
Minimal Collagen-Binding Epitope of Glycoprotein VI in Human and Mouse Platelets
 ,
,  ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
1.1. Collagen-Binding Sites of GPVI Receptor
1.2. CRP-Binding Sites of GPVI Receptor
1.3. Anti-Thrombotic Approaches Based on the Blockade of GPVI-Collagen Interaction
2. Materials and Methods
2.1. Computation Method and EA-20 Antibody Design
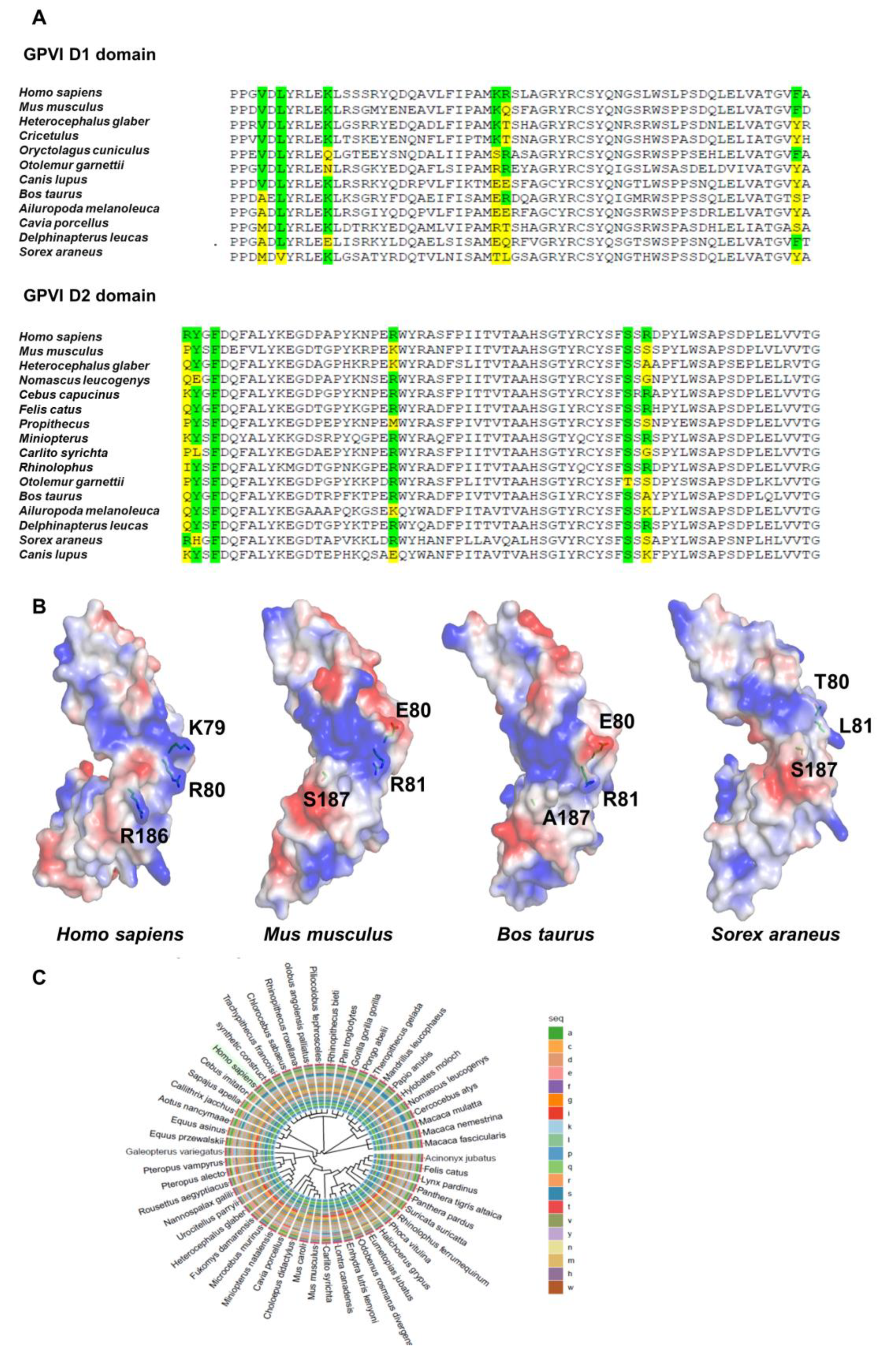
2.2. Homology Modeling and Phylogenetic Analysis
2.3. Generation of Humanized GPVI Mice
2.4. Platelet Isolation
2.5. Platelet Aggregation
2.6. Static Adhesion Assay
2.7. Platelet Adhesion under Flow
2.8. Measurement of Soluble GPVI
2.9. Statistical Analysis
3. Results
3.1. Collagen and CRP-Binding with GPVI Monomer in Mammalian Species
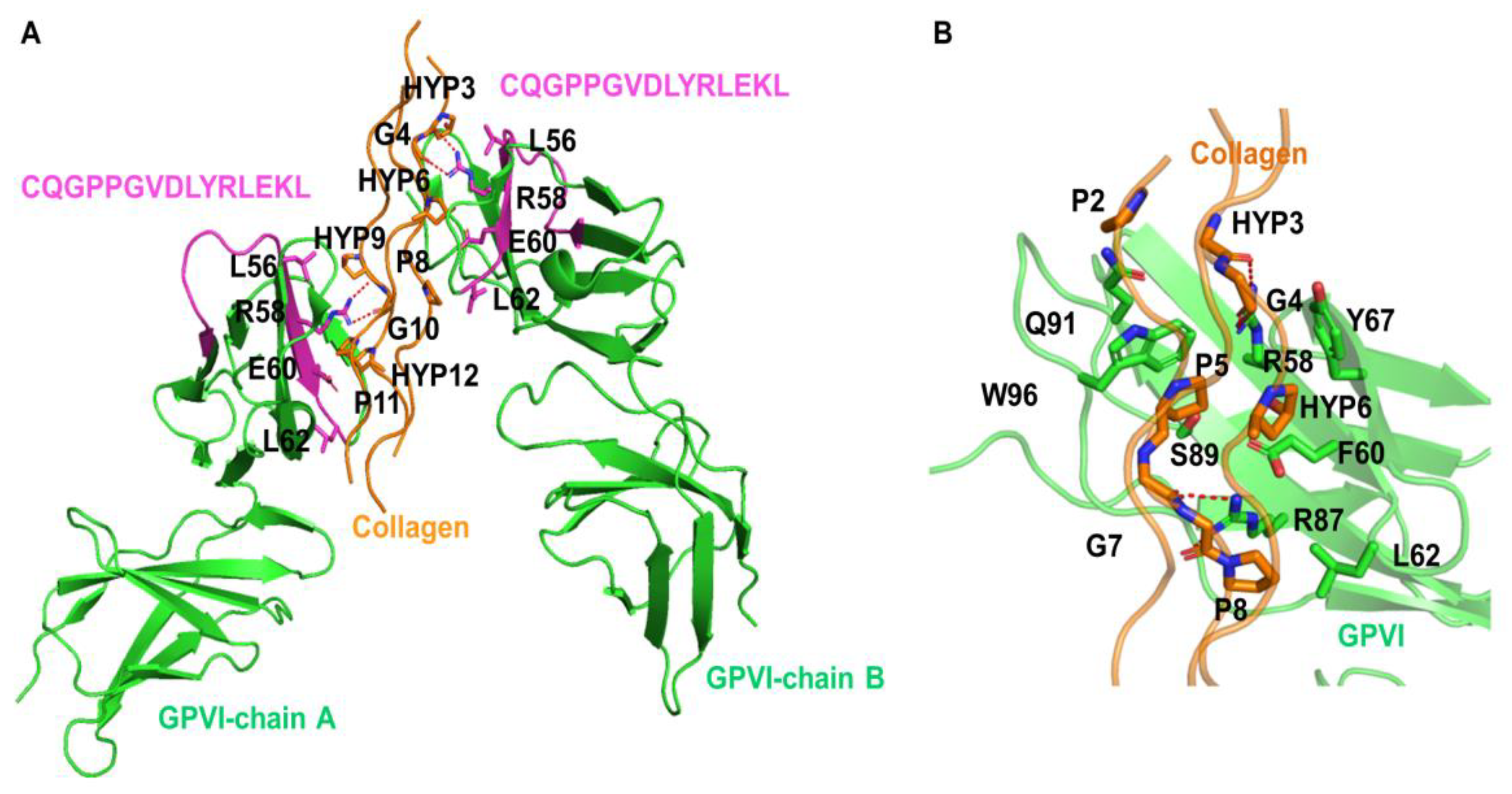
3.2. Collagen-Binding with GPVI Dimer in Humans
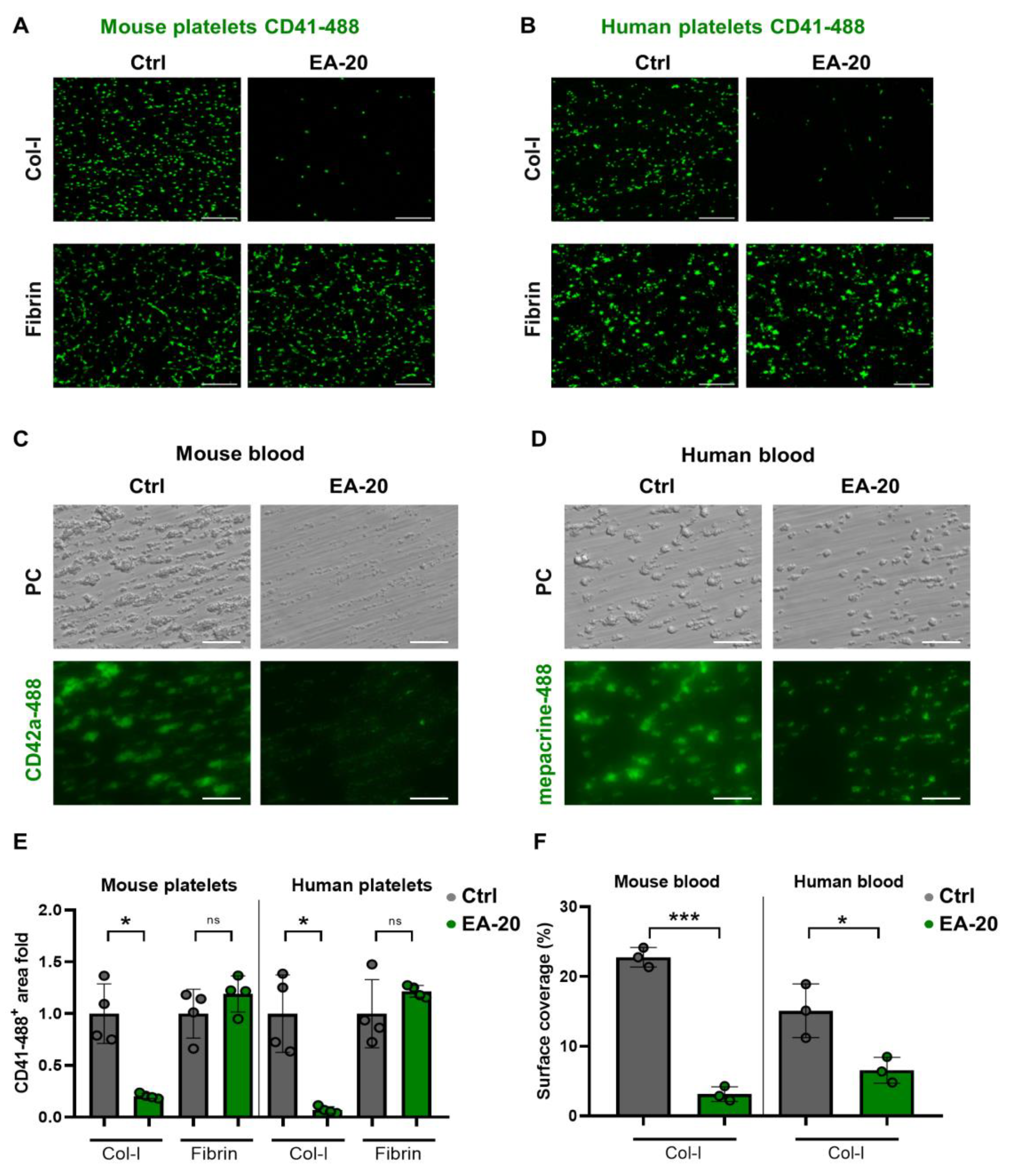
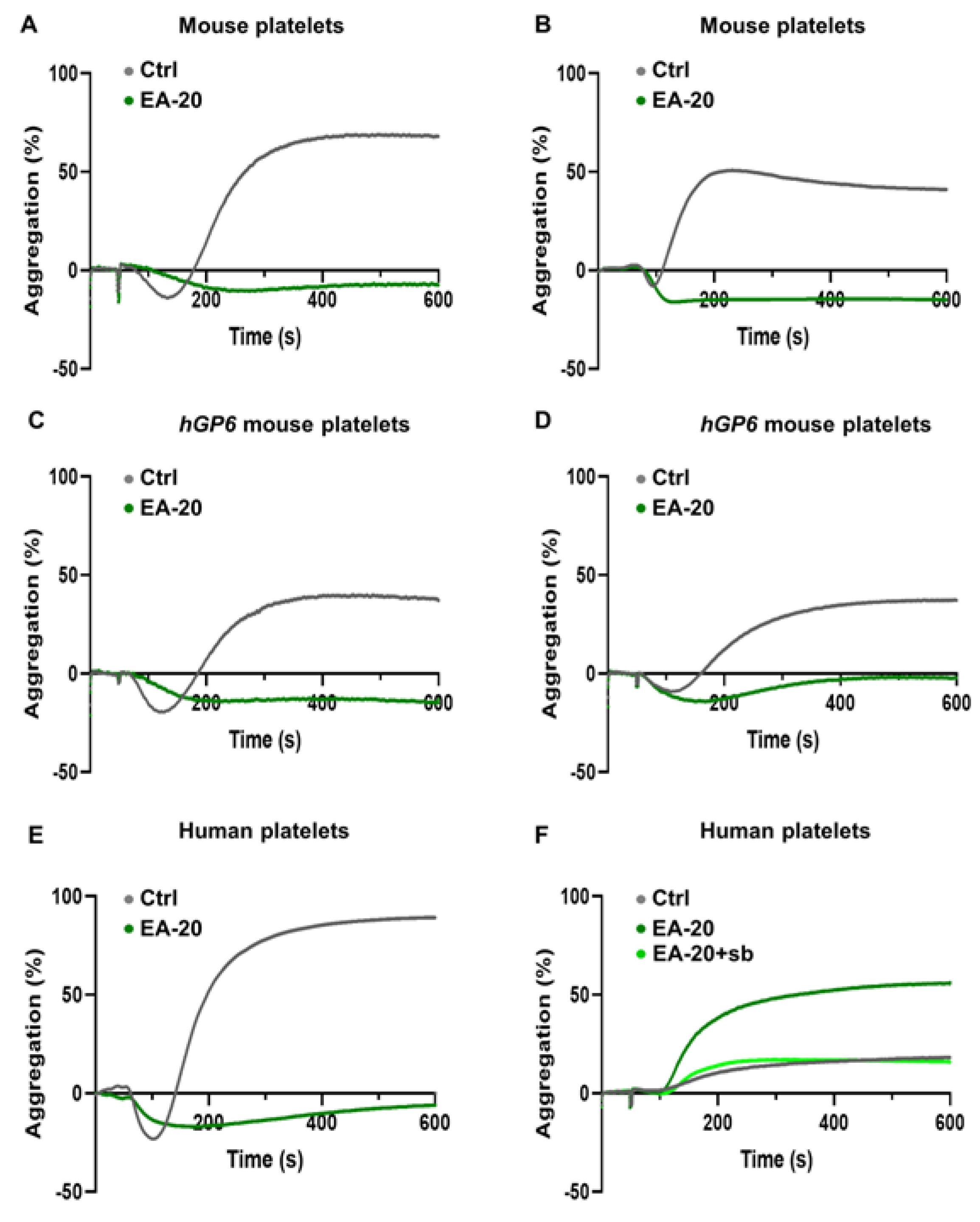
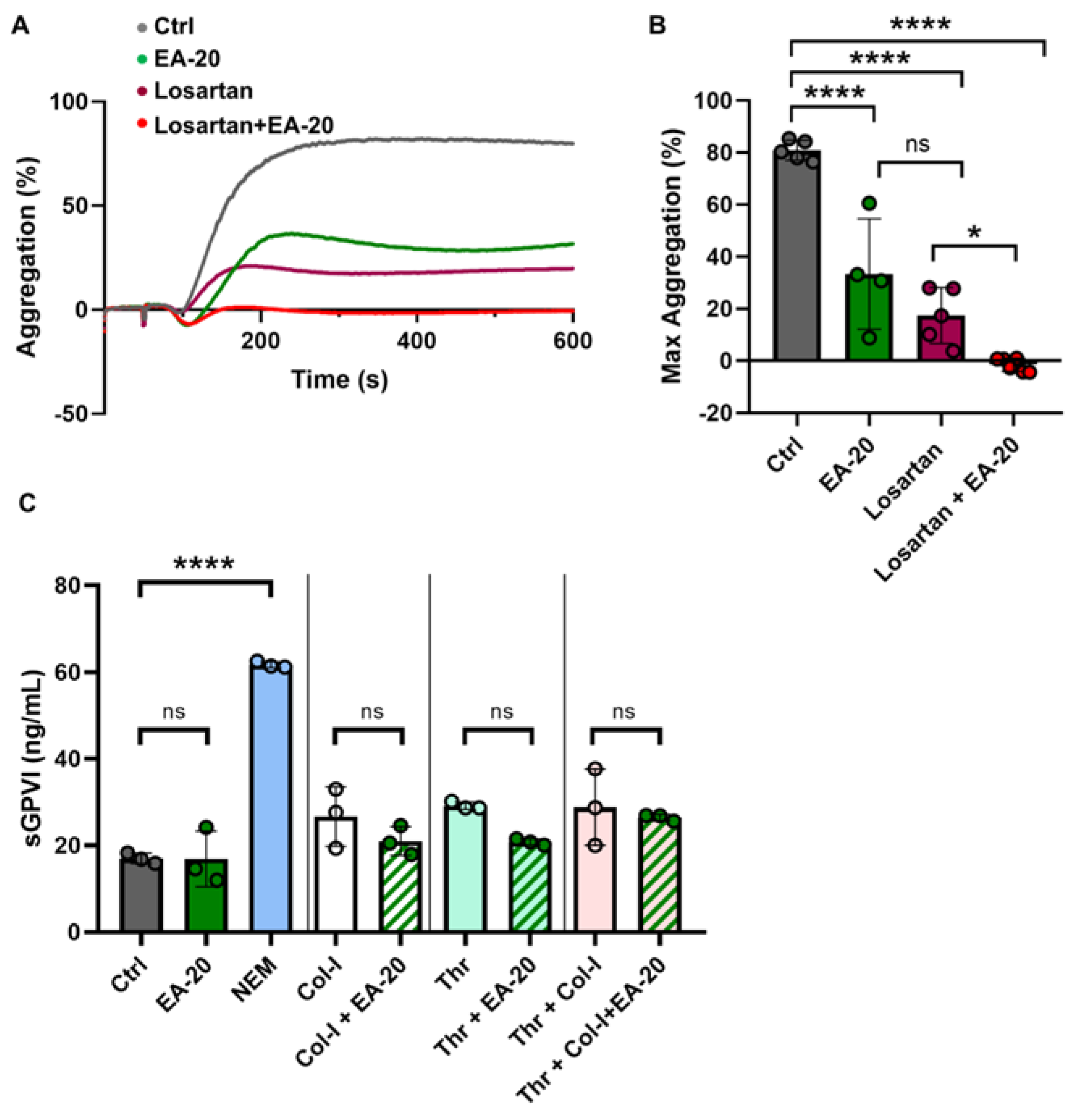
3.3. EA-20 Antibody Inhibits GPVI-Mediated Platelet Activation and Thrombus Formation on Collagen, but Not on Fibrin
3.4. Effect of EA-20 Antibody on GPVI-Clustering and Shedding in Human Platelets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scharf, R.E. Platelet Signaling in Primary Haemostasis and Arterial Thrombus Formation: Part 2. Hamostaseologie 2018, 38, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Scharf, R.E. Platelet Signaling in Primary Haemostasis and Arterial Thrombus Formation: Part 1. Hamostaseologie 2018, 38, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Jaeken, J.; Gudermann, T.; Braun, A. Platelets and Defective N-Glycosylation. Int. J. Mol. Sci. 2020, 21, 5630. [Google Scholar] [CrossRef]
- Badimon, L.; Badimon, J.J.; Turitto, V.T.; Vallabhajosula, S.; Fuster, V. Platelet thrombus formation on collagen type I. A model of deep vessel injury. Influence of blood rheology, von Willebrand factor, and blood coagulation. Circulation 1988, 78, 1431–1442. [Google Scholar] [CrossRef]
- Xu, X.R.; Carrim, N.; Neves, M.A.; McKeown, T.; Stratton, T.W.; Coelho, R.M.; Lei, X.; Chen, P.; Xu, J.; Dai, X.; et al. Platelets and platelet adhesion molecules: Novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb. J. 2016, 14, 29. [Google Scholar] [CrossRef]
- Jackson, S.P. Arterial thrombosis--insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef]
- Ruggeri, Z.M.; Mendolicchio, G.L. Adhesion mechanisms in platelet function. Circ. Res. 2007, 100, 1673–1685. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef]
- Moroi, M.; Jung, S.M. Platelet receptors for collagen. Thromb. Haemost. 1997, 78, 439–444. [Google Scholar] [CrossRef]
- Farndale, R.W.; Sixma, J.J.; Barnes, M.J.; de Groot, P.G. The role of collagen in thrombosis and hemostasis. J. Thromb. Haemost. 2004, 2, 561–573. [Google Scholar] [CrossRef]
- Nurden, A.T. Clinical significance of altered collagen-receptor functioning in platelets with emphasis on glycoprotein VI. Blood Rev. 2019, 38, 100592. [Google Scholar] [CrossRef]
- Horii, K.; Kahn, M.L.; Herr, A.B. Structural basis for platelet collagen responses by the immune-type receptor glycoprotein VI. Blood 2006, 108, 936–942. [Google Scholar] [CrossRef]
- Jung, S.M.; Tsuji, K.; Moroi, M. Glycoprotein (GP) VI dimer as a major collagen-binding site of native platelets: Direct evidence obtained with dimeric GPVI-specific Fabs. J. Thromb. Haemost. 2009, 7, 1347–1355. [Google Scholar] [CrossRef]
- Jung, S.M.; Moroi, M.; Soejima, K.; Nakagaki, T.; Miura, Y.; Berndt, M.C.; Gardiner, E.E.; Howes, J.M.; Pugh, N.; Bihan, D.; et al. Constitutive dimerization of glycoprotein VI (GPVI) in resting platelets is essential for binding to collagen and activation in flowing blood. J. Biol. Chem. 2012, 287, 30000–30013. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Takahashi, T.; Jung, S.M.; Moroi, M. Analysis of the interaction of platelet collagen receptor glycoprotein VI (GPVI) with collagen. A dimeric form of GPVI, but not the monomeric form, shows affinity to fibrous collagen. J. Biol. Chem. 2002, 277, 46197–46204. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.F.; Hargreaves, P.G.; Farndale, R.W.; Young, R.D.; Barnes, M.J. Integrin alpha 2 beta 1-independent activation of platelets by simple collagen-like peptides: Collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for alpha 2 beta 1-independent platelet reactivity. Biochem J 1995, 306 Pt 2, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Kamiguti, A.S.; Berlanga, O.; Leduc, M.; Theakston, R.D.; Watson, S.P. The use of snake venom toxins as tools to study platelet receptors for collagen and von Willebrand factor. Haemostasis 2001, 31, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Polgar, J.; Clemetson, J.M.; Kehrel, B.E.; Wiedemann, M.; Magnenat, E.M.; Wells, T.N.; Clemetson, K.J. Platelet activation and signal transduction by convulxin, a C-type lectin from Crotalus durissus terrificus (tropical rattlesnake) venom via the p62/GPVI collagen receptor. J. Biol. Chem. 1997, 272, 13576–13583. [Google Scholar] [CrossRef] [PubMed]
- Wijeyewickrema, L.C.; Gardiner, E.E.; Moroi, M.; Berndt, M.C.; Andrews, R.K. Snake venom metalloproteinases, crotarhagin and alborhagin, induce ectodomain shedding of the platelet collagen receptor, glycoprotein VI. Thromb. Haemost. 2007, 98, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Horii, K.; Brooks, M.T.; Herr, A.B. Convulxin forms a dimer in solution and can bind eight copies of glycoprotein VI: Implications for platelet activation. Biochemistry 2009, 48, 2907–2914. [Google Scholar] [CrossRef]
- Smethurst, P.A.; Joutsi-Korhonen, L.; O’Connor, M.N.; Wilson, E.; Jennings, N.S.; Garner, S.F.; Zhang, Y.; Knight, C.G.; Dafforn, T.R.; Buckle, A.; et al. Identification of the primary collagen-binding surface on human glycoprotein VI by site-directed mutagenesis and by a blocking phage antibody. Blood 2004, 103, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.A.; Onley, D.J.; Jarvis, G.E.; O’Connor, M.N.; Knight, C.G.; Herr, A.B.; Ouwehand, W.H.; Farndale, R.W. Structural basis for the platelet-collagen interaction: The smallest motif within collagen that recognizes and activates platelet Glycoprotein VI contains two glycine-proline-hydroxyproline triplets. J. Biol. Chem. 2007, 282, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Lecut, C.; Arocas, V.; Ulrichts, H.; Elbaz, A.; Villeval, J.L.; Lacapère, J.J.; Deckmyn, H.; Jandrot-Perrus, M. Identification of residues within human glycoprotein VI involved in the binding to collagen: Evidence for the existence of distinct binding sites. J. Biol. Chem. 2004, 279, 52293–52299. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Ollivier, V.; Loyau, S.; Schaff, M.; Dumont, B.; Favier, R.; Freyburger, G.; Latger-Cannard, V.; Nieswandt, B.; Gachet, C.; et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015, 126, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, O.M.; Hughes, C.E.; Montague, S.; Watson, S.K.; Frampton, J.; Bender, M.; Watson, S.P. Fibrin activates GPVI in human and mouse platelets. Blood 2015, 126, 1601–1608. [Google Scholar] [CrossRef]
- Induruwa, I.; Moroi, M.; Bonna, A.; Malcor, J.D.; Howes, J.M.; Warburton, E.A.; Farndale, R.W.; Jung, S.M. Platelet collagen receptor Glycoprotein VI-dimer recognizes fibrinogen and fibrin through their D-domains, contributing to platelet adhesion and activation during thrombus formation. J. Thromb. Haemost. 2018, 16, 389–404. [Google Scholar] [CrossRef]
- Moroi, M.; Induruwa, I.; Farndale, R.W.; Jung, S.M. Dimers of the platelet collagen receptor glycoprotein VI bind specifically to fibrin fibers during clot formation, but not to intact fibrinogen. J. Thromb. Haemost. 2021, 19, 2056–2067. [Google Scholar] [CrossRef]
- Fuentes, E. Modulation of Glycoprotein VI and Its Downstream Signaling Pathways as an Antiplatelet Target. Int. J. Mol. Sci. 2022, 23, 9882. [Google Scholar] [CrossRef]
- Jandrot-Perrus, M.; Hermans, C.; Mezzano, D. Platelet glycoprotein VI genetic quantitative and qualitative defects. Platelets 2019, 30, 708–713. [Google Scholar] [CrossRef]
- Feitsma, L.J.; Brondijk, H.C.; Jarvis, G.E.; Hagemans, D.; Bihan, D.; Jerah, N.; Versteeg, M.; Farndale, R.W.; Huizinga, E.G. Structural insights into collagen binding by platelet receptor glycoprotein VI. Blood 2022, 139, 3087–3098. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Xia, D.; Syed, A.A.S.; Wang, Z.; Shi, Y. Highly accurate protein structure prediction and drug screen of monkeypox virus proteome. J. Infect. 2022, 86, 66–117. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Bodenhofer, U.; Bonatesta, E.; Horejš-Kainrath, C.; Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Stritt, S.; Nurden, P.; Favier, R.; Favier, M.; Ferioli, S.; Gotru, S.K.; van Eeuwijk, J.M.; Schulze, H.; Nurden, A.T.; Lambert, M.P.; et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg2+ homeostasis and cytoskeletal architecture. Nat Commun 2016, 7, 11097. [Google Scholar] [CrossRef]
- Cazenave, J.P.; Ohlmann, P.; Cassel, D.; Eckly, A.; Hechler, B.; Gachet, C. Preparation of washed platelet suspensions from human and rodent blood. Methods Mol. Biol. 2004, 272, 13–28. [Google Scholar] [CrossRef]
- Jiang, P.; Loyau, S.; Tchitchinadze, M.; Ropers, J.; Jondeau, G.; Jandrot-Perrus, M. Inhibition of Glycoprotein VI Clustering by Collagen as a Mechanism of Inhibiting Collagen-Induced Platelet Responses: The Example of Losartan. PLoS ONE 2015, 10, e0128744. [Google Scholar] [CrossRef]
- Al-Tamimi, M.; Mu, F.T.; Moroi, M.; Gardiner, E.E.; Berndt, M.C.; Andrews, R.K. Measuring soluble platelet glycoprotein VI in human plasma by ELISA. Platelets 2009, 20, 143–149. [Google Scholar] [CrossRef]
- Nieswandt, B.; Schulte, V.; Bergmeier, W.; Mokhtari-Nejad, R.; Rackebrandt, K.; Cazenave, J.P.; Ohlmann, P.; Gachet, C.; Zirngibl, H. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J. Exp. Med. 2001, 193, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.; Starke, A.; Heemskerk, J.W.M.; Kuijpers, M.J.E.; Stegner, D.; Nieswandt, B. Targeting of a Conserved Epitope in Mouse and Human GPVI Differently Affects Receptor Function. Int. J. Mol. Sci. 2022, 23, 8610. [Google Scholar] [CrossRef] [PubMed]
- Perret, S.; Eble, J.A.; Siljander, P.R.; Merle, C.; Farndale, R.W.; Theisen, M.; Ruggiero, F. Prolyl hydroxylation of collagen type I is required for efficient binding to integrin alpha 1 beta 1 and platelet glycoprotein VI but not to alpha 2 beta 1. J. Biol. Chem. 2003, 278, 29873–29879. [Google Scholar] [CrossRef] [PubMed]
- Schulte, V.; Snell, D.; Bergmeier, W.; Zirngibl, H.; Watson, S.P.; Nieswandt, B. Evidence for two distinct epitopes within collagen for activation of murine platelets. J. Biol. Chem. 2001, 276, 364–368. [Google Scholar] [CrossRef]
- Lecut, C.; Feeney, L.A.; Kingsbury, G.; Hopkins, J.; Lanza, F.; Gachet, C.; Villeval, J.L.; Jandrot-Perrus, M. Human platelet glycoprotein VI function is antagonized by monoclonal antibody-derived Fab fragments. J. Thromb. Haemost 2003, 1, 2653–2662. [Google Scholar] [CrossRef]
- Kunicki, T.J.; Cheli, Y.; Moroi, M.; Furihata, K. The influence of N-linked glycosylation on the function of platelet glycoprotein VI. Blood 2005, 106, 2744–2749. [Google Scholar] [CrossRef]
- Wichaiyo, S.; Parichatikanond, W.; Rattanavipanon, W. Glenzocimab: A GPVI (Glycoprotein VI)-Targeted Potential Antiplatelet Agent for the Treatment of Acute Ischemic Stroke. Stroke 2022, 53, 3506–3513. [Google Scholar] [CrossRef]
- Billiald, P.; Slater, A.S.; Welin, M.; Clark, J.C.; Loyau, S.; Pugniere, M.; Jiacomini, I.G.; Rose, N.; Lebozec, K.; Toledano, E.; et al. Targeting platelet GPVI with glenzocimab: A novel mechanism for inhibition. Blood Adv. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Mammadova-Bach, E.; Gil-Pulido, J.; Sarukhanyan, E.; Burkard, P.; Shityakov, S.; Schonhart, C.; Stegner, D.; Remer, K.; Nurden, P.; Nurden, A.T.; et al. Platelet glycoprotein VI promotes metastasis through interaction with cancer cell-derived galectin-3. Blood 2020, 135, 1146–1160. [Google Scholar] [CrossRef]
- Goebel, S.; Li, Z.; Vogelmann, J.; Holthoff, H.P.; Degen, H.; Hermann, D.M.; Gawaz, M.; Ungerer, M.; Munch, G. The GPVI-Fc fusion protein Revacept improves cerebral infarct volume and functional outcome in stroke. PLoS ONE 2013, 8, e66960. [Google Scholar] [CrossRef]
- Saha, B.; Mathur, T.; Tronolone, J.J.; Chokshi, M.; Lokhande, G.K.; Selahi, A.; Gaharwar, A.K.; Afshar-Kharghan, V.; Sood, A.K.; Bao, G.; et al. Human tumor microenvironment chip evaluates the consequences of platelet extravasation and combinatorial antitumor-antiplatelet therapy in ovarian cancer. Sci Adv 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Groschel, K.; Uphaus, T.; Loftus, I.; Poppert, H.; Diener, H.C.; Zobel, J.; Munch, G. Revacept, an Inhibitor of Platelet Adhesion in Symptomatic Carotid Artery Stenosis: Design and Rationale of a Randomized Phase II Clinical Trial. TH Open 2020, 4, e393–e399. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Closest Residues | Distance, Å | Specific Interactions |
|---|---|---|---|
| GPVI:58:Arg * | collagen-1 : 4:Gly | 2.7 | 1x hb to collagen-1 : 4:Gly |
| collagen-1 : 3:Hyp | 3.0 | 1x hb to collagen-1 : 3:Hyp | |
| collagen-1 : 6:Hyp | 3.2 | ||
| collagen-2 : 5:Pro | 3.3 | ||
| collagen-2 : 4:Gly | 3.9 | ||
| GPVI:60:Glu * | collagen-1 : 6:Hyp | 2.8 | 2x clash to collagen-1 : 6:Hyp |
| collagen-2 : 5:Pro | 3.4 | ||
| GPVI:62:Leu * | collagen-2 : 8:Pro | 4.0 | |
| GPVI:67:Tyr | collagen-1 : 6:Hyp | 3.3 | |
| collagen-1 : 3:Hyp | 3.7 | ||
| collagen-1 : 5:Pro | 3.7 | ||
| collagen-1 : 4:Gly | 4.0 | ||
| GPVI:87:Arg | collagen-2 : 6:Hyp | 3.1 | 2x hb to collagen-2 : 6:Hyp |
| collagen-2 : 7:Gly | 3.2 | ||
| collagen-2 : 8:Pro | 3.2 | ||
| GPVI:89:Ser | collagen-2 : 5:Pro | 3.7 | |
| GPVI:91:Gln | collagen-1 : 3:Hyp | 2.7 | 1x hb to collagen-1 : 3:Hyp |
| collagen-2 : 2:Pro | 3.9 | ||
| GPVI:96:Trp | collagen-2 : 3:Hyp | 3.2 | 1x hb to collagen-2 : 3:Hyp |
| collagen-2 : 5:Pro | 3.8 | ||
| collagen-2 : 4:Gly | 3.8 | ||
| collagen-1 : 3:Hyp | 3.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, C.; Ren, P.; Mamtimin, M.; Kruk, L.; Sarukhanyan, E.; Li, C.; Anders, H.-J.; Dandekar, T.; Krueger, I.; Elvers, M.; et al. Minimal Collagen-Binding Epitope of Glycoprotein VI in Human and Mouse Platelets. Biomedicines 2023, 11, 423. https://doi.org/10.3390/biomedicines11020423
Han C, Ren P, Mamtimin M, Kruk L, Sarukhanyan E, Li C, Anders H-J, Dandekar T, Krueger I, Elvers M, et al. Minimal Collagen-Binding Epitope of Glycoprotein VI in Human and Mouse Platelets. Biomedicines. 2023; 11(2):423. https://doi.org/10.3390/biomedicines11020423
Chicago/Turabian StyleHan, Chao, Pengxuan Ren, Medina Mamtimin, Linus Kruk, Edita Sarukhanyan, Chenyu Li, Hans-Joachim Anders, Thomas Dandekar, Irena Krueger, Margitta Elvers, and et al. 2023. "Minimal Collagen-Binding Epitope of Glycoprotein VI in Human and Mouse Platelets" Biomedicines 11, no. 2: 423. https://doi.org/10.3390/biomedicines11020423
APA StyleHan, C., Ren, P., Mamtimin, M., Kruk, L., Sarukhanyan, E., Li, C., Anders, H. -J., Dandekar, T., Krueger, I., Elvers, M., Goebel, S., Adler, K., Münch, G., Gudermann, T., Braun, A., & Mammadova-Bach, E. (2023). Minimal Collagen-Binding Epitope of Glycoprotein VI in Human and Mouse Platelets. Biomedicines, 11(2), 423. https://doi.org/10.3390/biomedicines11020423