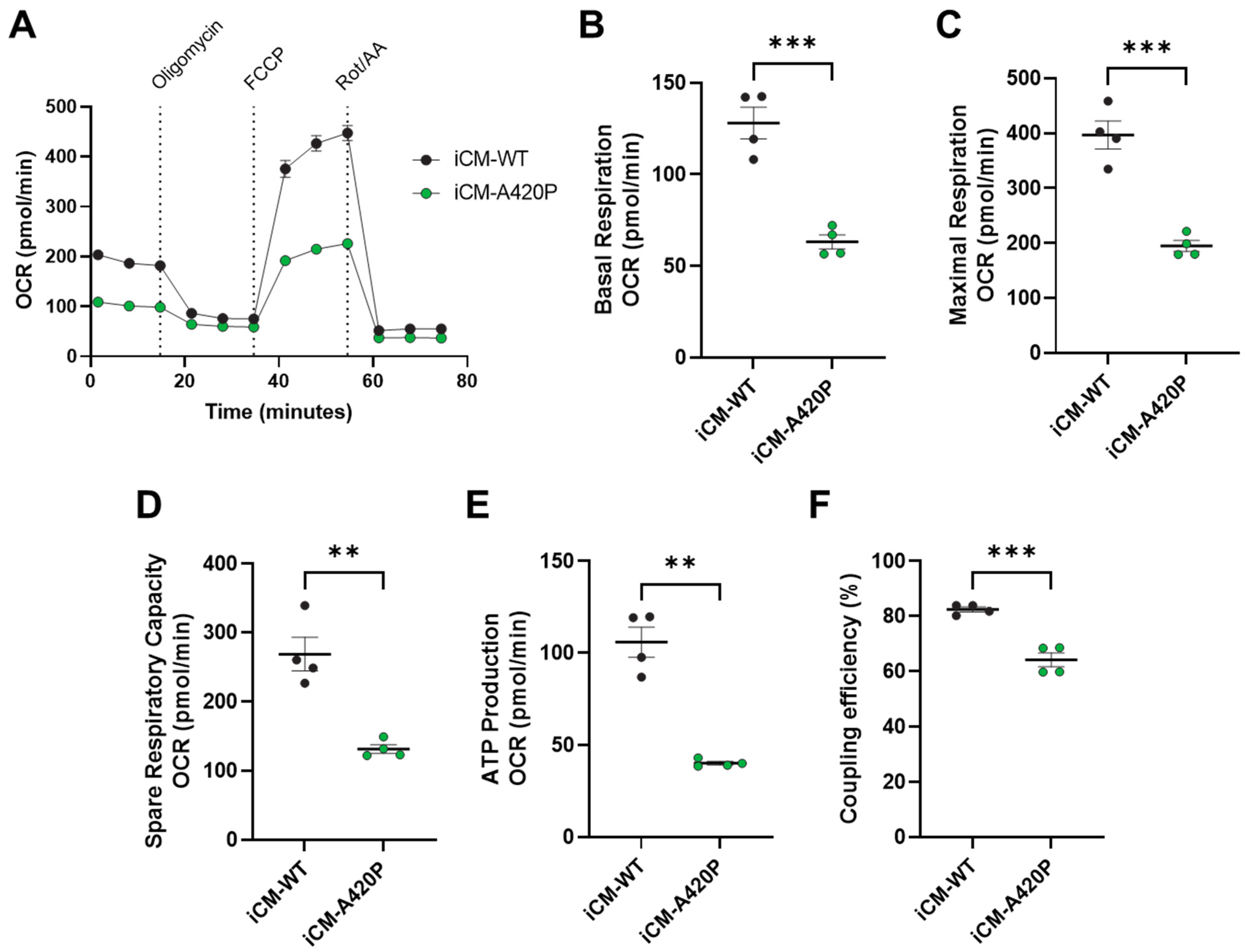
3.3. Mitochondrial Function in iCM-WT and iCM-A420P
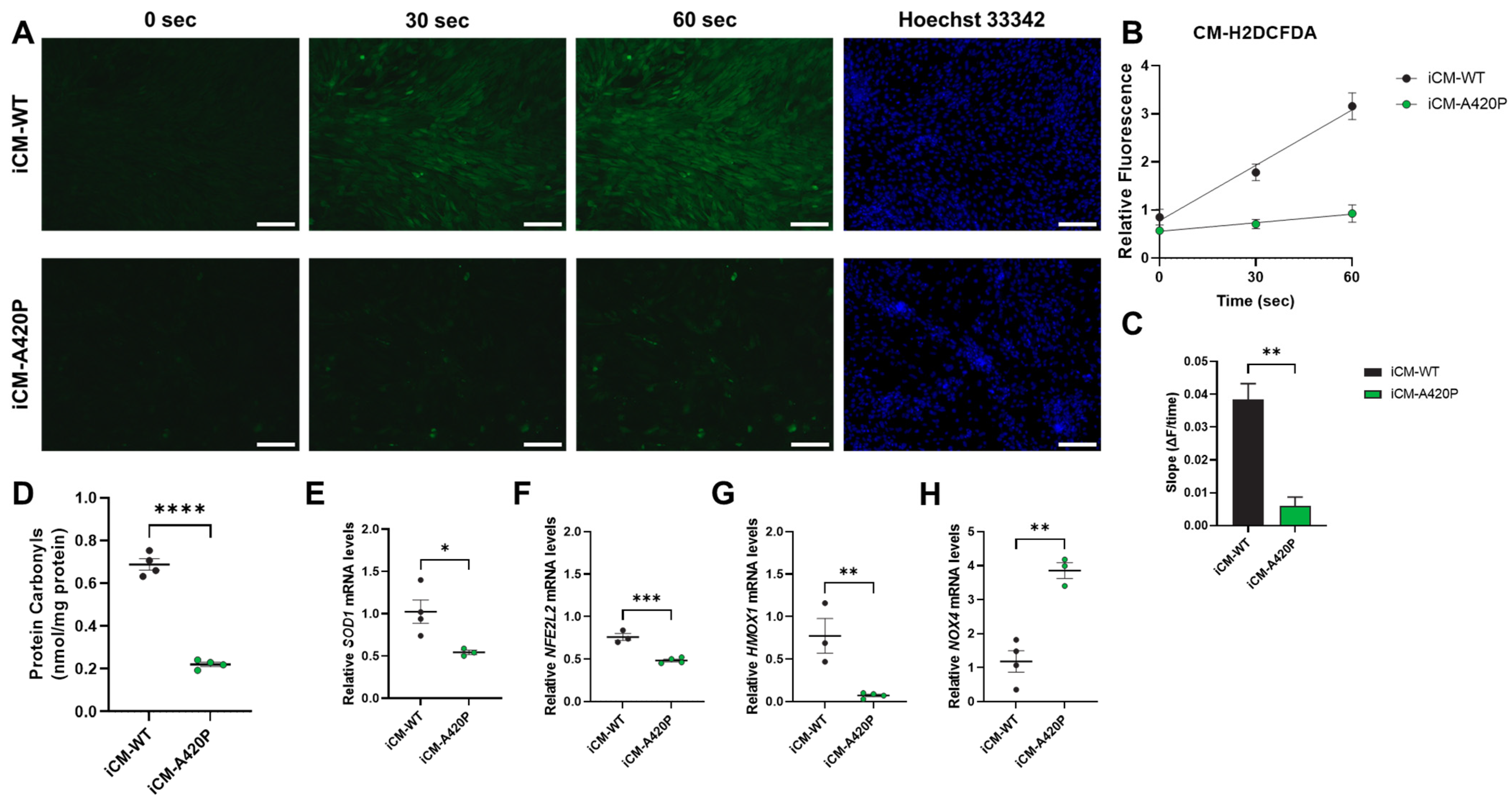
3.4. Decreased ROS Production and Regulation of Gene Markers of Cardiac Oxidative Stress in iCM-A420P
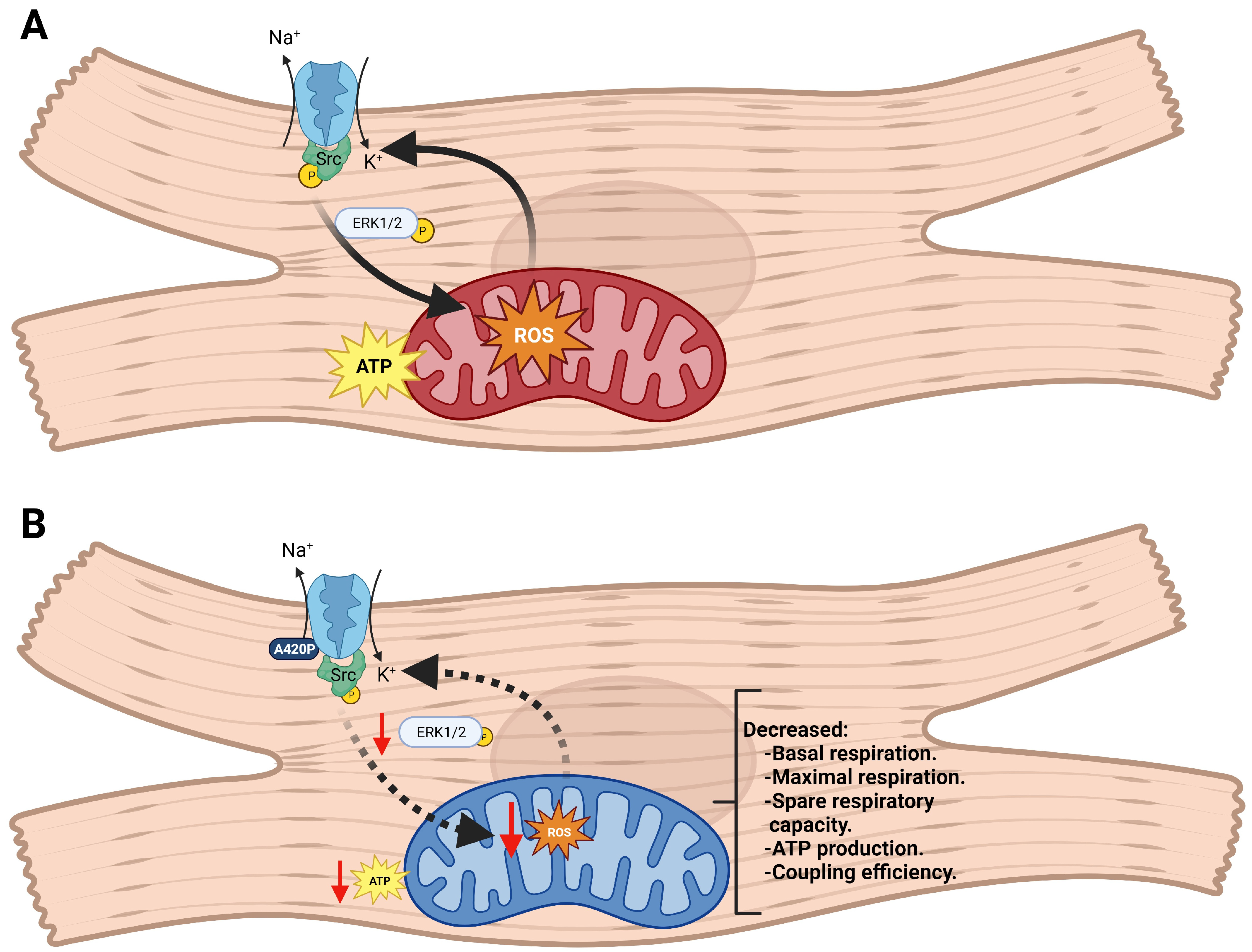
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Wu, J.; Kennedy, D.J. Regulation of Cardiac Remodeling by Cardiac Na(+)/K(+)-ATPase Isoforms. Front. Physiol. 2016, 7, 382. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. Physiol. 1998, 275, F633–F650. [Google Scholar] [CrossRef] [PubMed]
- McDonough, A.A.; Velotta, J.B.; Schwinger, R.H.; Philipson, K.D.; Farley, R.A. The cardiac sodium pump: Structure and function. Basic Res. Cardiol. 2002, 97 (Suppl. S1), I19–I24. [Google Scholar] [CrossRef] [PubMed]
- Akera, T.; Brody, T.M. The role of Na+,K+-ATPase in the inotropic action of digitalis. Pharmacol. Rev. 1977, 29, 187–220. [Google Scholar] [PubMed]
- Thomas, R.; Gray, P.; Andrews, J. Digitalis: Its Mode of Action, Receptor, and Structure–Activity Relationships. In Advances in Drug Research; Testa, B., Ed.; Academic Press: Cambridge, MA, USA, 1990; Volume 19, pp. 311–562. [Google Scholar]
- Peng, M.; Huang, L.; Xie, Z.; Huang, W.-H.; Askari, A. Partial Inhibition of Na/K-ATPase by Ouabain Induces the Ca-dependent Expressions of Early-response Genes in Cardiac Myocytes. J. Biol. Chem. 1996, 271, 10372–10378. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, H.; Xie, Z. Ouabain-induced hypertrophy in cultured cardiac myocytes is accompanied by changes in expression of several late response genes. J. Mol. Cell. Cardiol. 1997, 29, 429–437. [Google Scholar] [CrossRef]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides: Their roles in hypertension, salt metabolism, and cell growth. Am. J. Physiol. Cell Physiol. 2007, 293, C509–C536. [Google Scholar] [CrossRef]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J.C. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2001, 276, 46605–46611. [Google Scholar] [CrossRef]
- Kaunitz, J.D. Membrane transport proteins: Not just for transport anymore. Am. J. Physiol. Ren. Physiol. 2006, 290, F995–F996. [Google Scholar] [CrossRef]
- Li, J.; Zelenin, S.; Aperia, A.; Aizman, O. Low doses of ouabain protect from serum deprivation–triggered apoptosis and stimulate kidney cell proliferation via activation of NF-κB. J. Am. Soc. Nephrol. 2006, 17, 1848–1857. [Google Scholar] [CrossRef]
- Miyakawa-Naito, A.; Uhlén, P.; Lal, M.; Aizman, O.; Mikoshiba, K.; Brismar, H.; Zelenin, S.; Aperia, A. Cell signaling microdomain with Na, K-ATPase and inositol 1, 4, 5-trisphosphate receptor generates calcium oscillations. J. Biol. Chem. 2003, 278, 50355–50361. [Google Scholar] [CrossRef]
- Saunders, R.; Scheiner-Bobis, G. Ouabain stimulates endothelin release and expression in human endothelial cells without inhibiting the sodium pump. Eur. J. Biochem. 2004, 271, 1054–1062. [Google Scholar] [CrossRef]
- Kotova, O.; Al-Khalili, L.; Talia, S.; Hooke, C.; Fedorova, O.V.; Bagrov, A.Y.; Chibalin, A.V. Cardiotonic steroids stimulate glycogen synthesis in human skeletal muscle cells via a Src-and ERK1/2-dependent mechanism. J. Biol. Chem. 2006, 281, 20085–20094. [Google Scholar] [CrossRef]
- Khundmiri, S.J.; Metzler, M.A.; Ameen, M.; Amin, V.; Rane, M.J.; Delamere, N.A. Ouabain induces cell proliferation through calcium-dependent phosphorylation of Akt (protein kinase B) in opossum kidney proximal tubule cells. Am. J. Physiol.-Cell Physiol. 2006, 291, C1247–C1257. [Google Scholar] [CrossRef]
- Trevisi, L.; Visentin, B.; Cusinato, F.; Pighin, I.; Luciani, S. Antiapoptotic effect of ouabain on human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 321, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Kim, M.; Choi, S.; Kim, M.-J.; Suh, J.-k.; Choi, E.C.; Lee, K. Molecular mechanism of cofilin dephosphorylation by ouabain. Cell. Signal. 2006, 18, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; REN, Y.p.; LÜ, Z.r. Ouabain induces cardiac remodeling in rats independent of blood pressure 4. Acta Pharmacol. Sin. 2007, 28, 344–352. [Google Scholar] [CrossRef]
- Li, M.; Wang, Q.; Guan, L. Effects of ouabain on proliferation, intracellular free calcium and c-myc mRNA expression in vascular smooth muscle cells. J. Comp. Physiol. B 2007, 177, 589–595. [Google Scholar] [CrossRef]
- Skoumal, R.; Szokodi, I.; Aro, J.; Földes, G.; Göőz, M.; Seres, L.; Sármán, B.; Lakó-Futó, Z.; Papp, L.; Vuolteenaho, O. Involvement of endogenous ouabain-like compound in the cardiac hypertrophic process in vivo. Life Sci. 2007, 80, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Thundathil, J.C.; Anzar, M.; Buhr, M.M. Na+/K+ ATPase as a signaling molecule during bovine sperm capacitation. Biol. Reprod. 2006, 75, 308–317. [Google Scholar] [CrossRef]
- Larre, I.; Ponce, A.; Fiorentino, R.; Shoshani, L.; Contreras, R.G.; Cereijido, M. Contacts and cooperation between cells depend on the hormone ouabain. Proc. Natl. Acad. Sci. USA 2006, 103, 10911–10916. [Google Scholar] [CrossRef]
- Nguyen, A.-N.T.; Wallace, D.P.; Blanco, G. Ouabain binds with high affinity to the na, K-ATPase in human polycystic kidney cells and induces extracellular Signal–Regulated kinase activation and cell proliferation. J. Am. Soc. Nephrol. 2007, 18, 46–57. [Google Scholar] [CrossRef]
- Fontana, J.M.; Burlaka, I.; Khodus, G.; Brismar, H.; Aperia, A. Calcium oscillations triggered by cardiotonic steroids. FEBS J. 2013, 280, 5450–5455. [Google Scholar] [CrossRef] [PubMed]
- Wenceslau, C.F.; Rossoni, L.V. Rostafuroxin ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats: The role of Na+ K+-ATPase/cSRC pathway. J. Hypertens. 2014, 32, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Song, D.; Xu, J.; Li, B.; Hertz, L.; Peng, L. Ammonia-induced Na, K-ATPase/ouabain-mediated EGF receptor transactivation, MAPK/ERK and PI3K/AKT signaling and ROS formation cause astrocyte swelling. Neurochem. Int. 2013, 63, 610–625. [Google Scholar] [CrossRef]
- Aperia, A. 2011 Homer Smith Award: To serve and protect: Classic and novel roles for Na+, K+-adenosine triphosphatase. J. Am. Soc. Nephrol. 2012, 23, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Molinari, I.; Barassi, P.; Minotti, E.; Bianchi, G.; Ferrari, P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J. Biol. Chem. 2004, 279, 33306–33314. [Google Scholar] [CrossRef]
- Wang, Y.; Zhan, Y.; Xu, R.; Shao, R.; Jiang, J.; Wang, Z. Src mediates extracellular signal-regulated kinase 1/2 activation and autophagic cell death induced by cardiac glycosides in human non-small cell lung cancer cell lines. Mol. Carcinog. 2015, 54, E26–E34. [Google Scholar] [CrossRef]
- Dvela, M.; Rosen, H.; Ben-Ami, H.C.; Lichtstein, D. Endogenous ouabain regulates cell viability. Am. J. Physiol. Cell Physiol. 2011, 302, C442–C452. [Google Scholar] [CrossRef]
- Wansapura, A.N.; Lasko, V.M.; Lingrel, J.B.; Lorenz, J.N. Mice expressing ouabain-sensitive α1-Na, K-ATPase have increased susceptibility to pressure overload-induced cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2010, 300, H347–H355. [Google Scholar] [CrossRef]
- Arnaud-Batista, F.J.; Costa, G.T.; de Oliveira, I.M.B.; Costa, P.P.; Santos, C.F.; Fonteles, M.C.; Uchôa, D.E.; Silveira, E.R.; Cardi, B.A.; Carvalho, K.M. Natriuretic effect of bufalin in isolated rat kidneys involves activation of the Na+-K+-ATPase-Src kinase pathway. Am. J. Physiol. Ren. Physiol. 2012, 302, F959–F966. [Google Scholar] [CrossRef] [PubMed]
- Godinho, A.N.; Costa, G.T.; Oliveira, N.O.; Cardi, B.A.; Uchoa, D.E.A.; Silveira, E.R.; Quintas, L.E.M.; Noël, F.G.; Fonteles, M.C.; Carvalho, K.M. Effects of cardiotonic steroids on isolated perfused kidney and NHE3 activity in renal proximal tubules. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Barcroft, L.C.; Moseley, A.E.; Lingrel, J.B.; Watson, A.J. Deletion of the Na/K-ATPase alpha1-subunit gene (Atp1a1) does not prevent cavitation of the preimplantation mouse embryo. Mech. Dev. 2004, 121, 417–426. [Google Scholar] [CrossRef]
- Wang, X.; Cai, L.; Xie, J.X.; Cui, X.; Zhang, J.; Wang, J.; Chen, Y.; Larre, I.; Shapiro, J.I.; Pierre, S.V.; et al. A caveolin binding motif in Na/K-ATPase is required for stem cell differentiation and organogenesis in mammals and C. elegans. Sci. Adv. 2020, 6, eaaw5851. [Google Scholar] [CrossRef]
- Sweadner, K.J.; Herrera, V.L.; Amato, S.; Moellmann, A.; Gibbons, D.K.; Repke, K.R. Immunologic identification of Na+,K(+)-ATPase isoforms in myocardium. Isoform change in deoxycorticosterone acetate-salt hypertension. Circ. Res. 1994, 74, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Sweadner, K.J.; Farshi, S.K. Rat cardiac ventricle has two Na+,K+-ATPases with different affinities for ouabain: Developmental changes in immunologically different catalytic subunits. Proc. Natl. Acad. Sci. USA 1987, 84, 8404–8407. [Google Scholar] [CrossRef]
- Lucchesi, P.A.; Sweadner, K.J. Postnatal changes in Na,K-ATPase isoform expression in rat cardiac ventricle. Conservation of biphasic ouabain affinity. J. Biol. Chem. 1991, 266, 9327–9331. [Google Scholar] [CrossRef]
- Sweadner, K.J. Isozymes of the Na+/K+-ATPase. Biochim. Biophys. Acta 1989, 988, 185–220. [Google Scholar] [CrossRef]
- Schwinger, R.H.; Bundgaard, H.; Müller-Ehmsen, J.; Kjeldsen, K. The Na, K-ATPase in the failing human heart. Cardiovasc. Res. 2003, 57, 913–920. [Google Scholar] [CrossRef]
- James, P.F.; Grupp, I.L.; Grupp, G.; Woo, A.L.; Askew, G.R.; Croyle, M.L.; Walsh, R.A.; Lingrel, J.B. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol. Cell 1999, 3, 555–563. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elimban, V.; Bartekova, M.; Adameova, A. Involvement of Oxidative Stress in the Development of Subcellular Defects and Heart Disease. Biomedicines 2022, 10, 393. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Hamlyn, J.M. Ouabain, endogenous ouabain and ouabain-like factors: The Na(+) pump/ouabain receptor, its linkage to NCX, and its myriad functions. Cell Calcium 2020, 86, 102159. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Wang, J.; Frank, K.; Müller-Ehmsen, J.; Brixius, K.; McDonough, A.A.; Erdmann, E. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 1999, 99, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Shamraj, O.I.; Grupp, I.L.; Grupp, G.; Melvin, D.; Gradoux, N.; Kremers, W.; Lingrel, J.B.; De Pover, A. Characterisation of Na/K-ATPase, its isoforms, and the inotropic response to ouabain in isolated failing human hearts. Cardiovasc. Res. 1993, 27, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Swift, F.; Birkeland, J.A.; Tovsrud, N.; Enger, U.H.; Aronsen, J.M.; Louch, W.E.; Sjaastad, I.; Sejersted, O.M. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc. Res. 2008, 78, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Staehr, C.; Rohde, P.D.; Krarup, N.T.; Ringgaard, S.; Laustsen, C.; Johnsen, J.; Nielsen, R.; Beck, H.C.; Morth, J.P.; Lykke-Hartmann, K.; et al. Migraine-Associated Mutation in the Na,K-ATPase Leads to Disturbances in Cardiac Metabolism and Reduced Cardiac Function. J. Am. Heart Assoc. 2022, 11, e021814. [Google Scholar] [CrossRef] [PubMed]
- Biondo, E.D.; Spontarelli, K.; Ababioh, G.; Mendez, L.; Artigas, P. Diseases caused by mutations in the Na(+)/K(+) pump alpha1 gene ATP1A1. Am. J. Physiol. Cell Physiol. 2021, 321, C394–C408. [Google Scholar] [CrossRef]
- Sweadner, K.J.; Arystarkhova, E.; Penniston, J.T.; Swoboda, K.J.; Brashear, A.; Ozelius, L.J. Genotype-structure-phenotype relationships diverge in paralogs ATP1A1, ATP1A2, and ATP1A3. Neurol. Genet. 2019, 5, e303. [Google Scholar] [CrossRef]
- Cellini, A.; Hofler, D.; Arias-Loza, P.A.; Bandleon, S.; Langsenlehner, T.; Kohlhaas, M.; Maack, C.; Bauer, W.R.; Eder-Negrin, P. The alpha2-isoform of the Na(+)/K(+)-ATPase protects against pathological remodeling and beta-adrenergic desensitization after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H650–H662. [Google Scholar] [CrossRef]
- Boguslavskyi, A.; Pavlovic, D.; Aughton, K.; Clark, J.E.; Howie, J.; Fuller, W.; Shattock, M.J. Cardiac hypertrophy in mice expressing unphosphorylatable phospholemman. Cardiovasc. Res. 2014, 104, 72–82. [Google Scholar] [CrossRef]
- Rindler, T.N.; Lasko, V.M.; Nieman, M.L.; Okada, M.; Lorenz, J.N.; Lingrel, J.B. Knockout of the Na,K-ATPase alpha2-isoform in cardiac myocytes delays pressure overload-induced cardiac dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1147–H1158. [Google Scholar] [CrossRef] [PubMed]
- Correll, R.N.; Eder, P.; Burr, A.R.; Despa, S.; Davis, J.; Bers, D.M.; Molkentin, J.D. Overexpression of the Na+/K+ ATPase alpha2 but not alpha1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ. Res. 2014, 114, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Dostanic-Larson, I.; Lorenz, J.N.; Van Huysse, J.W.; Neumann, J.C.; Moseley, A.E.; Lingrel, J.B. Physiological role of the alpha1- and alpha2-isoforms of the Na+-K+-ATPase and biological significance of their cardiac glycoside binding site. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R524–R528. [Google Scholar] [CrossRef] [PubMed]
- Marck, P.V.; Pessoa, M.T.; Xu, Y.; Kutz, L.C.; Collins, D.M.; Yan, Y.; King, C.; Wang, X.; Duan, Q.; Cai, L.; et al. Cardiac Oxidative Signaling and Physiological Hypertrophy in the Na/K-ATPase alpha1(s/s)alpha2(s/s) Mouse Model of High Affinity for Cardiotonic Steroids. Int. J. Mol. Sci. 2021, 22, 3462. [Google Scholar] [CrossRef] [PubMed]
- Kutz, L.C.; Cui, X.; Xie, J.X.; Mukherji, S.T.; Terrell, K.C.; Huang, M.; Wang, X.; Wang, J.; Martin, A.J.; Pessoa, M.T.; et al. The Na/K-ATPase alpha1/Src interaction regulates metabolic reserve and Western diet intolerance. Acta Physiol. 2021, 232, e13652. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wang, X.; Banerjee, M.; Mukherji, S.T.; Kutz, L.C.; Zhao, A.; Sepanski, M.; Fan, C.M.; Zhu, G.Z.; Tian, J.; et al. Regulation of Myogenesis by a Na/K-ATPase α1 Caveolin-Binding Motif. Stem Cells 2022, 40, 133–148. [Google Scholar] [CrossRef]
- Lai, F.; Madan, N.; Ye, Q.; Duan, Q.; Li, Z.; Wang, S.; Si, S.; Xie, Z. Identification of a mutant α1 Na/K-ATPase that pumps but is defective in signal transduction. J. Biol. Chem. 2013, 288, 13295–13304. [Google Scholar] [CrossRef]
- Yu, H.; Cui, X.; Zhang, J.; Xie, J.X.; Banerjee, M.; Pierre, S.V.; Xie, Z. Heterogeneity of signal transduction by Na-K-ATPase α-isoforms: Role of Src interaction. Am. J. Physiol. Cell Physiol. 2018, 314, C202–C210. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- D'Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxidative Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef] [PubMed]
- De Nicolo, B.; Cataldi-Stagetti, E.; Diquigiovanni, C.; Bonora, E. Calcium and Reactive Oxygen Species Signaling Interplays in Cardiac Physiology and Pathologies. Antioxidants 2023, 12, 353. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [CrossRef] [PubMed]
- Pratt, R.D.; Brickman, C.R.; Cottrill, C.L.; Shapiro, J.I.; Liu, J. The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species. Int. J. Mol. Sci. 2018, 19, 2600. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lilly, M.N.; Shapiro, J.I. Targeting Na/K-ATPase Signaling: A New Approach to Control Oxidative Stress. Curr. Pharm. Des. 2018, 24, 359–364. [Google Scholar] [CrossRef] [PubMed]
- De Geest, B.; Mishra, M. Role of Oxidative Stress in Heart Failure: Insights from Gene Transfer Studies. Biomedicines 2021, 9, 1645. [Google Scholar] [CrossRef]
- Madan, N.; Xu, Y.; Duan, Q.; Banerjee, M.; Larre, I.; Pierre, S.V.; Xie, Z. Src-independent ERK signaling through the rat α3 isoform of Na/K-ATPase. Am. J. Physiol. Cell Physiol. 2017, 312, C222–C232. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Santos, C.X.; Anilkumar, N.; Zhang, M.; Brewer, A.C.; Shah, A.M. Redox signaling in cardiac myocytes. Free Radic. Biol. Med. 2011, 50, 777–793. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, Q.; Liu, C.; Xie, J.X.; Yan, Y.; Lai, F.; Duan, Q.; Li, X.; Tian, J.; Xie, Z. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 2014, 71, 415–426. [Google Scholar] [CrossRef]
- Zhou, B.; Caudal, A.; Tang, X.; Chavez, J.D.; McMillen, T.S.; Keller, A.; Villet, O.; Zhao, M.; Liu, Y.; Ritterhoff, J.; et al. Upregulation of mitochondrial ATPase inhibitory factor 1 (ATPIF1) mediates increased glycolysis in mouse hearts. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Badolia, R.; Lettlova, S.; Parnell, K.M.; Shankar, T.S.; Diakos, N.A.; Olson, K.A.; Taleb, I.; Tatum, S.M.; Berg, J.A.; et al. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. 2021, 33, 629–648. [Google Scholar] [CrossRef]
- Wang, C.; Qiao, S.; Zhao, Y.; Tian, H.; Yan, W.; Hou, X.; Wang, R.; Zhang, B.; Yang, C.; Zhu, F.; et al. The KLF7/PFKL/ACADL axis modulates cardiac metabolic remodelling during cardiac hypertrophy in male mice. Nat. Commun. 2023, 14, 959. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Keenan, J.E.; Piantadosi, C.A. Mitochondrial quality-control dysregulation in conditional HO-1(-/-) mice. JCI Insight 2017, 2, e89676. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Koh, X.; Hua, F.; Li, G.; Larrick, J.W.; Bian, J.S. Cardioprotection induced by Na(+)/K(+)-ATPase activation involves extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/Akt pathway. Cardiovasc. Res. 2011, 89, 51–59. [Google Scholar] [CrossRef]
- Duan, Q.; Madan, N.D.; Wu, J.; Kalisz, J.; Doshi, K.Y.; Haldar, S.M.; Liu, L.; Pierre, S.V. Role of phosphoinositide 3-kinase IA (PI3K-IA) activation in cardioprotection induced by ouabain preconditioning. J. Mol. Cell Cardiol. 2015, 80, 114–125. [Google Scholar] [CrossRef]
- Li, H.; Yin, A.; Cheng, Z.; Feng, M.; Zhang, H.; Xu, J.; Wang, F.; Qian, L. Attenuation of Na/K-ATPase/Src/ROS amplification signal pathway with pNaktide ameliorates myocardial ischemia-reperfusion injury. Int. J. Biol. Macromol. 2018, 118, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chaudhry, M.; Bai, F.; Chuang, J.; Chaudhry, H.; Al-Astal, A.Y.; Nie, Y.; Sollars, V.; Sodhi, K.; Seligman, P.; et al. Blockage of the Na-K-ATPase signaling-mediated oxidant amplification loop elongates red blood cell half-life and ameliorates uremic anemia induced by 5/6th PNx in C57BL/6 mice. Am. J. Physiol. Renal. Physiol. 2022, 322, F655–F666. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, L.; Pessoa, M.T.; Gao, Y.; Strause, S.; Banerjee, M.; Tian, J.; Xie, Z.; Pierre, S.V. The Na/K-ATPase α1/Src Signaling Axis Regulates Mitochondrial Metabolic Function and Redox Signaling in Human iPSC-Derived Cardiomyocytes. Biomedicines 2023, 11, 3207. https://doi.org/10.3390/biomedicines11123207
Cai L, Pessoa MT, Gao Y, Strause S, Banerjee M, Tian J, Xie Z, Pierre SV. The Na/K-ATPase α1/Src Signaling Axis Regulates Mitochondrial Metabolic Function and Redox Signaling in Human iPSC-Derived Cardiomyocytes. Biomedicines. 2023; 11(12):3207. https://doi.org/10.3390/biomedicines11123207
Chicago/Turabian StyleCai, Liquan, Marco T. Pessoa, Yingnyu Gao, Sidney Strause, Moumita Banerjee, Jiang Tian, Zijian Xie, and Sandrine V. Pierre. 2023. "The Na/K-ATPase α1/Src Signaling Axis Regulates Mitochondrial Metabolic Function and Redox Signaling in Human iPSC-Derived Cardiomyocytes" Biomedicines 11, no. 12: 3207. https://doi.org/10.3390/biomedicines11123207
APA StyleCai, L., Pessoa, M. T., Gao, Y., Strause, S., Banerjee, M., Tian, J., Xie, Z., & Pierre, S. V. (2023). The Na/K-ATPase α1/Src Signaling Axis Regulates Mitochondrial Metabolic Function and Redox Signaling in Human iPSC-Derived Cardiomyocytes. Biomedicines, 11(12), 3207. https://doi.org/10.3390/biomedicines11123207