
Differential Roles of CD36 in Regulating Muscle Insulin Response Depend on Palmitic Acid Load

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal
2.2. Cell Culture
2.3. RNA Interference
2.4. CD36 Plasmids Construction
2.5. Transcriptome Sequencing
2.6. Oil Red O Staining
2.7. Triglyceride (TG) Content Assay
2.8. Reactive Oxygen Species (ROS) Measurement
2.9. Adenosine Triphosphatase (ATPase) Activity Assay
2.10. Transmission Electron Microscopy (TEM)
2.11. Quantitative RT-PCR
2.12. Western Blot Analysis
2.13. Statistical Analysis
3. Results
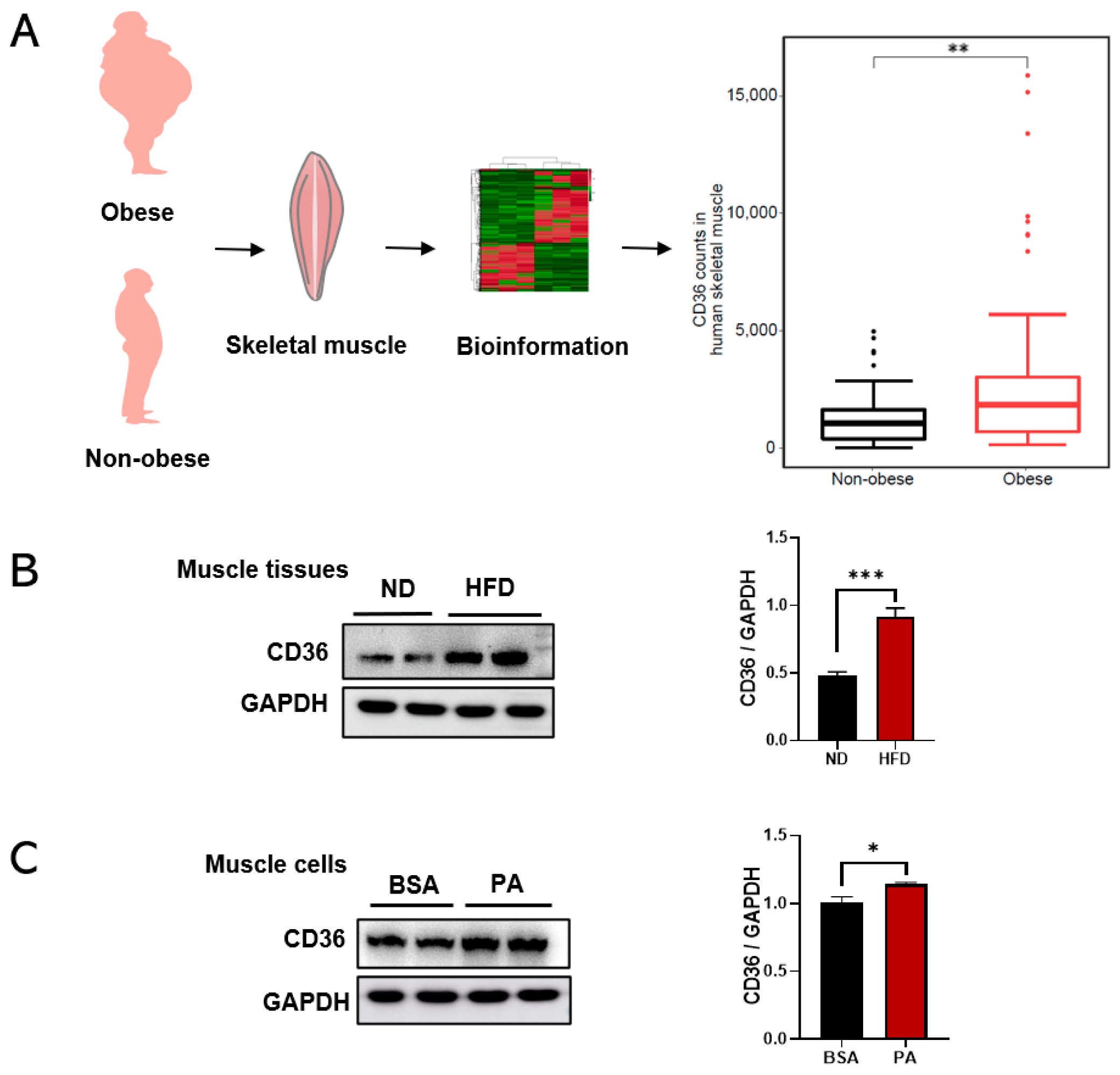
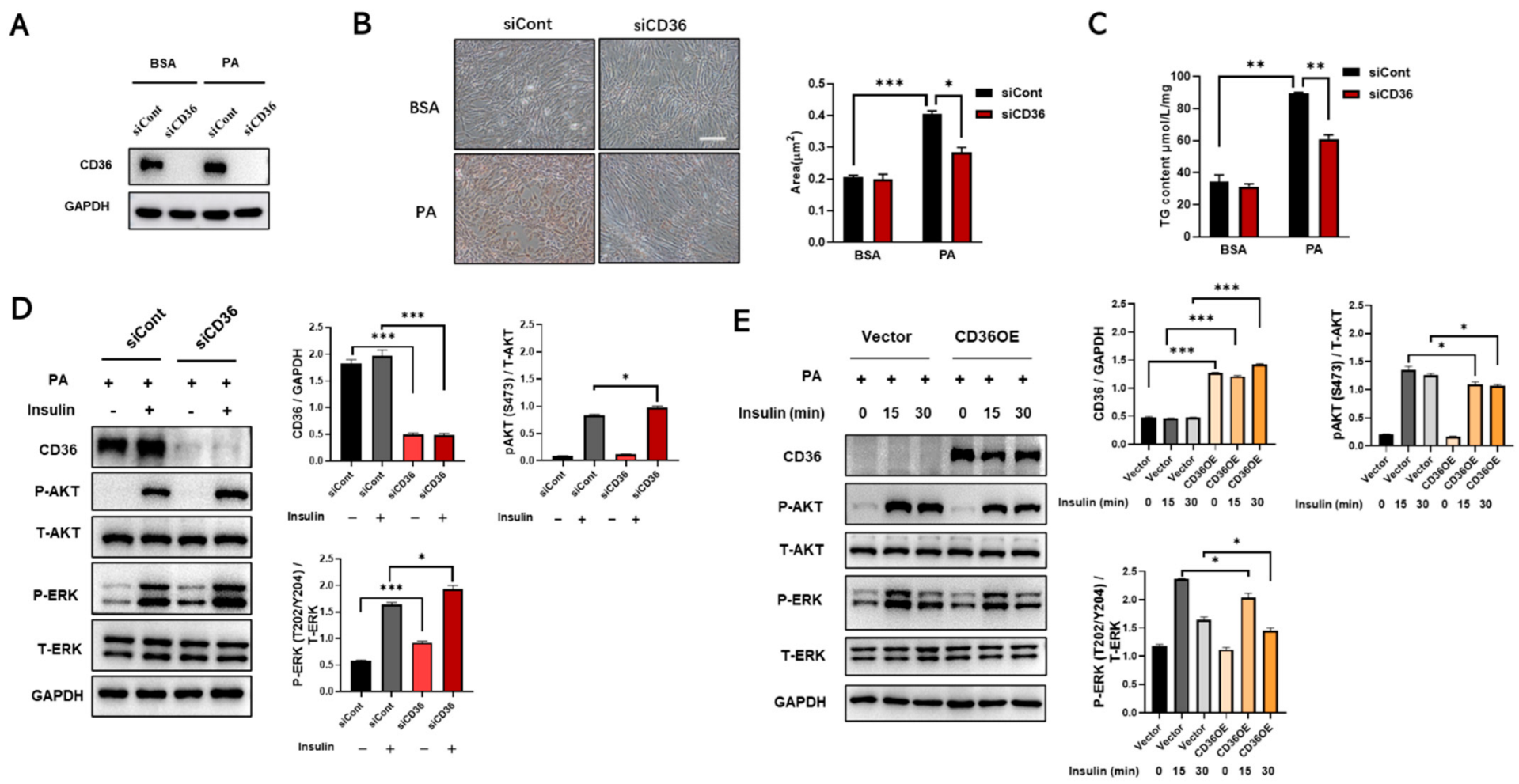
3.1. CD36 Is Highly Expressed in Skeletal Muscle under the Condition of HFD and PA Treatment
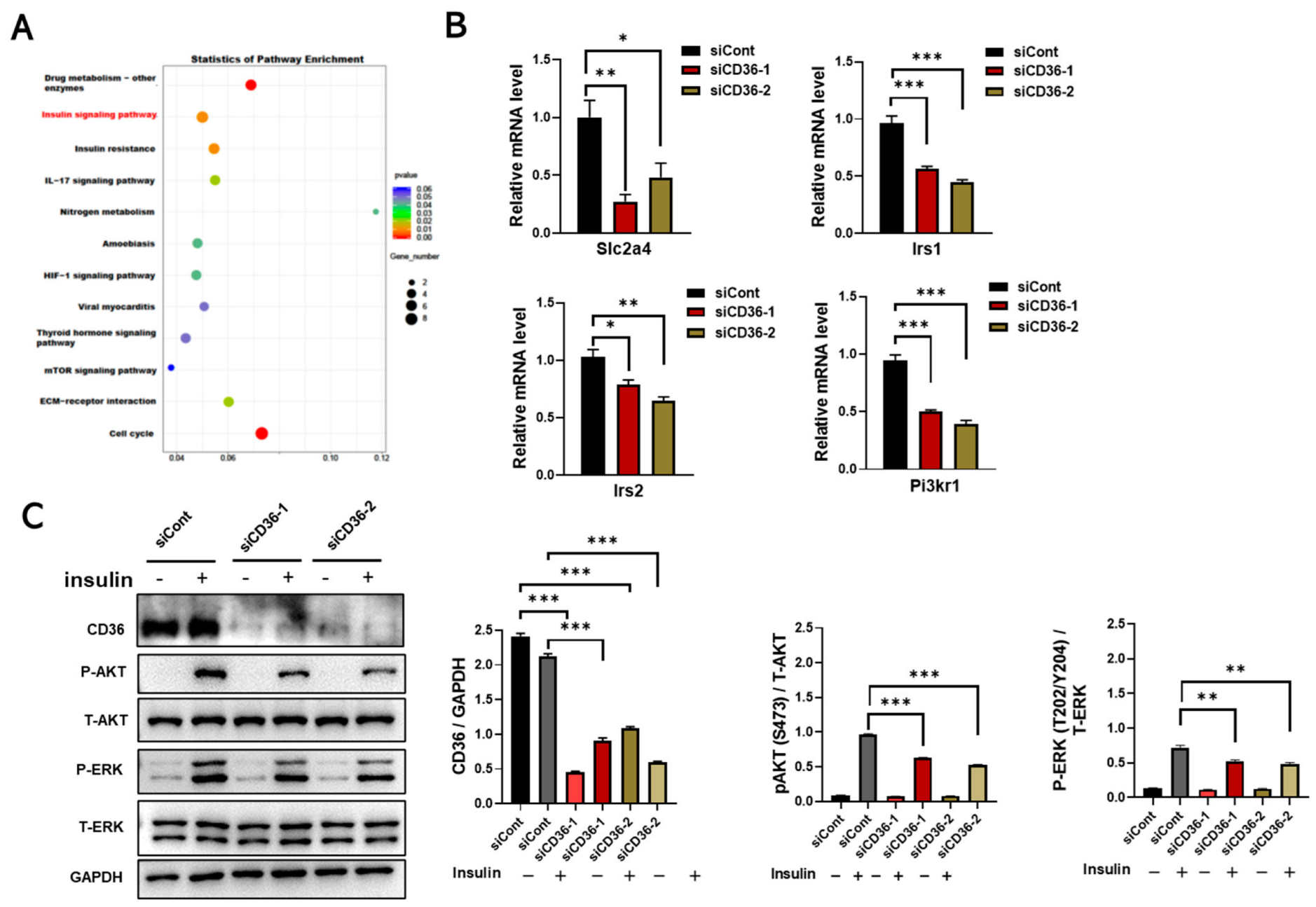
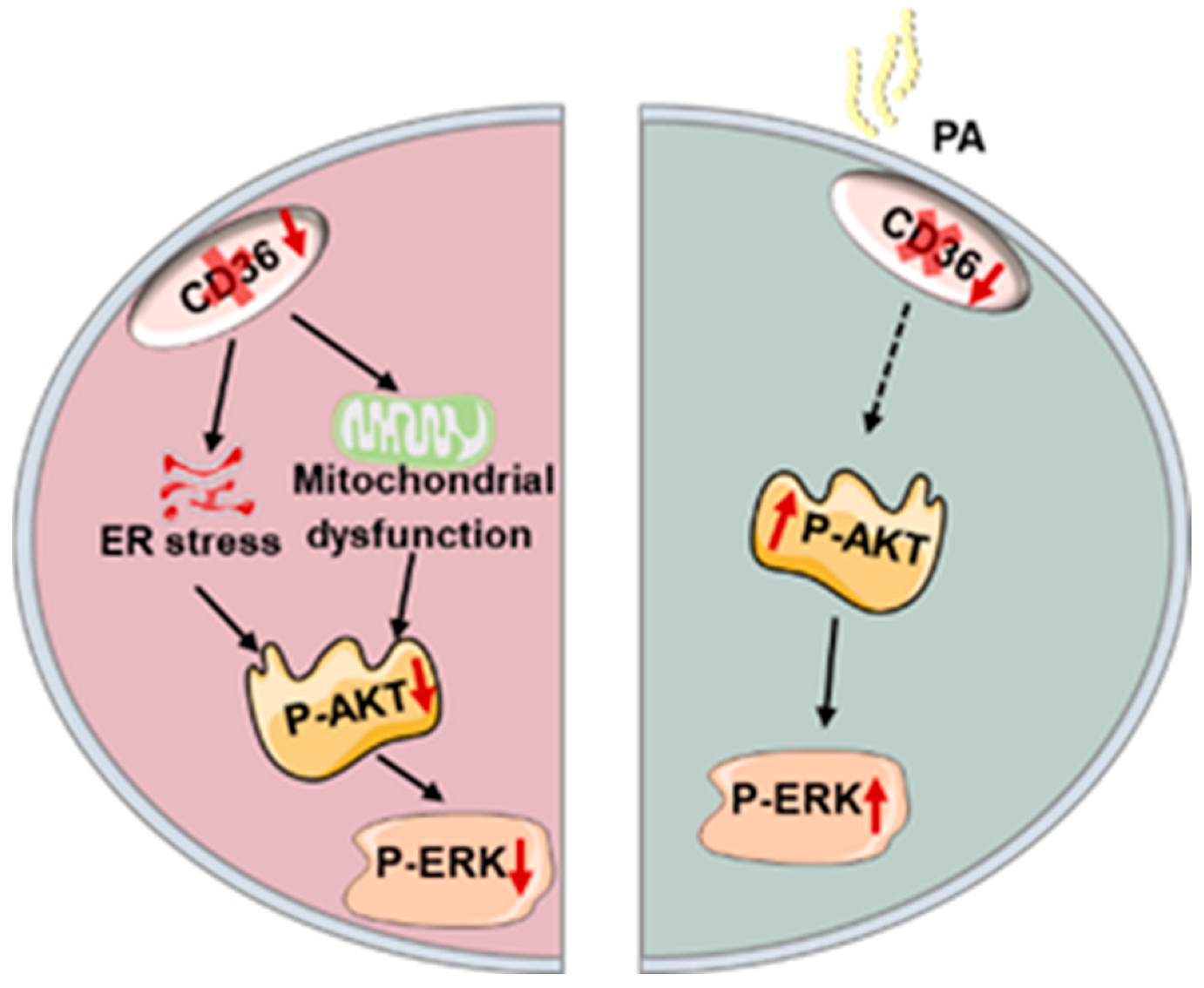
3.2. Loss of CD36 Impairs Insulin Signaling in C2C12 Myotubes in the Absence of PA Loading
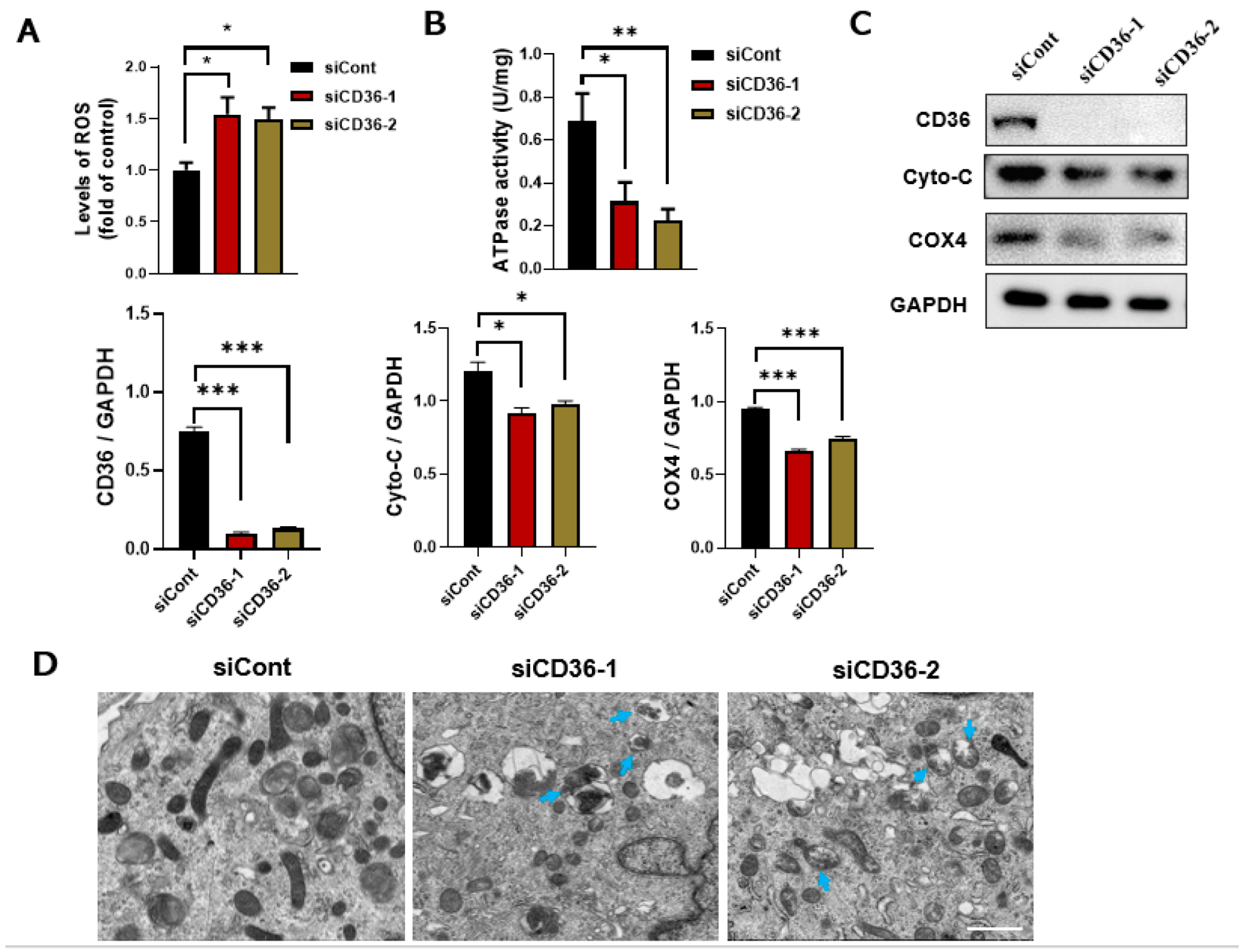
3.3. Loss of CD36 Induces Mitochondrial Dysfunction in C2C12 Myotubes in the Absence of PA Loading
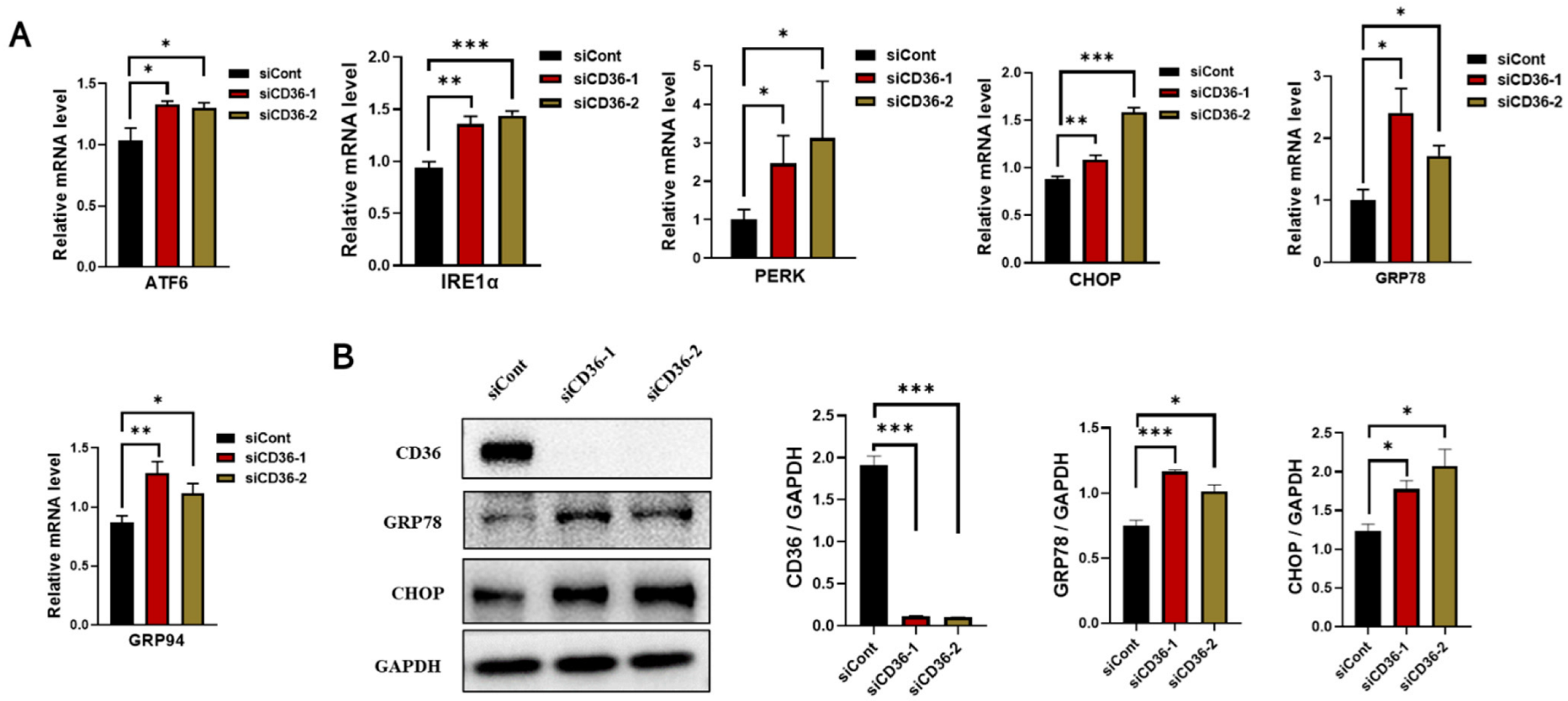
3.4. Loss of CD36 Enhances ER Stress in C2C12 Myotubes in the Absence of PA Loading
3.5. Loss of CD36 Protects against Insulin Resistance in the Presence of PA Overload
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramos, P.A.; Lytle, K.A.; Delivanis, D.; Nielsen, S.; LeBrasseur, N.K.; Jensen, M.D. Insulin-Stimulated Muscle Glucose Uptake and Insulin Signaling in Lean and Obese Humans. J. Clin. Endocrinol. Metab. 2021, 106, e1631–e1646. [Google Scholar] [CrossRef]
- Meng, Z.X.; Gong, J.; Chen, Z.; Sun, J.; Xiao, Y.; Wang, L.; Li, Y.; Liu, J.; Xu, X.Z.S.; Lin, J.D. Glucose Sensing by Skeletal Myocytes Couples Nutrient Signaling to Systemic Homeostasis. Mol. Cell 2017, 66, 332–344.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B., 3rd; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.K.; Zierath, J.R.; Kane, S.; Krook, A.; Lienhard, G.E.; Wallberg-Henriksson, H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes 2005, 54, 1692–1697. [Google Scholar] [CrossRef] [Green Version]
- Belfort, R.; Mandarino, L.; Kashyap, S.; Wirfel, K.; Pratipanawatr, T.; Berria, R.; Defronzo, R.A.; Cusi, K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 2005, 54, 1640–1648. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M., 2nd; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef] [Green Version]
- Samovski, D.; Dhule, P.; Pietka, T.; Jacome-Sosa, M.; Penrose, E.; Son, N.H.; Flynn, C.R.; Shoghi, K.I.; Hyrc, K.L.; Goldberg, I.J.; et al. Regulation of Insulin Receptor Pathway and Glucose Metabolism by CD36 Signaling. Diabetes 2018, 67, 1272–1284. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Zeng, H.; Tan, W.; Luo, X.; Zheng, E.; Zhao, L.; Wei, L.; Ruan, X.Z.; Chen, Y.; Chen, Y. Loss of CD36 impairs hepatic insulin signaling by enhancing the interaction of PTP1B with IR. FASEB J. 2020, 34, 5658–5672. [Google Scholar] [CrossRef] [Green Version]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sánchez-Campos, S.; García-Mediavilla, M.V.; Fernández-Bermejo, M.; Lozano-Rodríguez, T.; Vargas-Castrillón, J.; Buqué, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 2011, 60, 1394–1402. [Google Scholar] [CrossRef]
- Kuwasako, T.; Hirano, K.; Sakai, N.; Ishigami, M.; Hiraoka, H.; Yakub, M.J.; Yamauchi-Takihara, K.; Yamashita, S.; Matsuzawa, Y. Lipoprotein abnormalities in human genetic CD36 deficiency associated with insulin resistance and abnormal fatty acid metabolism. Diabetes Care 2003, 26, 1647–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajri, T.; Han, X.X.; Bonen, A.; Abumrad, N.A. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J. Clin. Investig. 2002, 109, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ha, T.Y.; Jung, C.H.; Nirmala, F.S.; Park, S.Y.; Huh, Y.H.; Ahn, J. Mitochondrial dysfunction in skeletal muscle contributes to the development of acute insulin resistance in mice. J. Cachexia Sarcopenia Muscle 2021, 12, 1925–1939. [Google Scholar] [CrossRef]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Heilbronn, L.K. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol. Metab. 2008, 19, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Hajduch, E.; Ferré, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef]
- Lepretti, M.; Martucciello, S.; Burgos Aceves, M.A.; Putti, R.; Lionetti, L. Omega-3 Fatty Acids and Insulin Resistance: Focus on the Regulation of Mitochondria and Endoplasmic Reticulum Stress. Nutrients 2018, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Su, Y.; Xu, Y.; Qin, D.; He, Q.; Qiu, H.; Zhuo, J.; Li, W. CD36 deficiency inhibits proliferation by cell cycle control in skeletal muscle cells. Front. Physiol. 2022, 13, 947325. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Jeromson, S.; Levesque-Damphousse, P.; Secco, B.; Laplante, M.; Estall, J.L. PGC1A regulates the IRS1:IRS2 ratio during fasting to influence hepatic metabolism downstream of insulin. Proc. Natl. Acad. Sci. USA 2019, 116, 4285–4290. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhu, J.; Wang, Y.H.; Hang, C.H. ROS-Mediated Mitochondrial Dysfunction and ER Stress Contribute to Compression-Induced Neuronal Injury. Neuroscience 2019, 416, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzefovych, L.V.; Solodushko, V.A.; Wilson, G.L.; Rachek, L.I. Protection from palmitate-induced mitochondrial DNA damage prevents from mitochondrial oxidative stress, mitochondrial dysfunction, apoptosis, and impaired insulin signaling in rat L6 skeletal muscle cells. Endocrinology 2012, 153, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, N.K.; Tokunaga, D.S.; Ha, H.Y.; Polgar, N. The Exocyst Is Required for CD36 Fatty Acid Translocase Trafficking and Free Fatty Acid Uptake in Skeletal Muscle Cells. Cells 2022, 11, 2440. [Google Scholar] [CrossRef] [PubMed]
- Coen, P.M.; Goodpaster, B.H. Role of intramyocelluar lipids in human health. Trends Endocrinol. Metab. 2012, 23, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Charytoniuk, T.; Ilowska, N.; Berk, K.; Drygalski, K.; Chabowski, A.; Konstantynowicz-Nowicka, K. The effect of enterolactone on sphingolipid pathway and hepatic insulin resistance development in HepG2 cells. Life Sci. 2019, 217, 1–7. [Google Scholar] [CrossRef]
- De Sanctis, P.; Filardo, G.; Abruzzo, P.M.; Astolfi, A.; Bolotta, A.; Indio, V.; Di Martino, A.; Hofer, C.; Kern, H.; Löfler, S.; et al. Non-Coding RNAs in the Transcriptional Network That Differentiates Skeletal Muscles of Sedentary from Long-Term Endurance- and Resistance-Trained Elderly. Int. J. Mol. Sci. 2021, 22, 1539. [Google Scholar] [CrossRef]
- Hajri, T.; Hall, A.M.; Jensen, D.R.; Pietka, T.A.; Drover, V.A.; Tao, H.; Eckel, R.; Abumrad, N.A. CD36-facilitated fatty acid uptake inhibits leptin production and signaling in adipose tissue. Diabetes 2007, 56, 1872–1880. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, D.J.; Kuchibhotla, S.; Westfall, K.M.; Silverstein, R.L.; Morton, R.E.; Febbraio, M. A CD36-dependent pathway enhances macrophage and adipose tissue inflammation and impairs insulin signalling. Cardiovasc. Res. 2011, 89, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Love-Gregory, L.; Sherva, R.; Schappe, T.; Qi, J.S.; McCrea, J.; Klein, S.; Connelly, M.A.; Abumrad, N.A. Common CD36 SNPs reduce protein expression and may contribute to a protective atherogenic profile. Hum. Mol. Genet. 2011, 20, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Rieusset, J. Contribution of mitochondria and endoplasmic reticulum dysfunction in insulin resistance: Distinct or interrelated roles? Diabetes Metab. 2015, 41, 358–368. [Google Scholar] [CrossRef]
- Szendroedi, J.; Schmid, A.I.; Chmelik, M.; Toth, C.; Brehm, A.; Krssak, M.; Nowotny, P.; Wolzt, M.; Waldhausl, W.; Roden, M. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 2007, 4, e154. [Google Scholar] [CrossRef]
- Fiorentino, T.V.; Monroy, A.; Kamath, S.; Sotero, R.; Cas, M.D.; Daniele, G.; Chavez, A.O.; Abdul-Ghani, M.; Hribal, M.L.; Sesti, G.; et al. Pioglitazone corrects dysregulation of skeletal muscle mitochondrial proteins involved in ATP synthesis in type 2 diabetes. Metabolism 2021, 114, 154416. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Ritov, V.B.; Menshikova, E.V.; Azuma, K.; Wood, R.; Toledo, F.G.; Goodpaster, B.H.; Ruderman, N.B.; Kelley, D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E49–E58. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Rao, Z.; Guo, Y.; Chen, P.; Xiao, W. High-Intensity Interval Training Restores Glycolipid Metabolism and Mitochondrial Function in Skeletal Muscle of Mice With Type 2 Diabetes. Front. Endocrinol. 2020, 11, 561. [Google Scholar] [CrossRef]
- Chaudhry, A.; Shi, R.; Luciani, D.S. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E87–E101. [Google Scholar] [CrossRef]
- Hatmal, M.M.; Alshaer, W.; Mahmoud, I.S.; Al-Hatamleh, M.A.I.; Al-Ameer, H.J.; Abuyaman, O.; Zihlif, M.; Mohamud, R.; Darras, M.; Al Shhab, M.; et al. Investigating the association of CD36 gene polymorphisms (rs1761667 and rs1527483) with T2DM and dyslipidemia: Statistical analysis, machine learning based prediction, and meta-analysis. PLoS ONE 2021, 16, e0257857. [Google Scholar] [CrossRef]
- Csordas, G.; Weaver, D.; Hajnoczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense (5′–3′) | Anti-Sense (5′–3′) |
|---|---|---|
| Slc2a4 | GTGACTGGAACACTGGTCCTA | CCAGCCACGTTGCATTGTAG |
| Irs1 | AGC GCG CCT GGA GTA TTA TGA GAA | GTC AGC CCG CTT GTT GAT GTT GAA |
| Irs2 | AAA GTG GCC TAC AAC CCT TAC CCA | TCA TCG CTC TTG CAG CTA TTG GG |
| Pi3kr1 | AAG GAG CTG GTG CTA CAT TAT C | CGC CTC TGT TGT GCA TAT ACT |
| ATF6 | GACTCACCCATCCGAGTTGTG | CTCCCAGTCTTCATCTGGTCC |
| IRE1α | ACACTGCCTGAGACCTTGTTG | GGAGCCCGTCCTCTTGCTA |
| PERK | GCGTCGGAGACAGTGTTTG | CGTCCATCTAAAGTGCTGATGAT |
| CHOP | CTGGAAGCCTGGTATGAGGAT | CAGGGTCAAGAGTAGTGAAGGT |
| GRP78 | ACTTGGGGACCACCTATTCCT | ATCGCCAATCAGACGCTCC |
| GRP94 | TCGTCAGAGCTGATGATGAAGT | GCGTTTAACCCATCCAACTGAAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Su, Y.; Chen, J.; Qin, D.; Xu, Y.; Chu, H.; Lu, T.; Dong, J.; Qin, L.; Li, W. Differential Roles of CD36 in Regulating Muscle Insulin Response Depend on Palmitic Acid Load. Biomedicines 2023, 11, 729. https://doi.org/10.3390/biomedicines11030729
Sun J, Su Y, Chen J, Qin D, Xu Y, Chu H, Lu T, Dong J, Qin L, Li W. Differential Roles of CD36 in Regulating Muscle Insulin Response Depend on Palmitic Acid Load. Biomedicines. 2023; 11(3):729. https://doi.org/10.3390/biomedicines11030729
Chicago/Turabian StyleSun, Jingyu, Yajuan Su, Jiajia Chen, Duran Qin, Yaning Xu, Hang Chu, Tianfeng Lu, Jingmei Dong, Lili Qin, and Weida Li. 2023. "Differential Roles of CD36 in Regulating Muscle Insulin Response Depend on Palmitic Acid Load" Biomedicines 11, no. 3: 729. https://doi.org/10.3390/biomedicines11030729
APA StyleSun, J., Su, Y., Chen, J., Qin, D., Xu, Y., Chu, H., Lu, T., Dong, J., Qin, L., & Li, W. (2023). Differential Roles of CD36 in Regulating Muscle Insulin Response Depend on Palmitic Acid Load. Biomedicines, 11(3), 729. https://doi.org/10.3390/biomedicines11030729