
The Anti-Epileptic Drugs Lamotrigine and Valproic Acid Reduce the Cardiac Sodium Current



Abstract
:1. Introduction
2. Materials and Methods
2.1. HEK293 Cell Culture
2.2. Rabbit Ventricular Myocyte Preparation
2.3. Patch-Clamp Recording
2.4. Sodium Current Measurements
2.5. Action Potential Measurement
2.6. Preparation of Antiepileptic Drugs
2.7. Statistical Analysis
3. Results
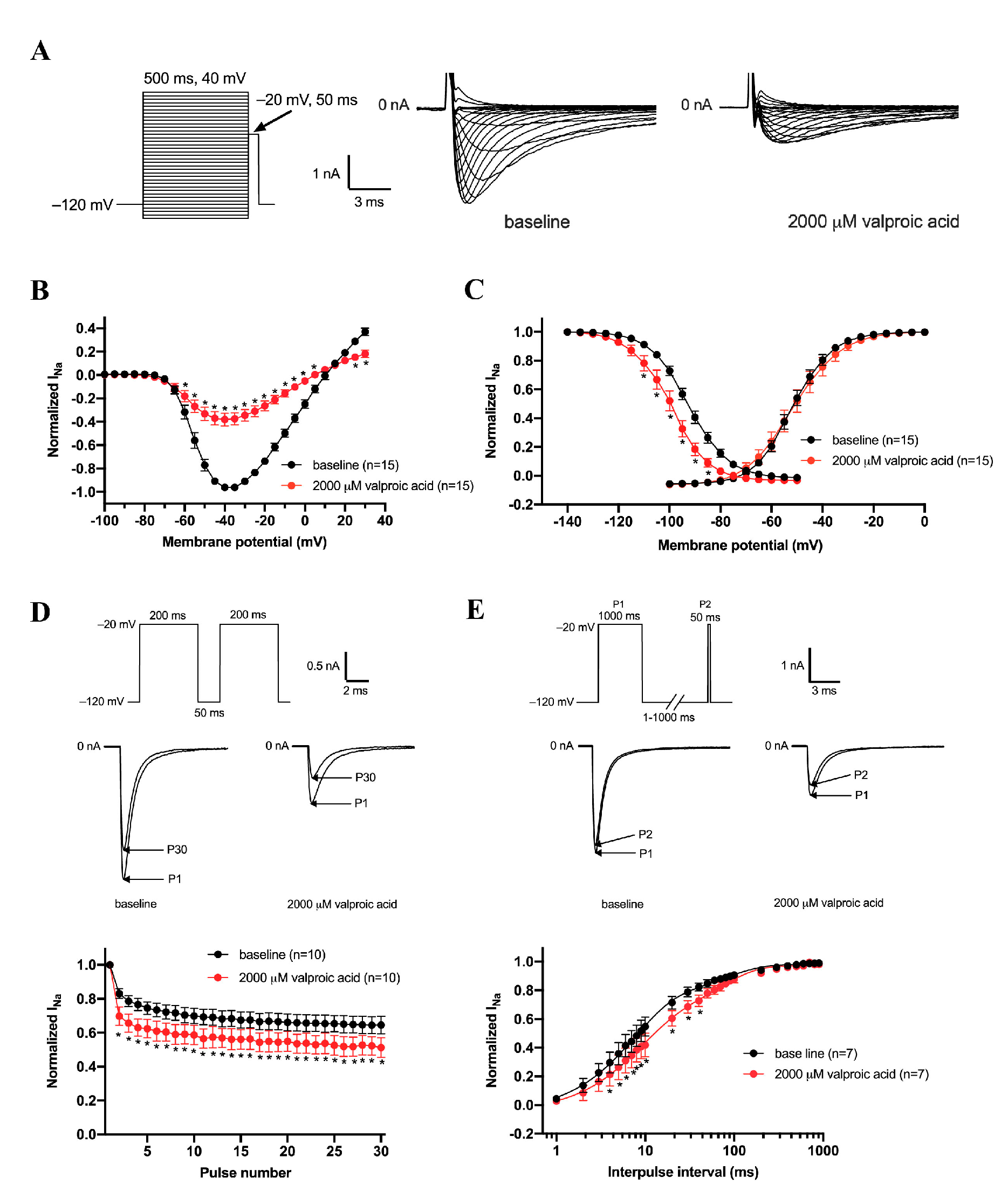
3.1. Inhibition of the Nav1.5 Current by Lamotrigine and Valproic Acid in a Concentration-Dependent Manner
3.2. Effects of Lamotrigine (100 μM) on Gating Properties of Nav1.5 Channels
3.3. Effects of Valproic Acid (2000 μM) on Gating Properties of Nav1.5 Channels
3.4. Effects of Lamotrigine and Valproic acid on Action Potentials Properties
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacob, S.; Nair, A.B. An updated overview on therapeutic drug monitoring of recent antiepileptic drugs. Drugs R&D 2016, 16, 303–316. [Google Scholar] [CrossRef]
- Perucca, E. An introduction to antiepileptic drugs. Epilepsia 2005, 46 (Suppl. S4), 31–37. [Google Scholar] [CrossRef] [PubMed]
- Bardai, A.; Blom, M.T.; van Noord, C.; Verhamme, K.M.; Sturkenboom, M.C.; Tan, H.L. Sudden cardiac death is associated both with epilepsy and with use of antiepileptic medications. Heart 2015, 101, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Eroglu, T.E.; Wilders, R.; Verkerk, A.O.; Tan, H.L. Carbamazepine increases the risk of sudden cardiac arrest by a reduction of the cardiac sodium current. Front. Cell Dev. Biol. 2022, 10, 891996. [Google Scholar] [CrossRef]
- Kléber, A.G.; Rudy, Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 2004, 84, 431–488. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Eroglu, T.E.; Blom, M.T.; Souverein, P.C.; de Boer, A.; Tan, H.L. Non-cardiac depolarization-blocking drugs are associated with increased risk of out-of-hospital cardiac arrest in the community. Pharmacoepidemiology 2022, 1, 64–75. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Amin, A.S.; Remme, C.A. Disease modifiers of inherited SCN5A channelopathy. Front. Cardiovasc. Med. 2018, 5, 137. [Google Scholar] [CrossRef]
- Amin, A.S.; Asghari-Roodsari, A.; Tan, H.L. Cardiac sodium channelopathies. Pflügers Arch. -Eur. J. Physiol. 2010, 460, 223–237. [Google Scholar] [CrossRef]
- Ruskin, J.N. The cardiac arrhythmia suppression trial (CAST). N. Engl. J. Med. 1989, 321, 386–388. [Google Scholar] [CrossRef]
- Heinemann, S.H.; Schlief, T.; Mori, Y.; Imoto, K. Molecular pore structure of voltage-gated sodium and calcium channels. Braz. J. Med. Biol. Res. 1994, 27, 2781–2802. [Google Scholar]
- Fozzard, H.A.; Hanck, D.A. Structure and function of voltage-dependent sodium channels: Comparison of brain II and cardiac isoforms. Physiol. Rev. 1996, 76, 887–926. [Google Scholar] [CrossRef]
- GIPdatabank.nl, Z.N.G. Available online: https://www.gipdatabank.nl/databank?infotype=g&label=00-totaal&tabel=B_01-basis&geg=ddd&item=N03AF (accessed on 26 July 2022).
- Portero, V.; Wilders, R.; Casini, S.; Charpentier, F.; Verkerk, A.O.; Remme, C.A. K(V)4.3 Expression Modulates Na(V)1.5 Sodium Current. Front. Physiol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Den Ruijter, H.M.; Verkerk, A.O.; Coronel, R. Incorporated fish oil fatty acids prevent action potential shortening induced by circulating fish oil fatty acids. Front. Physiol. 2010, 1, 149. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.H.; Lynch, J.W. Liquid junction potentials and small cell effects in patch-clamp analysis. J. Membr. Biol. 1991, 121, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Man, J.C.K.; Bosada, F.M.; Scholman, K.T.; Offerhaus, J.A.; Walsh, R.; van Duijvenboden, K.; van Eif, V.W.W.; Bezzina, C.R.; Verkerk, A.O.; Boukens, B.J.; et al. Variant Intronic Enhancer Controls SCN10A-short Expression and Heart Conduction. Circulation 2021, 144, 229–242. [Google Scholar] [CrossRef]
- Matsuki, N.; Quandt, F.N.; Ten Eick, R.E.; Yeh, J. Characterization of the block of sodium channels by phenytoin in mouse neuroblastoma cells. J. Pharmacol. Exp. Ther. 1984, 228, 523–530. [Google Scholar]
- Theile, J.W.; Cummins, T.R. Inhibition of Navβ4 peptide-mediated resurgent sodium currents in Nav1. 7 channels by carbamazepine, riluzole, and anandamide. Mol. Pharmacol. 2011, 80, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, F.L.; Mena, O.; Gary, R.D.; McIntyre, I.M. An acute gabapentin fatality: A case report with postmortem concentrations. Int. J. Legal. Med. 2015, 129, 771–775. [Google Scholar] [CrossRef]
- Armijo, J.A.; Pena, M.A.; Adín, J.; Vega-Gil, N. Association between patient age and gabapentin serum concentration-to-dose ratio: A preliminary multivariate analysis. Ther. Drug Monit. 2004, 26, 633–637. [Google Scholar] [CrossRef]
- Ito, S.; Yano, I.; Hashi, S.; Tsuda, M.; Sugimoto, M.; Yonezawa, A.; Ikeda, A.; Matsubara, K. Population pharmacokinetic modeling of levetiracetam in pediatric and adult patients with epilepsy by using routinely monitored data. Ther. Drug Monit. 2016, 38, 371–378. [Google Scholar] [CrossRef] [PubMed]
- May, T.W.; Rambeck, B.; Neb, R.; Jürgens, U. Serum concentrations of pregabalin in patients with epilepsy: The influence of dose, age, and comedication. Ther. Drug Monit. 2007, 29, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Kriikku, P.; Wilhelm, L.; Rintatalo, J.; Hurme, J.; Kramer, J.; Ojanperä, I. Pregabalin serum levels in apprehended drivers. Forensic. Sci. Int. 2014, 243, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard Khinchi, M.; Nielsen, K.A.; Dahl, M.; Wolf, P. Lamotrigine therapeutic thresholds. Seizure 2008, 17, 391–395. [Google Scholar] [CrossRef]
- Douglas-Hall, P.; Dzahini, O.; Gaughran, F.; Bile, A.; Taylor, D. Variation in dose and plasma level of lamotrigine in patients discharged from a mental health trust. Ther. Adv. Psychopharmacol. 2017, 7, 17–24. [Google Scholar] [CrossRef]
- Rahman, M.; Nguyen, H. Valproic Acid. 2020. StatPearls Publishing. Available online: http://www.ncbi.nlm.nih.gov/books/NBK559112/ (accessed on 24 June 2022).
- Ingleby-Talecki, L.; van Dijkman, S.C.; Oosterholt, S.P.; Della Pasqua, O.; Winter, C.; Cunnington, M.; Rebar, L.; Forero-Schwanhaeuser, S.; Patel, V.; Cooper, J.A.; et al. Cardiac sodium channel inhibition by lamotrigine: In vitro characterization and clinical implications. Clin. Transl. Sci. 2022, 15, 1978–1989. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Lancaster, B.; Peakman, T.; Garthwaite, J. Interaction of the antiepileptic drug lamotrigine with recombinant rat brain type IIA Na+ channels and with native Na+ channels in rat hippocampal neurones. Pflügers Arch. -Eur. J. Physiol. 1995, 430, 437–446. [Google Scholar] [CrossRef]
- Zona, C.; Avoli, M. Lamotrigine reduces voltage-gated sodium currents in rat central neurons in culture. Epilepsia 1997, 38, 522–525. [Google Scholar] [CrossRef]
- VanDongen, A.M.; VanErp, M.G.; Voskuyl, R.A. Valproate reduces excitability by blockage of sodium and potassium conductance. Epilepsia 1986, 27, 177–182. [Google Scholar] [CrossRef]
- Vreugdenhil, M.; van Veelen, C.W.; van Rijen, P.C.; Lopes da Silva, F.H.; Wadman, W.J. Effect of valproic acid on sodium currents in cortical neurons from patients with pharmaco-resistant temporal lobe epilepsy. Epilepsy Res. 1998, 32, 309–320. [Google Scholar] [CrossRef]
- Van den Berg, R.J.; Kok, P.; Voskuyl, R.A. Valproate and sodium currents in cultured hippocampal neurons. Exp. Brain Res. 1993, 93, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Martella, G.; Picconi, B.; Prosperetti, C.; Pisani, A.; Di Filippo, M.; Pisani, F.; Bernardi, G.; Calabresi, P. Multiple mechanisms underlying the neuroprotective effects of antiepileptic drugs against in vitro ischemia. Stroke 2006, 37, 1319–1326. [Google Scholar] [CrossRef]
- Taverna, S.; Mantegazza, M.; Franceschetti, S.; Avanzini, G. Valproate selectively reduces the persistent fraction of Na+ current in neocortical neurons. Epilepsy Res. 1998, 32, 304–308. [Google Scholar] [CrossRef]
- Harmer, A.R.; Valentin, J.P.; Pollard, C.E. On the relationship between block of the cardiac Na⁺ channel and drug-induced prolongation of the QRS complex. Br. J. Pharmacol. 2011, 164, 260–273. [Google Scholar] [CrossRef]
- Alyahya, B.; Friesen, M.; Nauche, B.; Laliberté, M. Acute lamotrigine overdose: A systematic review of published adult and pediatric cases. Clin. Toxicol. 2018, 56, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L.; et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef]
- Amin, A.S.; Reckman, Y.J.; Arbelo, E.; Spanjaart, A.M.; Postema, P.G.; Tadros, R.; Tanck, M.W.; Van den Berg, M.P.; Wilde, A.A.M.; Tan, H.L. SCN5A mutation type and topology are associated with the risk of ventricular arrhythmia by sodium channel blockers. Int. J. Cardiol. 2018, 266, 128–132. [Google Scholar] [CrossRef]
- Tan, H.L.; Bink-Boelkens, M.T.; Bezzina, C.R.; Viswanathan, P.C.; Beaufort-Krol, G.C.; van Tintelen, P.J.; van den Berg, M.P.; Wilde, A.A.; Balser, J.R. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 2001, 409, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Soh, M.S.; Bagnall, R.D.; Semsarian, C.; Scheffer, I.E.; Berkovic, S.F.; Reid, C.A. Rare sudden unexpected death in epilepsy SCN5A variants cause changes in channel function implicating cardiac arrhythmia as a cause of death. Epilepsia 2022, 63, e57–e62. [Google Scholar] [CrossRef] [PubMed]
- Aurlien, D.; Leren, T.P.; Taubøll, E.; Gjerstad, L. New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure 2009, 18, 158–160. [Google Scholar] [CrossRef]
- Postema, P.G.; Wolpert, C.; Amin, A.S.; Probst, V.; Borggrefe, M.; Roden, D.M.; Priori, S.G.; Tan, H.L.; Hiraoka, M.; Brugada, J.; et al. Drugs and Brugada syndrome patients: Review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org). Heart Rhythm 2009, 6, 1335–1341. [Google Scholar] [CrossRef] [Green Version]
- Dekker, L.R.; Bezzina, C.R.; Henriques, J.P.; Tanck, M.W.; Koch, K.T.; Alings, M.W.; Arnold, A.E.; de Boer, M.J.; Gorgels, A.P.; Michels, H.R.; et al. Familial sudden death is an important risk factor for primary ventricular fibrillation: A case-control study in acute myocardial infarction patients. Circulation 2006, 114, 1140–1145. [Google Scholar] [CrossRef]
- Eroglu, T.E.; Folke, F.; Tan, H.L.; Torp-Pedersen, C.; Gislason, G.H. Risk of out-of-hospital cardiac arrest in patients with epilepsy and users of antiepileptic drugs. Br. J. Clin. Pharmacol. 2022, 88, 3709–3715. [Google Scholar] [CrossRef] [PubMed]
- Hookana, E.; Ansakorpi, H.; Kortelainen, M.L.; Junttila, M.J.; Kaikkonen, K.S.; Perkiömäki, J.; Huikuri, H.V. Antiepileptic medications and the risk for sudden cardiac death caused by an acute coronary event: A prospective case-control study. Ann. Med. 2016, 48, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Bardai, A.; Lamberts, R.J.; Blom, M.T.; Spanjaart, A.M.; Berdowski, J.; van der Staal, S.R.; Brouwer, H.J.; Koster, R.W.; Sander, J.W.; Thijs, R.D.; et al. Epilepsy is a risk factor for sudden cardiac arrest in the general population. PLoS ONE 2012, 7, e42749. [Google Scholar] [CrossRef]
- Bunschoten, J.W.; Husein, N.; Devinsky, O.; French, J.A.; Sander, J.W.; Thijs, R.D.; Keezer, M.R. Sudden Death and Cardiac Arrythmia With Lamotrigine: A Rapid Systematic Review. Neurology 2022, 98, e1748–e1760. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, N.; Reitz, T.; Wang, Y.; Flores, C.M. Inhibition of the rat brain sodium channel Nav1.2 after prolonged exposure to gabapentin. Epilepsy Res. 2006, 70, 263–268. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activation | Inactivation | Recovery from Inactivation | ||||||
|---|---|---|---|---|---|---|---|---|
| V1/2 (mV) | k (mV) | V1/2 (mV) | k (mV) | τf (ms) | τs (ms) | As/(As + Af) (ms) | ||
| lamotrigine | baseline | −52.7 ± 1.7 (n = 9) | 5.7 ± 0.4 (n = 9) | −92.9 ± 1.5 (n = 9) | 6.0 ± 0.4 (n = 9) | 11.2 ± 1.7 (n = 7) | 134.8 ± 21.5 (n = 7) | 0.18 ± 0.02 (n = 7) |
| wash-in | −56.7 ± 1.5 * (n = 9) | 5.1 ± 0.3 (n = 9) | −99.5 ± 2.6 * (n = 9) | 7.7 ± 0.6 * (n = 9) | 17.1 ± 3.6 * (n = 7) | 657.7 ± 125.1 * (n = 7) | 0.44 ± 0.03 * (n = 7) | |
| valproic acid | baseline | −50.6 ± 1.5 (n = 15) | 5.9 ± 0.3 (n = 15) | −92.7 ± 1.4 (n = 15) | 6.6 ± 0.2 (n = 15) | 11.3 ± 2.1 (n = 7) | 165.0 ± 18.0 (n = 7) | 0.16 ± 0.02 (n = 7) |
| wash-in | −50.5 ± 2.7 (n = 15) | 5.8 ± 0.3 (n = 15) | −99.0 ± 1.9 * (n = 15) | 5.9 ± 0.3 * (n = 15) | 17.5 ± 3.6 * (n = 7) | 200.8 ± 34.5 (n = 7) | 0.2 ± 0.02 (n = 7) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, L.; Verkerk, A.O.; Tan, H.L. The Anti-Epileptic Drugs Lamotrigine and Valproic Acid Reduce the Cardiac Sodium Current. Biomedicines 2023, 11, 477. https://doi.org/10.3390/biomedicines11020477
Jia L, Verkerk AO, Tan HL. The Anti-Epileptic Drugs Lamotrigine and Valproic Acid Reduce the Cardiac Sodium Current. Biomedicines. 2023; 11(2):477. https://doi.org/10.3390/biomedicines11020477
Chicago/Turabian StyleJia, Lixia, Arie O. Verkerk, and Hanno L. Tan. 2023. "The Anti-Epileptic Drugs Lamotrigine and Valproic Acid Reduce the Cardiac Sodium Current" Biomedicines 11, no. 2: 477. https://doi.org/10.3390/biomedicines11020477
APA StyleJia, L., Verkerk, A. O., & Tan, H. L. (2023). The Anti-Epileptic Drugs Lamotrigine and Valproic Acid Reduce the Cardiac Sodium Current. Biomedicines, 11(2), 477. https://doi.org/10.3390/biomedicines11020477