
Design and Prediction of Aptamers Assisted by In Silico Methods


Abstract
:1. Introduction
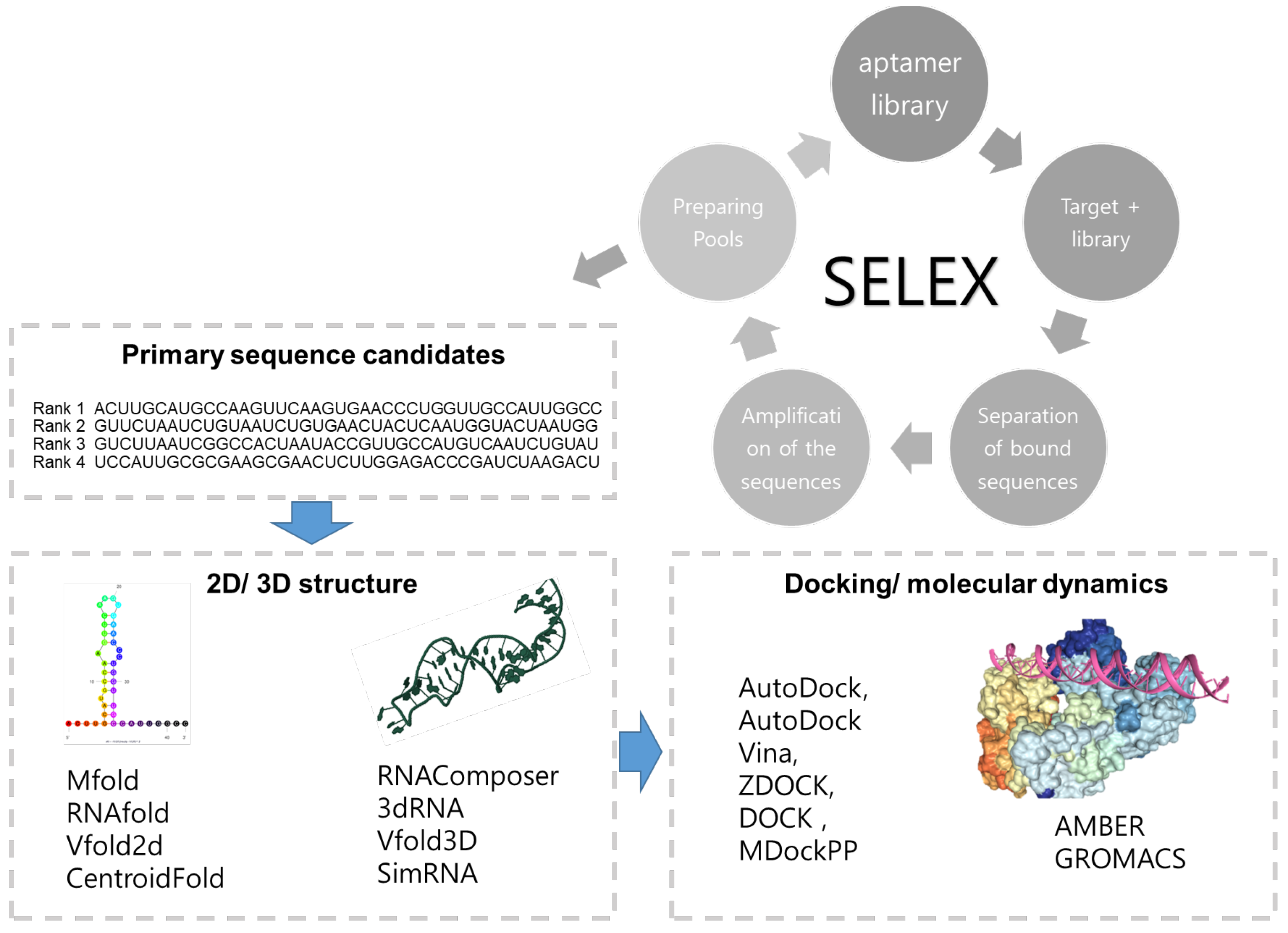
2. Prediction of the Aptamer Based on Its Structure
2.1. 2D Structure Prediction of Aptamers
2.2. 3D Structure Prediction of Aptamers
2.3. G4 Structure of Aptamers
2.4. Molecular Docking
2.5. Molecular Dynamics
2.6. Others Affecting Affinity
3. Application of an In Silico Method for the Development of the Aptamer
3.1. Aptamers Binding Proteins
3.1.1. Thrombin Binding Aptamers (TBA)
3.1.2. Infectious Disease Marker Binding Aptamers
3.1.3. Cancer Marker Binding Aptamers
3.1.4. Other Protein-Binding Aptamers
3.2. Aptamers Binding Small Molecules
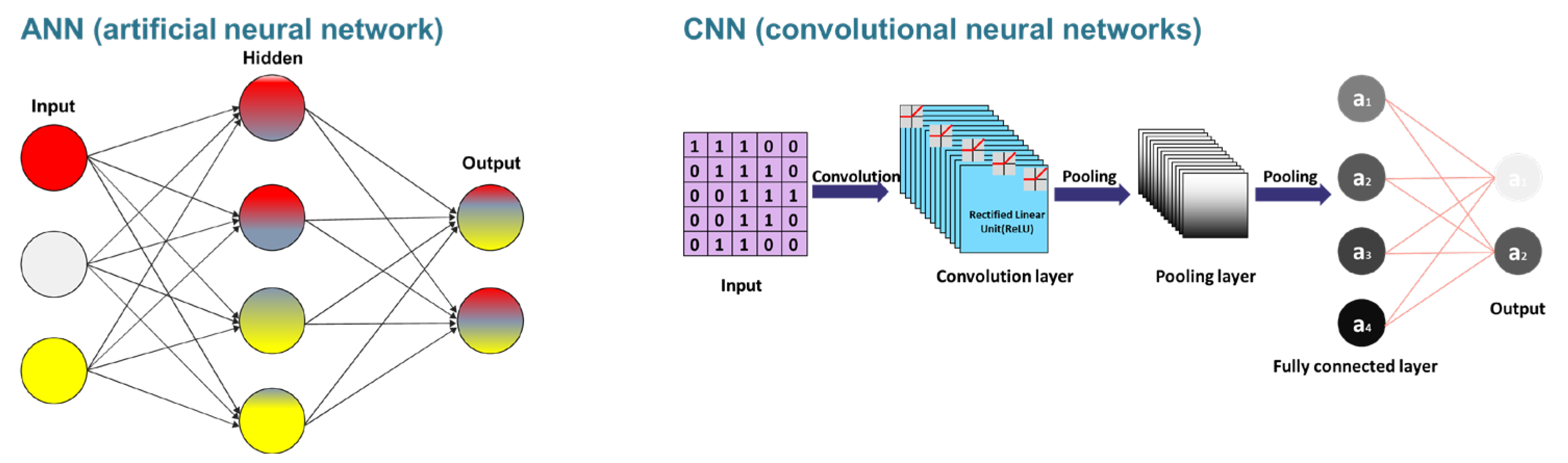
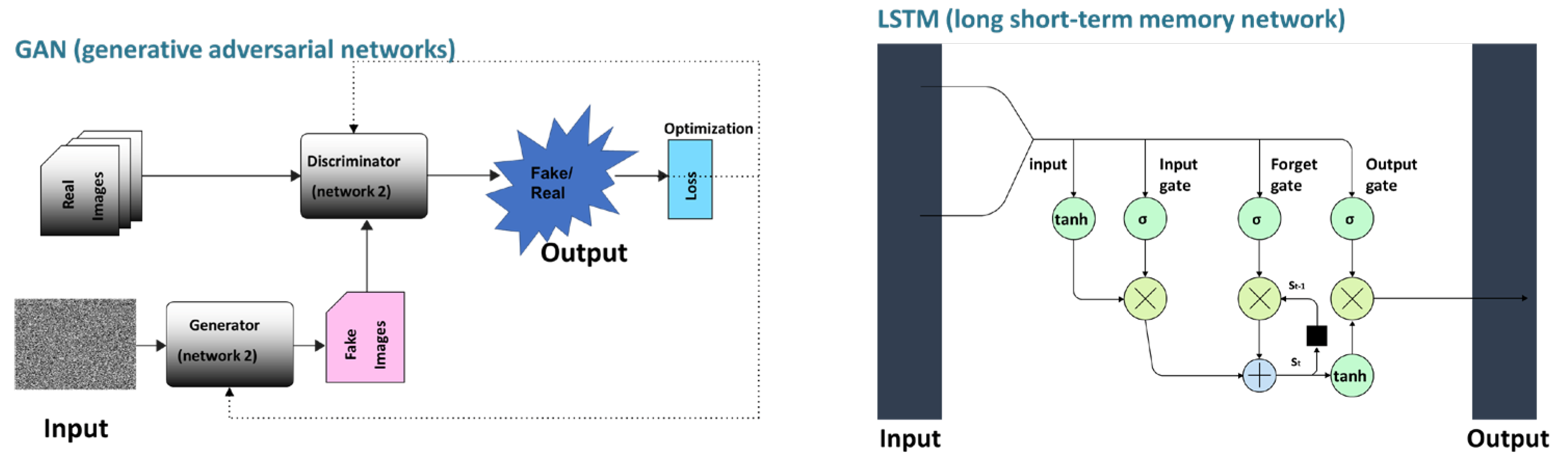
4. Machine/Deep Learning for Designing the Aptamer
4.1. Clustering for the Development of Aptamers Based on Machine Learning
4.1.1. Sequence-Based Clustering
4.1.2. Structure-Based Clustering
4.2. Machine/Deep Learning for the Prediction of the Structure of Aptamers
4.2.1. Machine/Deep Learning for Prediction of 2D Structure
4.2.2. Machine/Deep Learning for the Prediction of 3D Aptamer Structure
4.3. Trait-Based Machine Learning
4.4. Deep Learnings for Developing Aptamers
5. Application of Machine/Deep Learning for Aptamer Prediction
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as Targeted Therapeutics: Current Potential and Challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, U.S.; Hong, J.C. Recent Advances in Aptameric Biosensors Designed to Detect Toxic Contaminants from Food, Water, Human Fluids, and the Environment. Trends Environ. Anal. Chem. 2022, 36, e00184. [Google Scholar] [CrossRef]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent Advances in Aptamer Discovery and Applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [Green Version]
- Kinghorn, A.B.; Fraser, L.A.; Liang, S.; Shiu, S.C.; Tanner, J.A. Aptamer Bioinformatics. Int. J. Mol. Sci. 2017, 18, 2516. [Google Scholar] [CrossRef] [Green Version]
- Chushak, Y.; Stone, M.O. In Silico Selection of RNA Aptamers. Nucleic Acids Res. 2009, 37, e87. [Google Scholar] [CrossRef] [Green Version]
- Hofacker, I.L. Vienna RNA Secondary Structure Server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [Green Version]
- Ahirwar, R.; Nahar, S.; Aggarwal, S.; Ramachandran, S.; Maiti, S.; Nahar, P. In Silico Selection of an Aptamer to Estrogen Receptor Alpha Using Computational Docking Employing Estrogen Response Elements as Aptamer-Alike Molecules. Sci. Rep. 2016, 6, 21285. [Google Scholar] [CrossRef] [Green Version]
- Thafar, M.; Raies, A.B.; Albaradei, S.; Essack, M.; Bajic, V.B. Comparison Study of Computational Prediction Tools for Drug-Target Binding Affinities. Front. Chem. 2019, 7, 782. [Google Scholar] [CrossRef] [Green Version]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-Learning Scoring Functions to Improve Structure-Based Binding Affinity Prediction and Virtual Screening. WIREs Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef]
- Zhuo, Z.; Wan, Y.; Guan, D.; Ni, S.; Wang, L.; Zhang, Z.; Liu, J.; Liang, C.; Yu, Y.; Lu, A.; et al. A Loop-Based and AGO-Incorporated Virtual Screening Model Targeting AGO-Mediated MiRNA–MRNA Interactions for Drug Discovery to Rescue Bone Phenotype in Genetically Modified Mice. Adv. Sci. 2020, 7, 1903451. [Google Scholar] [CrossRef]
- Hamada, M. In Silico Approaches to RNA Aptamer Design. Biochimie 2018, 145, 8–14. [Google Scholar] [CrossRef]
- Hoinka, J.; Przytycka, T. AptaPLEX – A Dedicated, Multithreaded Demultiplexer for HT-SELEX Data. Methods 2016, 106, 82–85. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, J. SPA-LN: A Scoring Function of Ligand–Nucleic Acid Interactions via Optimizing Both Specificity and Affinity. Nucleic Acids Res. 2017, 45, e110. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Nucleobase Sequence Based Building up of Reliable QSAR Models with the Index of Ideality Correlation Using Monte Carlo Method. J. Biomol. Struct. Dyn. 2020, 38, 3296–3306. [Google Scholar] [CrossRef]
- Buglak, A.A.; Samokhvalov, A.V.; Zherdev, A.V.; Dzantiev, B.B. Methods and Applications of in Silico Aptamer Design and Modeling. Int. J. Mol. Sci. 2020, 21, 8420. [Google Scholar] [CrossRef]
- Sullivan, R.; Adams, M.C.; Naik, R.R.; Milam, V.T. Analyzing Secondary Structure Patterns in DNA Aptamers Identified via CompELS. Mol. 2019, 24, 1572. [Google Scholar] [CrossRef] [Green Version]
- Pagba, C.V.; Lane, S.M.; Cho, H.; Wachsmann-Hogiu, S. Direct Detection of Aptamer-Thrombin Binding via Surface-Enhanced Raman Spectroscopy. J. Biomed. Opt. 2010, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Jeddi, I.; Saiz, L. Three-Dimensional Modeling of Single Stranded DNA Hairpins for Aptamer-Based Biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Xu, X.; Chen, S.-J. Predicting RNA Structure with Vfold BT—Functional Genomics: Methods and Protocols; Kaufmann, M., Klinger, C., Savelsbergh, A., Eds.; Springer: New York, NY, USA, 2017; pp. 3–15. ISBN 978-1-4939-7231-9. [Google Scholar]
- Zuker, M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Domin, G.; Findeiß, S.; Wachsmuth, M.; Will, S.; Stadler, P.F.; Mörl, M. Applicability of a Computational Design Approach for Synthetic Riboswitches. Nucleic Acids Res. 2017, 45, 4108–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.J.; Gloor, J.W.; Mathews, D.H. Improved RNA Secondary Structure Prediction by Maximizing Expected Pair Accuracy. RNA 2009, 15, 1805–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Lawrence, C.E. A Statistical Sampling Algorithm for RNA Secondary Structure Prediction. Nucleic Acids Res. 2003, 31, 7280–7301. [Google Scholar] [CrossRef] [Green Version]
- Bellaousov, S.; Mathews, D.H. ProbKnot: Fast Prediction of RNA Secondary Structure Including Pseudoknots. RNA 2010, 16, 1870–1880. [Google Scholar] [CrossRef] [Green Version]
- Hilder, T.A.; Hodgkiss, J.M. The Bound Structures of 17β-Estradiol-Binding Aptamers. ChemPhysChem 2017, 18, 1881–1887. [Google Scholar] [CrossRef]
- Rockey, W.M.; Hernandez, F.J.; Huang, S.-Y.; Cao, S.; Howell, C.A.; Thomas, G.S.; Liu, X.Y.; Lapteva, N.; Spencer, D.M.; McNamara, J.O.; et al. Rational Truncation of an RNA Aptamer to Prostate-Specific Membrane Antigen Using Computational Structural Modeling. Nucleic Acid Ther. 2011, 21, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhao, P.; Chen, S.-J. Vfold: A Web Server for RNA Structure and Folding Thermodynamics Prediction. PLoS ONE 2014, 9, e107504. [Google Scholar] [CrossRef]
- Sato, K.; Hamada, M.; Asai, K.; Mituyama, T. CentroidFold: A Web Server for RNA Secondary Structure Prediction. Nucleic Acids Res. 2009, 37, W277–W280. [Google Scholar] [CrossRef] [Green Version]
- Biesiada, M.; Pachulska-Wieczorek, K.; Adamiak, R.W.; Purzycka, K.J. RNAComposer and RNA 3D Structure Prediction for Nanotechnology. Methods 2016, 103, 120–127. [Google Scholar] [CrossRef]
- Hu, W.-P.; Kumar, J.V.; Huang, C.-J.; Chen, W.-Y. Computational Selection of RNA Aptamer against Angiopoietin-2 and Experimental Evaluation. Biomed Res. Int. 2015, 2015, 658712. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, J.; Huang, Y.; Xiao, Y. 3dRNA v2.0: An Updated Web Server for RNA 3D Structure Prediction. Int. J. Mol. Sci. 2019, 20, 4116. [Google Scholar] [CrossRef] [Green Version]
- Soon, S.; Aina Nordin, N. In Silico Predictions and Optimization of Aptamers against Streptococcus Agalactiae Surface Protein Using Computational Docking. Mater. Today Proc. 2019, 16, 2096–2100. [Google Scholar] [CrossRef]
- Xu, X.; Dickey, D.D.; Chen, S.-J.; Giangrande, P.H. Structural Computational Modeling of RNA Aptamers. Methods 2016, 103, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Boniecki, M.J.; Lach, G.; Dawson, W.K.; Tomala, K.; Lukasz, P.; Soltysinski, T.; Rother, K.M.; Bujnicki, J.M. SimRNA: A Coarse-Grained Method for RNA Folding Simulations and 3D Structure Prediction. Nucleic Acids Res. 2016, 44, e63. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, L.; Zhang, B.T.; Lu, A.; Wang, Y.; Yu, Y.; Zhang, G. Artificial Intelligence in Aptamer–Target Binding Prediction. Int. J. Mol. Sci. 2021, 22, 3605. [Google Scholar] [CrossRef]
- Sabri, M.Z.; Abdul Hamid, A.A.; Sayed Hitam, S.M.; Abdul Rahim, M.Z. In Silico Screening of Aptamers Configuration against Hepatitis B Surface Antigen. Adv. Bioinformatics 2019, 2019, 6912914. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-L.; Cui, H.-F.; Du, J.-F.; Lv, Q.-Y.; Song, X. In Silico Post-SELEX Screening and Experimental Characterizations for Acquisition of High Affinity DNA Aptamers against Carcinoembryonic Antigen. RSC Adv. 2019, 9, 6328–6334. [Google Scholar] [CrossRef] [Green Version]
- Zavyalova, E.; Golovin, A.; Reshetnikov, R.; Mudrik, N.; Panteleyev, D.; Kopylov, G.P. and A. Novel Modular DNA Aptamer for Human Thrombin with High Anticoagulant Activity. Curr. Med. Chem. 2011, 18, 3343–3350. [Google Scholar] [CrossRef]
- Riccardi, C.; Napolitano, E.; Platella, C.; Musumeci, D.; Montesarchio, D. G-Quadruplex-Based Aptamers Targeting Human Thrombin: Discovery, Chemical Modifications and Antithrombotic Effects. Pharmacol. Ther. 2021, 217, 107649. [Google Scholar] [CrossRef]
- Roxo, C.; Kotkowiak, W.; Pasternak, A. G-Quadruplex-Forming Aptamers—Characteristics, Applications, and Perspectives. Molecules 2019, 24, 3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webba da Silva, M. NMR Methods for Studying Quadruplex Nucleic Acids. Methods 2007, 43, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.H.; Parkinson, G.N. Crystallographic Studies of Quadruplex Nucleic Acids. Methods 2007, 43, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, E.P.; Londoño-Vallejo, A. A Guide to Computational Methods for G-Quadruplex Prediction. Nucleic Acids Res. 2020, 48, 1603. [Google Scholar] [CrossRef] [Green Version]
- Hon, J.; Martínek, T.; Zendulka, J.; Lexa, M. Pqsfinder: An Exhaustive and Imperfection-Tolerant Search Tool for Potential Quadruplex-Forming Sequences in R. Bioinformatics 2017, 33, 3373–3379. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [Green Version]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef] [Green Version]
- Cataldo, R.; Ciriaco, F.; Alfinito, E. A Validation Strategy for in Silico Generated Aptamers. Comput. Biol. Chem. 2018, 77, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK Server: Interactive Docking Prediction of Protein–Protein Complexes and Symmetric Multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating Protein Docking in ZDOCK Using an Advanced 3D Convolution Library. PLoS One 2011, 6, e24657. [Google Scholar] [CrossRef]
- Biesiada, J.; Porollo, A.; Velayutham, P.; Kouril, M.; Meller, J. Survey of Public Domain Software for Docking Simulations and Virtual Screening. Hum. Genomics 2011, 5, 497. [Google Scholar] [CrossRef] [Green Version]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining Techniques to Model RNA–Small Molecule Complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- Shcherbinin, D.S.; Gnedenko, O.V.; Khmeleva, S.A.; Usanov, S.A.; Gilep, A.A.; Yantsevich, A.V.; Shkel, T.V.; Yushkevich, I.V.; Radko, S.P.; Ivanov, A.S.; et al. Computer-Aided Design of Aptamers for Cytochrome P450. J. Struct. Biol. 2015, 191, 112–119. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. MDockPP: A Hierarchical Approach for Protein-Protein Docking and Its Application to CAPRI Rounds 15–19. Proteins Struct. Funct. Bioinforma. 2010, 78, 3096–3103. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham III, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Moccia, F.; Platella, C.; Musumeci, D.; Batool, S.; Zumrut, H.; Bradshaw, J.; Mallikaratchy, P.; Montesarchio, D. The Role of G-Quadruplex Structures of LIGS-Generated Aptamers R1.2 and R1.3 in IgM Specific Recognition. Int. J. Biol. Macromol. 2019, 133, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Tucker, O.W.; Shum, T.K.; Tanner, A.J. G-Quadruplex DNA Aptamers and Their Ligands: Structure, Function and Application. Curr. Pharm. Des. 2012, 18, 2014–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.-Y.; Ashrafuzzaman, M.; Mane, J.Y.; Kapty, J.; Mercer, J.R.; Tuszynski, J.A. Entropic Fragment-Based Approach to Aptamer Design. Chem. Biol. Drug Des. 2011, 78, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lietard, J.; Assi, H.A.; Gómez-Pinto, I.; González, C.; Somoza, M.M.; Damha, M.J. Mapping the Affinity Landscape of Thrombin-Binding Aptamers on 2F-ANA/DNA Chimeric G-Quadruplex Microarrays. Nucleic Acids Res. 2017, 45, 1619–1632. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Olson, W.K. 3DNA: A Software Package for the Analysis, Rebuilding and Visualization of Three-dimensional Nucleic Acid Structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [Green Version]
- Varizhuk, A.M.; Tsvetkov, V.B.; Tatarinova, O.N.; Kaluzhny, D.N.; Florentiev, V.L.; Timofeev, E.N.; Shchyolkina, A.K.; Borisova, O.F.; Smirnov, I.P.; Grokhovsky, S.L.; et al. Synthesis, Characterization and in Vitro Activity of Thrombin-Binding DNA Aptamers with Triazole Internucleotide Linkages. Eur. J. Med. Chem. 2013, 67, 90–97. [Google Scholar] [CrossRef]
- Tsvetkov, V.B.; Varizhuk, A.M.; Pozmogova, G.E.; Smirnov, I.P.; Kolganova, N.A.; Timofeev, E.N. A Universal Base in a Specific Role: Tuning up a Thrombin Aptamer with 5-Nitroindole. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Mahmood, M.A.I.; Ali, W.; Adnan, A.; Iqbal, S.M. 3D Structural Integrity and Interactions of Single-Stranded Protein-Binding Dna in a Functionalized Nanopore. J. Phys. Chem. B 2014, 118, 5799–5806. [Google Scholar] [CrossRef]
- Rangnekar, A.; Nash, J.A.; Goodfred, B.; Yingling, Y.G.; LaBean, T.H. Design of Potent and Controllable Anticoagulants Using DNA Aptamers and Nanostructures. Mol. 2016, 21, 202. [Google Scholar] [CrossRef] [Green Version]
- Van Riesen, A.J.; Fadock, K.L.; Deore, P.S.; Desoky, A.; Manderville, R.A.; Sowlati-Hashjin, S.; Wetmore, S.D. Manipulation of a DNA Aptamer-Protein Binding Site through Arylation of Internal Guanine Residues. Org. Biomol. Chem. 2018, 16, 3831–3840. [Google Scholar] [CrossRef]
- Sgobba, M.; Olubiyi, O.; Ke, S.; Haider, S. Molecular Dynamics of HIV1-Integrase in Complex with 93del—A Structural Perspective on the Mechanism of Inhibition. J. Biomol. Struct. Dyn. 2012, 29, 863–877. [Google Scholar] [CrossRef]
- Nguyen, P.D.M.; Zheng, J.; Gremminger, T.J.; Qiu, L.; Zhang, D.; Tuske, S.; Lange, M.J.; Griffin, P.R.; Arnold, E.; Chen, S.-J.; et al. Binding Interface and Impact on Protease Cleavage for an RNA Aptamer to HIV-1 Reverse Transcriptase. Nucleic Acids Res. 2020, 48, 2709–2722. [Google Scholar] [CrossRef]
- Xu, X.; Qiu, L.; Yan, C.; Ma, Z.; Grinter, S.Z.; Zou, X. Performance of MDockPP in CAPRI Rounds 28-29 and 31-35 Including the Prediction of Water-Mediated Interactions. Proteins Struct. Funct. Bioinforma. 2017, 85, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Musafia, B.; Oren-Banaroya, R.; Noiman, S. Designing Anti-Influenza Aptamers: Novel Quantitative Structure Activity Relationship Approach Gives Insights into Aptamer – Virus Interaction. PLoS One 2014, 9, e97696. [Google Scholar] [CrossRef]
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, Z.; Yang, C. Discovery of Aptamers Targeting the Receptor-Binding Domain of the SARS-CoV-2 Spike Glycoprotein. Anal. Chem. 2020, 92, 9895–9900. [Google Scholar] [CrossRef]
- Song, J.; Zheng, Y.; Huang, M.; Wu, L.; Wang, W.; Zhu, Z.; Song, Y.; Yang, C. A Sequential Multidimensional Analysis Algorithm for Aptamer Identification Based on Structure Analysis and Machine Learning. Anal. Chem. 2020, 92, 3307–3314. [Google Scholar] [CrossRef]
- Gupta, A.; Anand, A.; Jain, N.; Goswami, S.; Ananthraj, A.; Patil, S.; Singh, R.; Kumar, A.; Shrivastava, T.; Bhatnagar, S.; et al. A Novel G-Quadruplex Aptamer-Based Spike Trimeric Antigen Test for the Detection of SARS-CoV-2. Mol. Ther.-Nucleic Acids 2021. [Google Scholar] [CrossRef]
- Bellaousov, S.; Reuter, J.S.; Seetin, M.G.; Mathews, D.H. RNAstructure: Web Servers for RNA Secondary Structure Prediction and Analysis. Nucleic Acids Res. 2013, 41, 471–474. [Google Scholar] [CrossRef] [Green Version]
- Bavi, R.; Liu, Z.; Han, Z.; Zhang, H.; Gu, Y. In Silico Designed RNA Aptamer against Epithelial Cell Adhesion Molecule for Cancer Cell Imaging. Biochem. Biophys. Res. Commun. 2019, 509, 937–942. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Bell, D.R.; Weber, J.K.; Yin, W.; Huynh, T.; Duan, W.; Zhou, R. In Silico Design and Validation of High-Affinity RNA Aptamers Targeting Epithelial Cellular Adhesion Molecule Dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 8486–8493. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, V.A.; Thompson, E.E.; Pique, M.E.; Perez, M.S.; Ten Eyck, L.F. DOT2: Macromolecular Docking with Improved Biophysical Models. J. Comput. Chem. 2013, 34, 1743–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, B.L.; Zúñiga-Bustos, M.; Vidal-Limon, A.; Alderete, J.B.; Águila, S.A.; Jiménez, V.A. In Silico Design of Novel Mutant Anti-MUC1 Aptamers for Targeted Cancer Therapy. J. Chem. Inf. Model. 2020, 60, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [Green Version]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK Web Server for Data-Driven Biomolecular Docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Heiat, M.; Najafi, A.; Ranjbar, R.; Latifi, A.M.; Rasaee, M.J. Computational Approach to Analyze Isolated SsDNA Aptamers against Angiotensin II. J. Biotechnol. 2016, 230, 34–39. [Google Scholar] [CrossRef]
- Rabal, O.; Pastor, F.; Villanueva, H.; Soldevilla, M.M.; Hervas-Stubbs, S.; Oyarzabal, J. In Silico Aptamer Docking Studies: From a Retrospective Validation to a Prospective Case Study’TIM3 Aptamers Binding. Mol. Ther.-Nucleic Acids 2016, 5, e376. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Chou, F.-C.; Das, R. Chapter Two—Modeling Complex RNA Tertiary Folds with Rosetta. In Computational Methods for Understanding Riboswitches; Chen, S.-J., Burke-Aguero, D.H., Eds.; Academic Press: Cambridge, MA, USA, 2015; ISBN 0076-6879. [Google Scholar]
- Huang, Y.; Liu, S.; Guo, D.; Li, L.; Xiao, Y. A Novel Protocol for Three-Dimensional Structure Prediction of RNA-Protein Complexes. Sci. Rep. 2013, 3, 1887. [Google Scholar] [CrossRef] [Green Version]
- Trinh, K.H.; Kadam, U.S.; Rampogu, S.; Cho, Y.; Yang, K.A.; Kang, C.H.; Lee, K.W.; Lee, K.O.; Chung, W.S.; Hong, J.C. Development of Novel Fluorescence-Based and Label-Free Noncanonical G4-Quadruplex-like DNA Biosensor for Facile, Specific, and Ultrasensitive Detection of Fipronil. J. Hazard. Mater. 2022, 427, 127939. [Google Scholar] [CrossRef]
- Kadam, U.S.; Trinh, K.H.; Kumar, V.; Lee, K.W.; Cho, Y.; Can, M.H.T.; Lee, H.; Kim, Y.; Kim, S.; Kang, J.; et al. Identification and Structural Analysis of Novel Malathion-Specific DNA Aptameric Sensors Designed for Food Testing. Biomaterials 2022, 287, 121617. [Google Scholar] [CrossRef]
- Mousivand, M.; Anfossi, L.; Bagherzadeh, K.; Barbero, N.; Mirzadi-Gohari, A.; Javan-Nikkhah, M. In Silico Maturation of Affinity and Selectivity of DNA Aptamers against Aflatoxin B1 for Biosensor Development. Anal. Chim. Acta 2020, 1105, 178–186. [Google Scholar] [CrossRef]
- Fukaya, T.; Abe, K.; Savory, N.; Tsukakoshi, K.; Yoshida, W.; Ferri, S.; Sode, K.; Ikebukuro, K. Improvement of the VEGF Binding Ability of DNA Aptamers through in Silico Maturation and Multimerization Strategy. J. Biotechnol. 2015, 212, 99–105. [Google Scholar] [CrossRef]
- Nonaka, Y.; Yoshida, W.; Abe, K.; Ferri, S.; Schulze, H.; Bachmann, T.T.; Ikebukuro, K. Affinity Improvement of a VEGF Aptamer by in Silico Maturation for a Sensitive VEGF-Detection System. Anal. Chem. 2013, 85, 1132–1137. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep Learning in Neural Networks: An Overview. Neural Networks 2015, 61, 85–117. [Google Scholar] [CrossRef] [Green Version]
- Pahikkala, T.; Airola, A.; Pietilä, S.; Shakyawar, S.; Szwajda, A.; Tang, J.; Aittokallio, T. Toward More Realistic Drug-Target Interaction Predictions. Brief. Bioinform. 2015, 16, 325–337. [Google Scholar] [CrossRef]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A Read-across Approach for Predicting Drug-Target Binding Affinities Using Gradient Boosting Machines. J. Cheminform. 2017, 9, 24. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, S.-P.; Yu, X.-L.; Yang, X.-H.; Guo, Q.-P.; Tang, L.-J.; Jiang, J.-H.; Yu, R.-Q. A Novel Nucleic Acid Sequence Encoding Strategy for High-Performance Aptamer Identification and the Aid of Sequence Design and Optimization. Chemom. Intell. Lab. Syst. 2017, 170, 32–37. [Google Scholar] [CrossRef]
- Hoinka, J.; Backofen, R.; Przytycka, T.M. AptaSUITE: A Full-Featured Bioinformatics Framework for the Comprehensive Analysis of Aptamers from HT-SELEX Experiments. Mol. Ther.-Nucleic Acids 2018, 11, 515–517. [Google Scholar] [CrossRef]
- Ishida, R.; Adachi, T.; Yokota, A.; Yoshihara, H.; Aoki, K.; Nakamura, Y.; Hamada, M. RaptRanker: In Silico RNA Aptamer Selection from HT-SELEX Experiment Based on Local Sequence and Structure Information. Nucleic Acids Res. 2020, 48. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Li, X.; Gu, J. PPAI: A Web Server for Predicting Protein-Aptamer Interactions. BMC Bioinformatics 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shieh, K.R.; Kratschmer, C.; Maier, K.E.; Greally, J.M.; Levy, M.; Golden, A. AptCompare: Optimized de Novo Motif Discovery of RNA Aptamers via HTS-SELEX. Bioinformatics 2020, 36, 2905–2906. [Google Scholar] [CrossRef] [PubMed]
- Caroli, J.; Forcato, M.; Bicciato, S. APTANI2: Update of Aptamer Selection through Sequence-Structure Analysis. Bioinformatics 2020, 36, 2266–2268. [Google Scholar] [CrossRef] [PubMed]
- Emami, N.; Ferdousi, R. AptaNet as a Deep Learning Approach for Aptamer–Protein Interaction Prediction. Sci. Rep. 2021, 11, 1–19. [Google Scholar] [CrossRef]
- Hoinka, J.; Berezhnoy, A.; Sauna, Z.E.; Gilboa, E.; Przytycka, T.M. AptaCluster—A Method to Cluster HT-SELEX Aptamer Pools and Lessons from Its Application BT—Research in Computational Molecular Biology; Sharan, R., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 115–128. [Google Scholar]
- Alam, K.K.; Chang, J.L.; Burke, D.H. FASTAptamer: A Bioinformatic Toolkit for High-Throughput Sequence Analysis of Combinatorial Selections. Mol. Ther.-Nucleic Acids 2015, 4, e230. [Google Scholar] [CrossRef]
- Dao, P.; Hoinka, J.; Takahashi, M.; Zhou, J.; Ho, M.; Wang, Y.; Costa, F.; Rossi, J.J.; Backofen, R.; Burnett, J.; et al. AptaTRACE Elucidates RNA Sequence-Structure Motifs from Selection Trends in HT-SELEX Experiments. Cell Syst. 2016, 3, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Caroli, J.; Taccioli, C.; De La Fuente, A.; Serafini, P.; Bicciato, S. APTANI: A Computational Tool to Select Aptamers through Sequence-Structure Motif Analysis of HT-SELEX Data. Bioinformatics 2016, 32, 161–164. [Google Scholar] [CrossRef]
- BINDEWALD, E.; SHAPIRO, B.A. RNA Secondary Structure Prediction from Sequence Alignments Using a Network of K-Nearest Neighbor Classifiers. RNA 2006, 12, 342–352. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Hanson, J.; Paliwal, K.; Zhou, Y. RNA Secondary Structure Prediction Using an Ensemble of Two-Dimensional Deep Neural Networks and Transfer Learning. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Fudenberg, G.; Kelley, D.R.; Pollard, K.S. Predicting 3D Genome Folding from DNA Sequence with Akita. Nat. Methods 2020, 17, 1111–1117. [Google Scholar] [CrossRef]
- Li, B.-Q.; Zhang, Y.-C.; Huang, G.-H.; Cui, W.-R.; Zhang, N.; Cai, Y.-D. Prediction of Aptamer-Target Interacting Pairs with Pseudo-Amino Acid Composition. PLoS One 2014, 9, e86729. [Google Scholar] [CrossRef] [Green Version]
- DING, C.; PENG, H. MINIMUM REDUNDANCY FEATURE SELECTION FROM MICROARRAY GENE EXPRESSION DATA. J. Bioinform. Comput. Biol. 2005, 03, 185–205. [Google Scholar] [CrossRef]
- Katakis, I.M.; Tsoumakas, G.; Vlahavas, I.P. Dynamic Feature Space and Incremental Feature Selection for the Classification of Textual Data Streams; Aristotle University of Thessaloniki: Thessaloniki, Greece, 2006. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Hong, Z.; Wenzhen, J.; Guocai, Y. An Effective Text Classification Model Based on Ensemble Strategy. J. Phys. Conf. Ser. 2019, 1229, 12058. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Wornow, M. Applying Deep Learning to Discover Highly Functionalized Nucleic Acid Polymers That Bind to Small Molecules. Ph.D. Thesis, Harvard University, Cambridge, MA, USA, 2020. [Google Scholar]
- Valueva, M.V.; Nagornov, N.N.; Lyakhov, P.A.; Valuev, G.V.; Chervyakov, N.I. Application of the Residue Number System to Reduce Hardware Costs of the Convolutional Neural Network Implementation. Math. Comput. Simul. 2020, 177, 232–243. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Y.; Yang, H.; Huang, X. Prediction of the Binding Affinity of Aptamers against the Influenza Virus. SAR QSAR Environ. Res. 2019, 30, 51–62. [Google Scholar] [CrossRef]
- van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian Interaction Profile Kernels for Predicting Drug–Target Interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [Green Version]
- Ashtawy, H.M.; Mahapatra, N.R. A Comparative Assessment of Ranking Accuracies of Conventional and Machine-Learning-Based Scoring Functions for Protein-Ligand Binding Affinity Prediction. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2012, 9, 1301–1313. [Google Scholar] [CrossRef]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and Evaluation of a Deep Learning Model for Protein–Ligand Binding Affinity Prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Shin, W.-H.; Ko, J.; Lee, J. AK-Score: Accurate Protein-Ligand Binding Affinity Prediction Using an Ensemble of 3D-Convolutional Neural Networks. Int. J. Mol. Sci. 2020, 21, 8424. [Google Scholar] [CrossRef]
- Ashtawy, H.M.; Mahapatra, N.R. BgN-Score and BsN-Score: Bagging and Boosting Based Ensemble Neural Networks Scoring Functions for Accurate Binding Affinity Prediction of Protein-Ligand Complexes. BMC Bioinformatics 2015, 16, S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. DeepAffinity: Interpretable Deep Learning of Compound–Protein Affinity through Unified Recurrent and Convolutional Neural Networks. Bioinformatics 2019, 35, 3329–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep Drug–Target Binding Affinity Prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [Green Version]
- Öztürk, H.; Ozkirimli, E.; Özgür, A. WideDTA: Prediction of Drug-Target Binding Affinity. arXiv Quant. Methods 2019. [Google Scholar]
- Ghimire, A.; Tayara, H.; Xuan, Z.; Chong, K.T. CSatDTA: Prediction of Drug–Target Binding Affinity Using Convolution Model with Self-Attention. Int. J. Mol. Sci. 2022, 23, 8453. [Google Scholar] [CrossRef]
- Deng, L.; Zeng, Y.; Liu, H.; Liu, Z.; Liu, X. DeepMHADTA: Prediction of Drug-Target Binding Affinity Using Multi-Head Self-Attention and Convolutional Neural Network. Curr. Issues Mol. Biol. 2022, 44, 2287–2299. [Google Scholar] [CrossRef]
- Saadat, M.; Behjati, A.; Zare-Mirakabad, F.; Gharaghani, S. Drug-Target Binding Affinity Prediction Using Transformers. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.; Pang, L.; Liu, Y.; Zhang, J. GANsDTA: Predicting Drug-Target Binding Affinity Using GANs. Front. Genet. 2020, 10, 1243. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Before In Silico Method | In Silico Method | References |
|---|---|---|---|
| Aflatoxin B1 | 38.5 pM | 4.02 pM | [95] |
| EpCAM | 39.89 nM | 10.78 nM | [82] |
| Vascular Endothelial Growth Factor | 200 nM | 52 nM | [96] |
| Vascular Endothelial Growth Factor | 4.7 nM | 300 pM | [97] |
| Tools | Features | Reference |
|---|---|---|
| Apta-loopEnc | Labels the candidates with high and low binding affinity. Predicts aptamer based on SVM | [101] |
| AptaSUITE | Framework analysis of data from HT-SELEX such as sequences and aptamer counts. | [102] |
| SMART-Aptamer | Predicts aptamers based on ranking of sequence abundance, stability of the secondary structure | [77] |
| RaptRanker | Predicts aptamer based on structure and frequency of sequence | [103] |
| PPAI (http://39.96.85.9/PPAI/, accessed on 30 December 2022) | Web server for prediction of aptamers and interaction between protein and aptamer | [104] |
| AptCompare | Meta-analysis platform for HT-SELEX | [105] |
| APTANI2 | GUI platform for aptamers based on frequency of sequence and stability of secondary structure | [106] |
| AptaNet | Predicts the affinity of aptamer-protein using a multi-layer perceptron as a classification model. | [107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.J.; Cho, J.; Lee, B.-H.; Hwang, D.; Park, J.-W. Design and Prediction of Aptamers Assisted by In Silico Methods. Biomedicines 2023, 11, 356. https://doi.org/10.3390/biomedicines11020356
Lee SJ, Cho J, Lee B-H, Hwang D, Park J-W. Design and Prediction of Aptamers Assisted by In Silico Methods. Biomedicines. 2023; 11(2):356. https://doi.org/10.3390/biomedicines11020356
Chicago/Turabian StyleLee, Su Jin, Junmin Cho, Byung-Hoon Lee, Donghwan Hwang, and Jee-Woong Park. 2023. "Design and Prediction of Aptamers Assisted by In Silico Methods" Biomedicines 11, no. 2: 356. https://doi.org/10.3390/biomedicines11020356
APA StyleLee, S. J., Cho, J., Lee, B. -H., Hwang, D., & Park, J. -W. (2023). Design and Prediction of Aptamers Assisted by In Silico Methods. Biomedicines, 11(2), 356. https://doi.org/10.3390/biomedicines11020356