
Identification of Natural Lead Compounds against Hemagglutinin-Esterase Surface Glycoprotein in Human Coronaviruses Investigated via MD Simulation, Principal Component Analysis, Cross-Correlation, H-Bond Plot and MMGBSA

 ,
,  , , and
, , and
Abstract
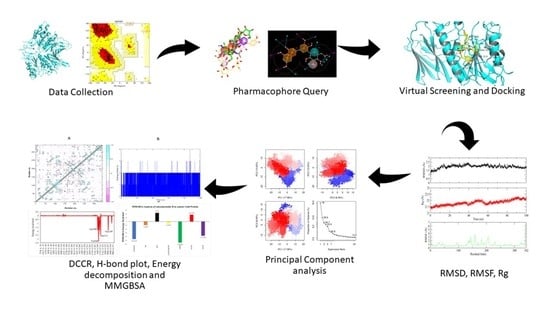
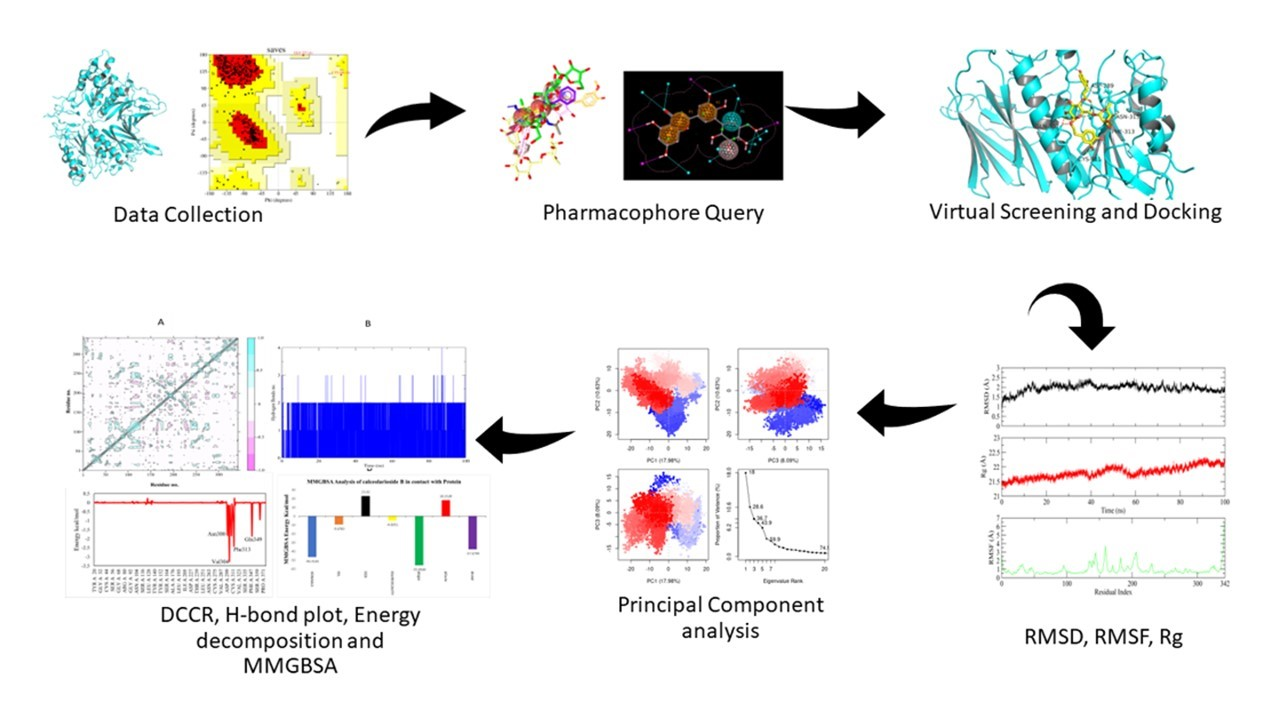
:
1. Introduction
2. Materials and Methods
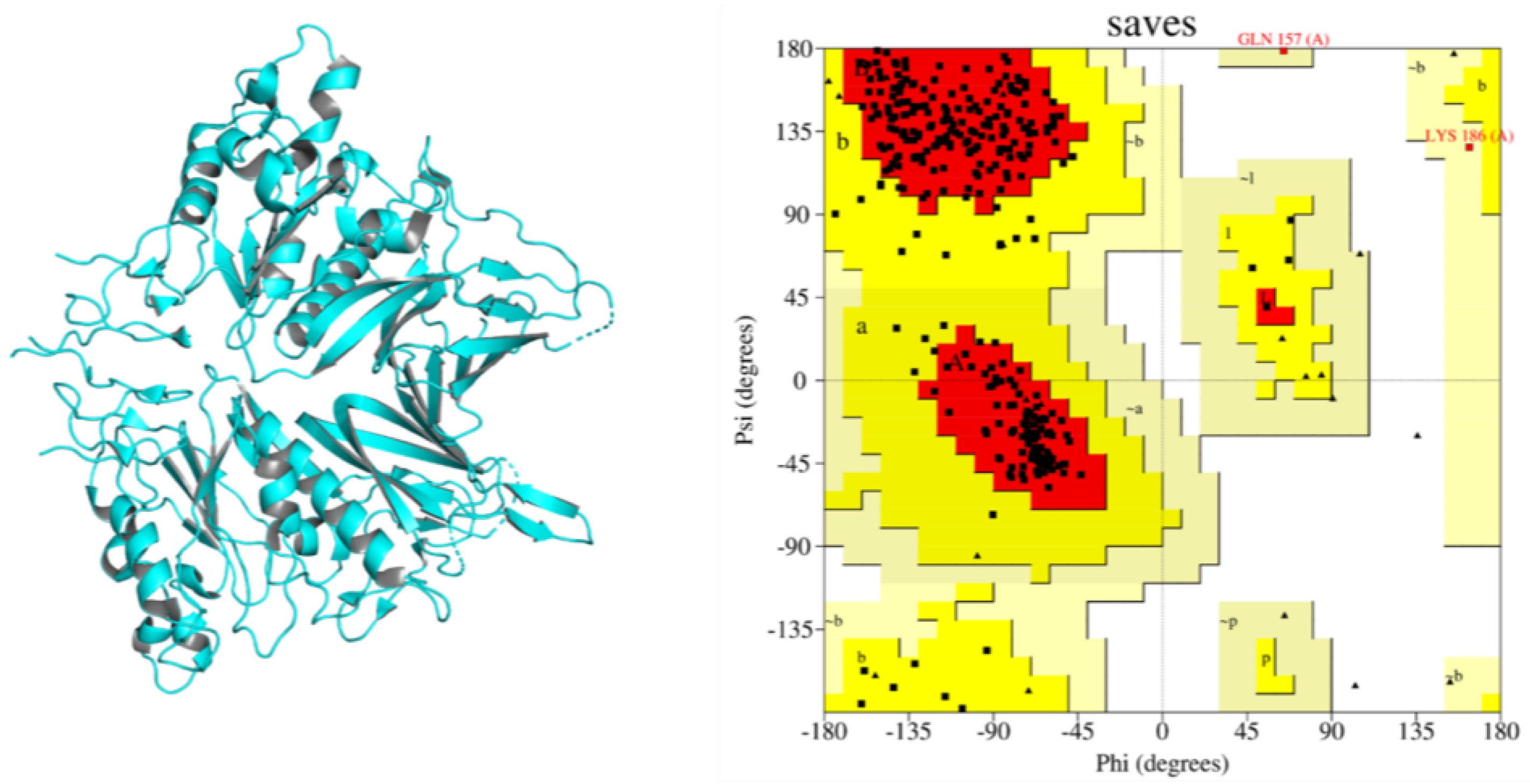
2.1. Structure Retrieval, Refinement and Evaluation
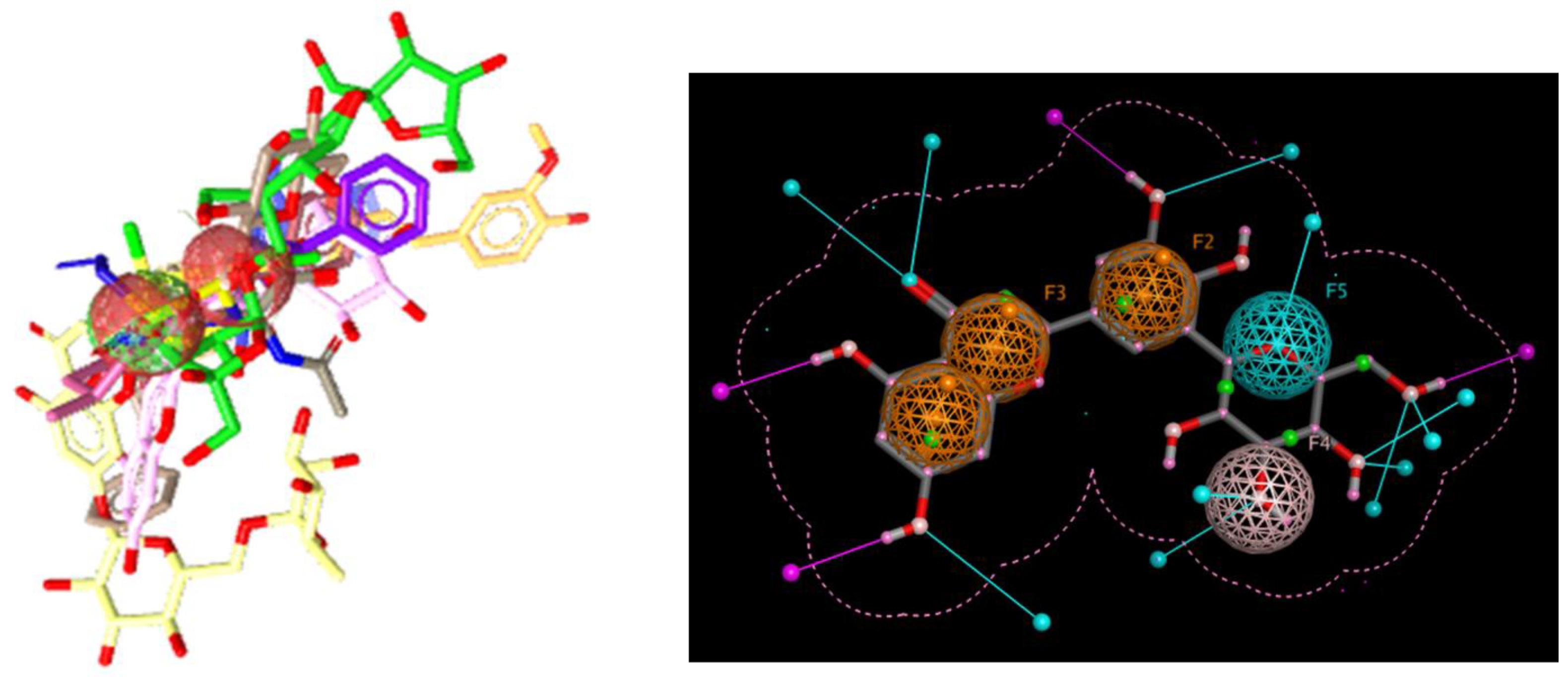
2.2. Selection of Ligands and Pharmacophore Generation
2.3. Library Preparation and Virtual Screening
2.4. Docking Calculation and Interaction
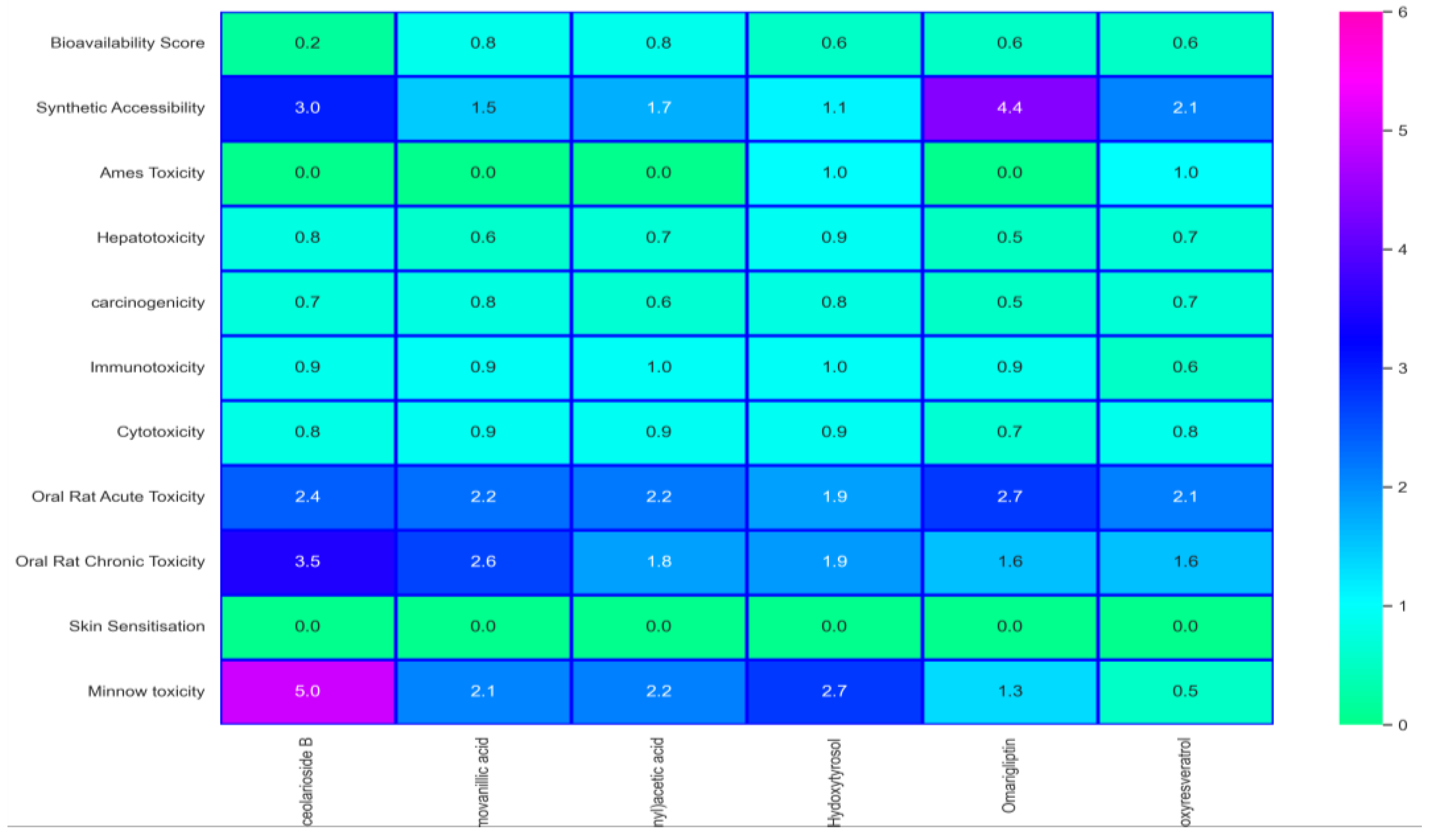
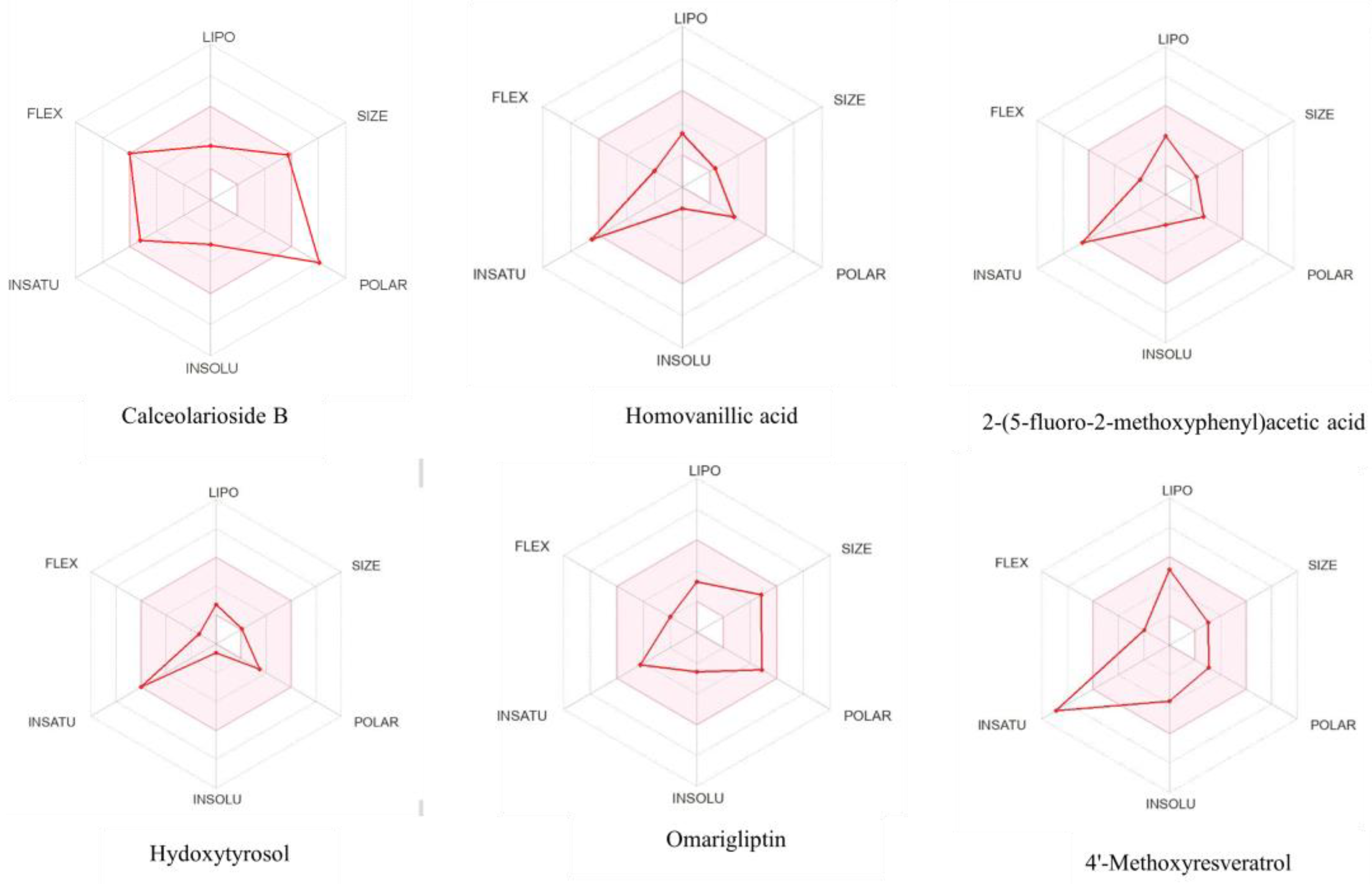
2.5. Toxicity Analysis and Bioactivity Prediction
2.6. Lead Identification
2.7. Molecular Dynamic (MD) Simulations
2.8. Molecular Mechanics/Generalized Born Surface Area (MMGBSA) Analysis
3. Results
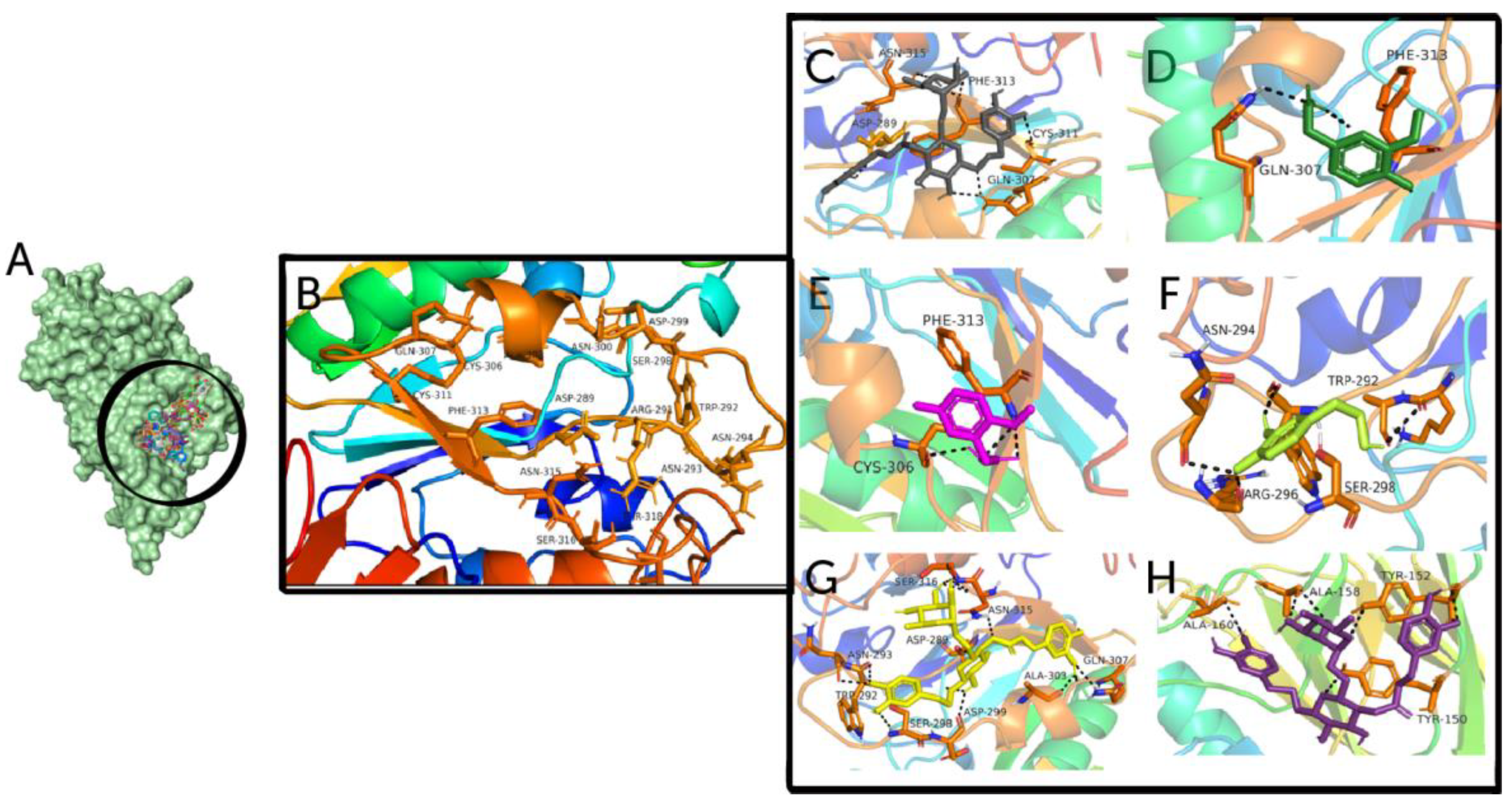
3.1. Ligand-Based Virtual Screening and Molecular Docking
3.2. Molecular Dynamics Simulations
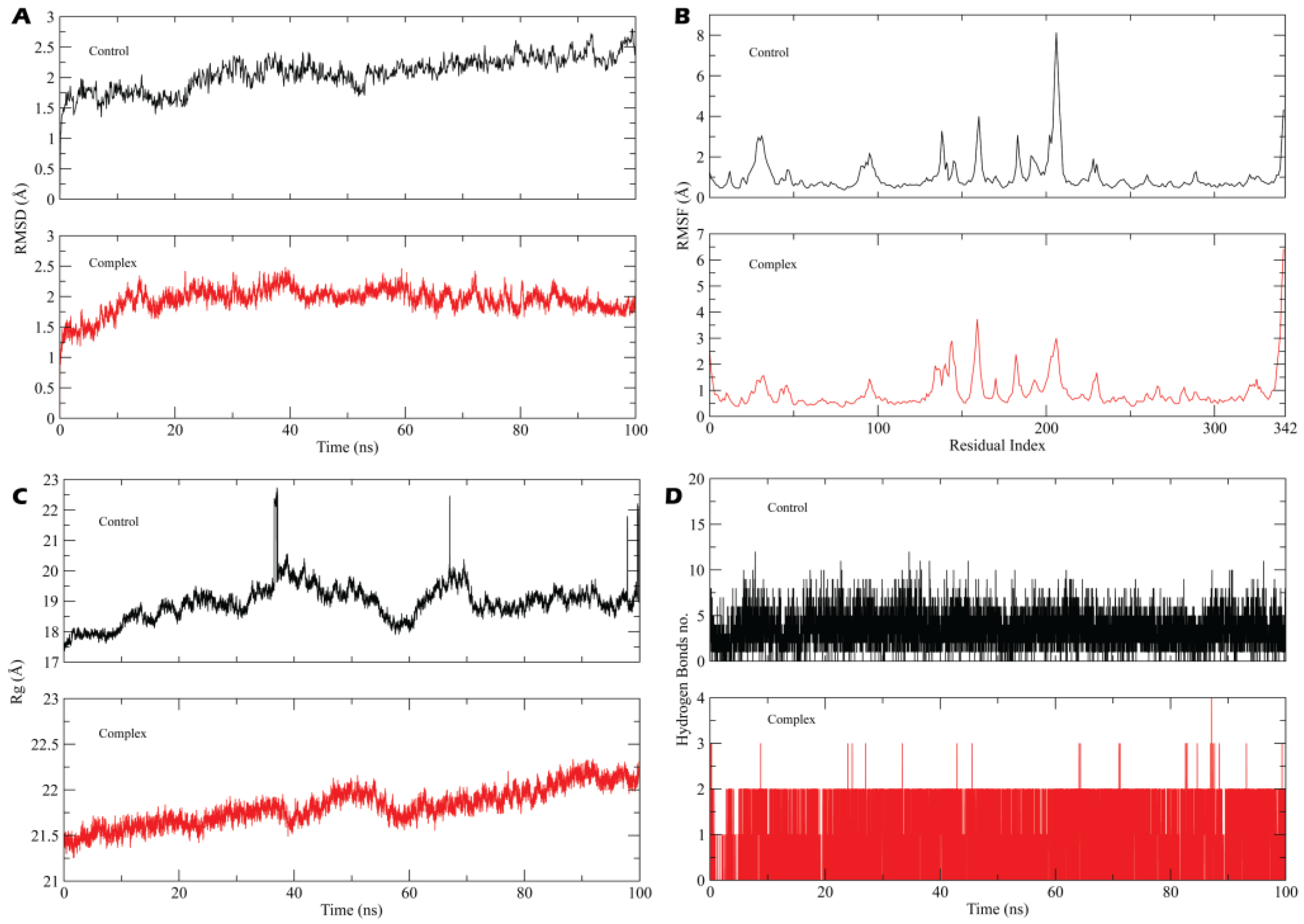
3.2.1. System Stability, Fluctuation and Radius of Gyration
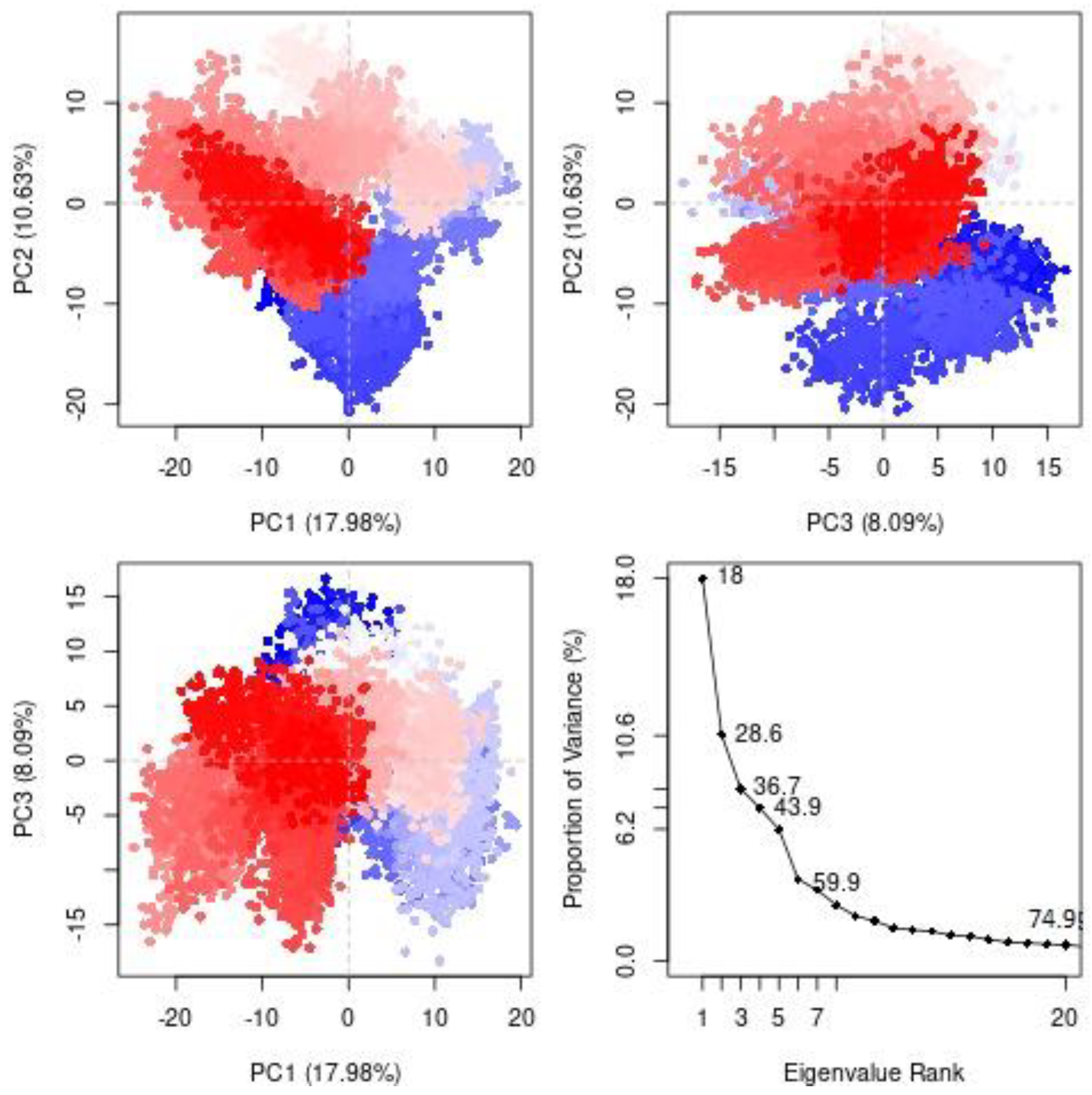
3.2.2. Principal Component Analysis (PCA)
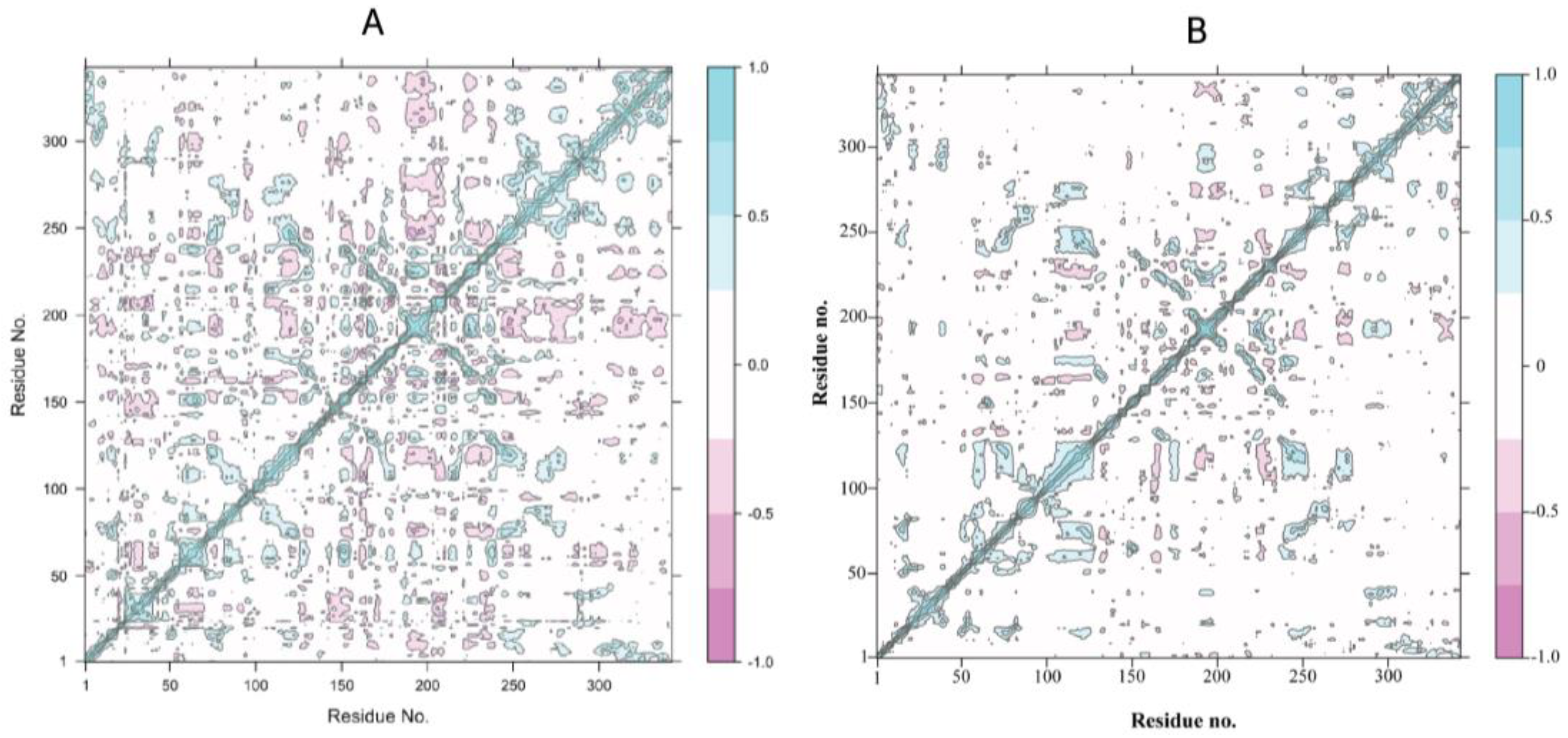
3.2.3. Positive-Negative Correlation Movements of Residues
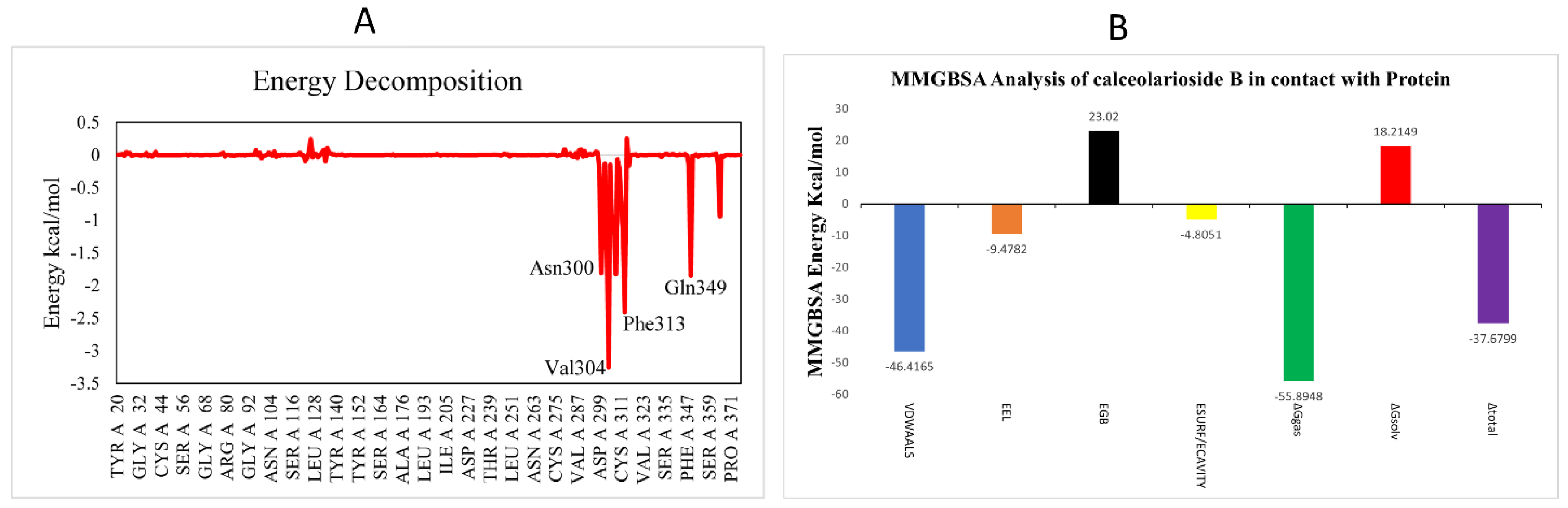
3.2.4. Binding Energy Landscape and Energy Decomposition Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 genome-encoded proteins. Sci. China Life Sci. 2022, 65, 280–294. [Google Scholar] [CrossRef]
- Ul Haq, I.; Krukiewicz, K.; Yahya, G.; Haq, M.U.; Maryam, S.; Mosbah, R.A.; Saber, S.; Alrouji, M. The Breadth of Bacteriophages Contributing to the Development of the Phage-Based Vaccines for COVID-19: An Ideal Platform to Design the Multiplex Vaccine. Int. J. Mol. Sci. 2023, 24, 1536. Available online: https://www.mdpi.com/1422-0067/24/2/1536/htm (accessed on 18 February 2023). [CrossRef] [PubMed]
- Maryam, S.; Ul Haq, I.; Yahya, G.; Ul Haq, M.; Algammal, A.M.; Saber, S.; Cavalu, S. COVID-19 surveillance in wastewater: An epidemiological tool for the monitoring of SARS-CoV-2. Front. Cell. Infect. Microbiol. 2023, 12, 1743. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard|WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/cited (accessed on 16 August 2022).
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The Species Severe Acute Respiratory syndrome-related Coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. Available online: https://www.nature.com/articles/s41564-020-0695-z (accessed on 17 August 2022). [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Xiao, Z.; Ye, K.; He, X.; Sun, B.; Qin, Z.; Yu, J.; Yao, J.; Wu, Q.; Bao, Z.; et al. Coronavirus Disease-19 (COVID-19): A Perspective of New Scenario: Volume 1; Bentham Science Publisher: Singapore, 2021; Available online: https://virologyj.biomedcentral.com/articles/10.1186/s12985-020-01369-z (accessed on 16 August 2022).
- Yang, Y.; Xiao, Z.; Ye, K.; He, X.; Sun, B.; Qin, Z.; Yu, J.; Yao, J.; Wu, Q.; Bao, Z.; et al. SARS-CoV-2: Characteristics and current advances in research. Virol. J. 2020, 17, 1–17. [Google Scholar] [CrossRef]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. Available online: https://link.springer.com/article/10.1007/s11427-020-1637-5 (accessed on 17 August 2022). [CrossRef] [Green Version]
- Banerjee, D. The impact of Covid-19 pandemic on elderly mental health. Int. J. Geriatr. Psychiatry 2020, 35, 1466. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. Available online: https://www.nature.com/articles/s41586-020-2286-9 (accessed on 16 August 2022). [CrossRef]
- El-Sokkary, M.; El-Baz, A.M.; El-Morsi, R.M.; Keuper, K.; El-Hawary, S.; Shata, A.; Yahya, G. Early Forecasting of COVID-19 Case Progression with Hematological and Biochemical Parameters of Patients in Egypt. Pak. J. Pharm. Sci. 2022, 35, 401–408. Available online: https://search.ebscohost.com/login.aspx?direct=true&profile=ehost&scope=site&authtype=crawler&jrnl=1011601X&AN=155566618&h=dx9mqQoSjdT3pPfugglCdC7kxM7S%2BCcm8g8cAB%2FMhqt7zgzQf2HGZqb6DITMi28iJeWLVPPTiKc95QgWrWlyJg%3D%3D&crl=c (accessed on 20 February 2023).
- Muhammad, M.A.; Ihtisham Ulhaq, K.R. Histopathologic Evaluation and Scoring of SARSCoV- 2 Infection. Coronavirus Dis. A Perspect. New Scenar. 2021, 01, 52–71. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 2698. [Google Scholar] [CrossRef]
- Zia, K.; Khan, S.A.; Ashraf, S.; Nur-e-Alam, M.; Ahmed, S.; Ul-Haq, Z. Probing CAS database as prospective antiviral agents against SARS-CoV-2 main protease. J. Mol. Struct. 2021, 1231, 129953. [Google Scholar] [CrossRef]
- Krafcikova, P.; Silhan, J.; Nencka, R.; Boura, E. Structural analysis of the SARS-CoV-2 methyltransferase complex involved in RNA cap creation bound to sinefungin. Nat. Commun. 2020, 11, 3717. Available online: https://www.nature.com/articles/s41467-020-17495-9 (accessed on 5 April 2022). [CrossRef]
- Khan, A.; Zia, K.; Altowyan, M.S.; Ul-Haq, Z. Deciphering the impact of mutations on binding efficacy of SARS-CoV-2 Omicron and Delta variants with human ACE2 receptor. Front. Chem. 2022, 566, 892093. [Google Scholar] [CrossRef]
- Zeng, Q.; Langereis, M.A.; Van Vliet, A.L.W.; Huizinga, E.G.; De Groot, R.J. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 9065–9069. Available online: https://www.pnas.org/doi/abs/10.1073/pnas.0800502105 (accessed on 3 September 2022). [CrossRef] [PubMed] [Green Version]
- Haq, I.U.; Khan, Z.I.; Aziz, I.; Basit, A.; Hussain, F.; Bibi, A.; Aqib, A.I.; Siddique, F.; Younas, U.; Rahim, K. Phages and SARS-CoV-2. In Applications of Natural Products and SARS-CoV-2; Academic Press: Amsterdam, The Netherlands, 2023; pp. 273–292. [Google Scholar] [CrossRef]
- Patel, C.N.; Kumar, S.P.; Pandya, H.A.; Rawal, R.M. Identification of potential inhibitors of coronavirus hemagglutinin-esterase using molecular docking, molecular dynamics simulation and binding free energy calculation. Mol. Divers. 2021, 25, 421. [Google Scholar] [CrossRef] [PubMed]
- Bakkers, M.J.G.; Lang, Y.; Feitsma, L.J.; Hulswit, R.J.G.; de Poot, S.A.H.; van Vliet, A.L.W.; Margine, I.; de Groot-Mijnes, J.D.F.; van Kuppeveld, F.J.M.; Langereis, M.A.; et al. Betacoronavirus Adaptation to Humans Involved Progressive Loss of Hemagglutinin-Esterase Lectin Activity. Cell Host Microbe 2017, 21, 356–366. [Google Scholar] [CrossRef] [Green Version]
- Haq, I.U.; Fayyaz, F.; Shafqat, A.; Basit, A.; Hussain, F.; Aziz, I.; Khan, Z.I.; Aqib, A.I.; Siddique, F.; Younas, U.; et al. Natural Products and SARS-CoV-2. In Applications of Natural Products and SARS-CoV-2; Academic Press: Amsterdam, The Netherlands, 2023; pp. 1–24. [Google Scholar] [CrossRef]
- Ul Haq, I.; Rahim, K.; Rafiq, M.; Asif, T.; Alvi, S.; Yaseen, K. Polyketides and SARS-CoV-2. In Applications of Natural Products and SARS-CoV-2; Academic Press: Amsterdam, The Netherlands, 2023; pp. 423–444. [Google Scholar] [CrossRef]
- Kumar, A.; Choudhir, G.; Shukla, S.K.; Sharma, M.; Tyagi, P.; Bhushan, A.; Rathore, M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2021, 39, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Balunas, M.J.; Kinghorn, A.D. Drug discovery from medicinal plants. Life Sci. 2005, 78, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Elmorsy, M.A.; El-Baz, A.M.; Mohamed, N.H.; Almeer, R.; Abdel-Daim, M.M.; Yahya, G. In silico screening of potent inhibitors against COVID-19 key targets from a library of FDA-approved drugs. Environ. Sci. Pollut. Res. Int. 2022, 29, 12336–12346. [Google Scholar] [CrossRef]
- Shaldam, M.A.; Yahya, G.; Mohamed, N.H.; Abdel-Daim, M.M.; Al Naggar, Y. In silico screening of potent bioactive compounds from honeybee products against COVID-19 target enzymes. Environ. Sci. Pollut. Res. 2021, 28, 40507–40514. Available online: https://link.springer.com/article/10.1007/s11356-021-14195-9 (accessed on 20 February 2023). [CrossRef] [PubMed]
- Al Naggar, Y.; Giesy, J.P.; Abdel-Daim, M.M.; Javed Ansari, M.; Al-Kahtani, S.N.; Yahya, G. Fighting against the second wave of COVID-19: Can honeybee products help protect against the pandemic? Saudi J. Biol. Sci. 2021, 28, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, A.; Mostafa, I.; Elshaier, Y.A.M.M.; Mahmoud, S.H.; Abo Shama, N.M.; Shehata, M.; Yahya, G.; Nasr, N.F.; El-Halawany, A.M.; Ali, M.A.; et al. Robust Antiviral Activity of Santonica Flower Extract (Artemisia cina) against Avian and Human Influenza A Viruses: In Vitro and Chemoinformatic Studies. ACS Omega 2022, 7, 41212–41223. Available online: https://pubs.acs.org/doi/full/10.1021/acsomega.2c04867 (accessed on 20 February 2023). [CrossRef]
- Al-Karmalawy, A.A.; Soltane, R.; Abo Elmaaty, A.; Tantawy, M.A.; Antar, S.A.; Yahya, G.; Chrouda, A.; Pashameah, R.A.; Mustafa, M.; Abu Mraheil, M.; et al. Coronavirus Disease (COVID-19) Control between Drug Repurposing and Vaccination: A Comprehensive Overview. Vaccines 2021, 9, 1317. Available online: https://www.mdpi.com/2076-393X/9/11/1317/htm (accessed on 20 February 2023). [CrossRef]
- BIOVIA San Diego: Dassault Systèmes. Available online: https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=BIOVIA++San+Diego%3A+Dassault+Systèmes%2C+%5BYear&btnG= (accessed on 10 August 2022).
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, T.; Wang, Y.; Bryant, S.H. PubChem as a public resource for drug discovery. Drug Discov. Today 2010, 15, 1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, X.; Lee, X.Y.; De Raeymaeker, J.; Tame, J.R.; Zhang, K.Y.; De Maeyer, M.; Voet, A.R. Pharmacophore modeling: Advances, limitations, and current utility in drug discovery. J. Receptor. Ligand Channel Res. 2014, 7, 81–92. Available online: https://www.dovepress.com/pharmacophore-modeling-advances-limitations-and-current-utility-in-dru-peer-reviewed-fulltext-article-JRLCR (accessed on 12 August 2022).
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2004, 45, 160–169. Available online: https://pubs.acs.org/doi/abs/10.1021/ci049885e (accessed on 12 August 2022). [CrossRef]
- Averill, F.W.; Painter, G.S. Steepest-descent determination of occupation numbers and energy minimization in the local-density approximation. Phys. Rev. B 1992, 46, 2498. [Google Scholar] [CrossRef]
- Yuan, G.; Li, T.; Hu, W. A conjugate gradient algorithm and its application in large-scale optimization problems and image restoration. J. Inequal. Appl. 2019, 2019, 247. Available online: https://journalofinequalitiesandapplications.springeropen.com/articles/10.1186/s13660-019-2192-6 (accessed on 12 August 2022). [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. Available online: https://www.nature.com/articles/srep42717 (accessed on 13 August 2022). [CrossRef] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. Available online: https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.5b00104 (accessed on 13 August 2022). [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, T.; OSIRIS Property Explorer. Organic Chemistry Portal.—References—Scientific Research Publishing. 2001. Available online: https://www.scirp.org/(S(351jmbntv-nsjt1aadkposzje))/reference/refeencespapers.aspx?referenceid=2628716 (accessed on 18 February 2023).
- Price, D. A Modified TIP3P Water Potential for Simulation with Ewald Summation. J. Chem. Phys. 2004, 121, 10096–10103. Available online: https://aip.scitation.org/doi/abs/10.1063/1.1808117 (accessed on 28 December 2022). [CrossRef] [PubMed]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Perez-Sanchez, H.; Lightstone, F.C. A Comprehensive Docking and MM/GBSA Rescoring Study of Ligand Recognition upon Binding Antithrombin. Curr. Top. Med. Chem. 2017, 17, 1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamri, M.A.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; Alqahtani, S.M. Structure-based virtual screening and molecular dynamics of phytochemicals derived from Saudi medicinal plants to identify potential COVID-19 therapeutics. Arab. J. Chem. 2020, 13, 7224–7234. [Google Scholar] [CrossRef] [PubMed]
- Bitencourt-Ferreira, G.; Veit-Acosta, M.; de Azevedo, W.F. Hydrogen bonds in protein-ligand complexes. Methods Mol. Biol. 2019, 2053, 93–107. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Names | Pharmacophore Fit Score | Structures |
|---|---|---|---|
| 1 | 5-Norbornene 2,3 dicarboxy-chloride | 32.99 |  |
| 2 | Levan | 34.31 |  |
| 3 | Caffeic Acid | 40.44 |  |
| 4 | S-Nitroso-N-Acetylpenicillamine | 41.00 |  |
| 5 | Curcumin | 41.75 |  |
| 6 | Quercetin | 41.60 |  |
| 7 | Diallyl disulphide | 33.22 |  |
| 8 | Pulegone | 37.29 |  |
| 9 | Flavylium | 13.27 |  |
| 10 | Pinocembrin | 34.61 |  |
| 11 | Gallic acid | 34.68 |  |
| 12 | Rosmeric acid | 41.50 |  |
| 13 | Luteolin | 34.67 |  |
| 14 | Hesperidin | 41.61 |  |
| Selected Compounds | Docking Energies | Pharmacophore Fit Score | HBA | HBD | Rotatable Bonds | M.W | logP |
|---|---|---|---|---|---|---|---|
| Calceolarioside B | −7.8 | 40.08 | 10 | 7 | 9 | 478.4 | 0.6 |
| Homovanillic acid | −7.7 | 40.05 | 4 | 2 | 3 | 182 | 0.4 |
| 2-(5-fluoro-2-methoxyphenyl)acetic acid | −7.7 | 40.00 | 4 | 1 | 3 | 184 | 1.5 |
| Hydroxytyrosol | −7.7 | 40.05 | 3 | 3 | 2 | 154.16 | 0.17 |
| Omarigliptin | −7.7 | 39.86 | 8 | 1 | 3 | 398.4 | 0.3 |
| 4′-Methoxyresveratrol | −7.7 | 39.86 | 3 | 2 | 3 | 242.27 | 3.5 |
| 12-Hydroxy-10,13-dimethyl-2,4,5,6,17-dione | −7.7 | 39.96 | 8 | 2 | 3 | 391.4 | 0.3 |
| AZ628 | −7.6 | 40.4 | 5 | 2 | 5 | 451.5 | 4.2 |
| Telaprevir | −7.5 | 39.99 | 8 | 4 | 14 | 679.8 | 4.2 |
| Verdinexor | −7.5 | 39.87 | 11 | 2 | 5 | 422.3 | 4.1 |
| 4-[3-(morpholine-4-carbonyl)-5-[4-(trifluoromethyl)phenyl]pyrazol-1-yl]benzenesulfonamide | −7.5 | 39.93 | 9 | 1 | 4 | 480.5 | 2.4 |
| 3,4 dihydroxyphenylacetic acid | −7.3 | 40.08 | 4 | 3 | 2 | 168.15 | 0.5 |
| aminomethyl(phenyl)phosphinic acid | −7.3 | 40.00 | 3 | 2 | 2 | 171.13 | −2.7 |
| 3-[2-(3-cyanatophenoxy)ethoxy]phenyl]cyanate | −7.0 | 39.89 | 6 | 0 | 7 | 296.28 | 3.9 |
| N-[(4,5-difluoro-1H-benzimidazol-2-yl)methyl]-9-(3-fluorophenyl)-2-morpholin-4-ylpurin-6-amine | −6.5 | 39.87 | 10 | 2 | 5 | 480.4 | 3.5 |
| N-(2-methyl-4-phenylbut-3-en-2-yl)-1-phenylmethanimine | −6.4 | 40.4 | 1 | 0 | 4 | 249.3 | 4.4 |
| Ruboxistaurin | −6.2 | 40.4 | 4 | 1 | 2 | 468.5 | 2.7 |
| Daunorubicin | −5.3 | 40.11 | 11 | 5 | 4 | 527.5 | 1.8 |
| Forsythoside A | −5.1 | 40.08 | 15 | 9 | 11 | 624.6 | −0.5 |
| Turofexorate Isopropyl | −5.1 | 40.4 | 5 | 1 | 4 | 438.5 | 5.0 |
| Phytochemicals | Calceolarioside B | Homovanillic Acid | 2-(5-fluoro-2-methoxyphenyl)acetic Acid | Hydroxytyrosol | Omarigliptin | 4′-Methoxyresveratrol |
|---|---|---|---|---|---|---|
| Formula | C23H26O11 | C9H10O4 | C9H9FO3 | C8H10O3 | C17H20F2N4O3S | C15H14O3 |
| Pfizer Rule | Accepted | Accepted | Accepted | Accepted | Accepted | Rejected |
| Golden Triangle | Accepted | Rejected | Rejected | Rejected | Accepted | Accepted |
| BBB Penetration | BBB+ | BBB+ | BBB+ | BBB+ | BBB+ | BBB+ |
| Fu | 5.8% | 18.71% | 5.98% | 61.31% | 73.074% | 1.403% |
| Density | 1.048 | 1.01 | 1.037 | 0.982 | 1.118 | 0.935 |
| ESOL Class | Soluble | Very soluble | Soluble | Very soluble | Soluble | Soluble |
| Ali Class | Moderately soluble | Very soluble | Soluble | Very soluble | Very soluble | Moderately soluble |
| Silicos-IT class | Soluble | Soluble | Soluble | Soluble | Soluble | Soluble |
| GI absorption | Low | High | High | High | High | High |
| Pgp substrate | Yes | No | No | No | Yes | No |
| log Kp (skin permeation) | −8.80 | −7.18 | −6.39 | −7.75 | −8.55 | −5.33 |
| Lipinski violations | 2 | 0 | 0 | 0 | 0 | 0 |
| Ghose violations | 1 | 0 | 0 | 1 | 0 | 0 |
| Veber violations | 1 | 0 | 0 | 0 | 0 | 0 |
| Acute Toxicity Alert | 0 | 0 | 0 | 0 | 0 | 0 |
| Genotoxic Carcinogenicity Alerts | 1 | 0 | 0 | 0 | 0 | 0 |
| SureChEMBL Rule Alert | 0 | 0 | 0 | 0 | 0 | 0 |
| Synthetic Accessibility | 2.96 | 1.49 | 1.71 | 1.08 | 4.40 | 2.08 |
| Drug-likeness | −0.05 | 0.17 | −2.0 | −1.3 | 3.65 | −3.1 |
| Drug Score | 0.56 | 0.75 | 0.54 | 0.59 | 0.85 | 0.27 |
| Mutagenicity | No | No | No | No | No | No |
| Tumorgenic | No | No | No | No | No | No |
| Irritant | No | No | No | No | No | No |
| Reproductive Effect | No | No | No | No | No | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, I.; Rasheed, M.A.; Cavalu, S.; Rahim, K.; Ijaz, S.; Yahya, G.; Goh, L.P.W.; Popoviciu, M.S. Identification of Natural Lead Compounds against Hemagglutinin-Esterase Surface Glycoprotein in Human Coronaviruses Investigated via MD Simulation, Principal Component Analysis, Cross-Correlation, H-Bond Plot and MMGBSA. Biomedicines 2023, 11, 793. https://doi.org/10.3390/biomedicines11030793
Ali I, Rasheed MA, Cavalu S, Rahim K, Ijaz S, Yahya G, Goh LPW, Popoviciu MS. Identification of Natural Lead Compounds against Hemagglutinin-Esterase Surface Glycoprotein in Human Coronaviruses Investigated via MD Simulation, Principal Component Analysis, Cross-Correlation, H-Bond Plot and MMGBSA. Biomedicines. 2023; 11(3):793. https://doi.org/10.3390/biomedicines11030793
Chicago/Turabian StyleAli, Iqra, Muhammad Asif Rasheed, Simona Cavalu, Kashif Rahim, Sana Ijaz, Galal Yahya, Lucky Poh Wah Goh, and Mihaela Simona Popoviciu. 2023. "Identification of Natural Lead Compounds against Hemagglutinin-Esterase Surface Glycoprotein in Human Coronaviruses Investigated via MD Simulation, Principal Component Analysis, Cross-Correlation, H-Bond Plot and MMGBSA" Biomedicines 11, no. 3: 793. https://doi.org/10.3390/biomedicines11030793
APA StyleAli, I., Rasheed, M. A., Cavalu, S., Rahim, K., Ijaz, S., Yahya, G., Goh, L. P. W., & Popoviciu, M. S. (2023). Identification of Natural Lead Compounds against Hemagglutinin-Esterase Surface Glycoprotein in Human Coronaviruses Investigated via MD Simulation, Principal Component Analysis, Cross-Correlation, H-Bond Plot and MMGBSA. Biomedicines, 11(3), 793. https://doi.org/10.3390/biomedicines11030793