

The Tryptophan-Kynurenine Metabolic System Is Suppressed in Cuprizone-Induced Model of Demyelination Simulating Progressive Multiple Sclerosis

 , , ,
, , ,  ,
, 
 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
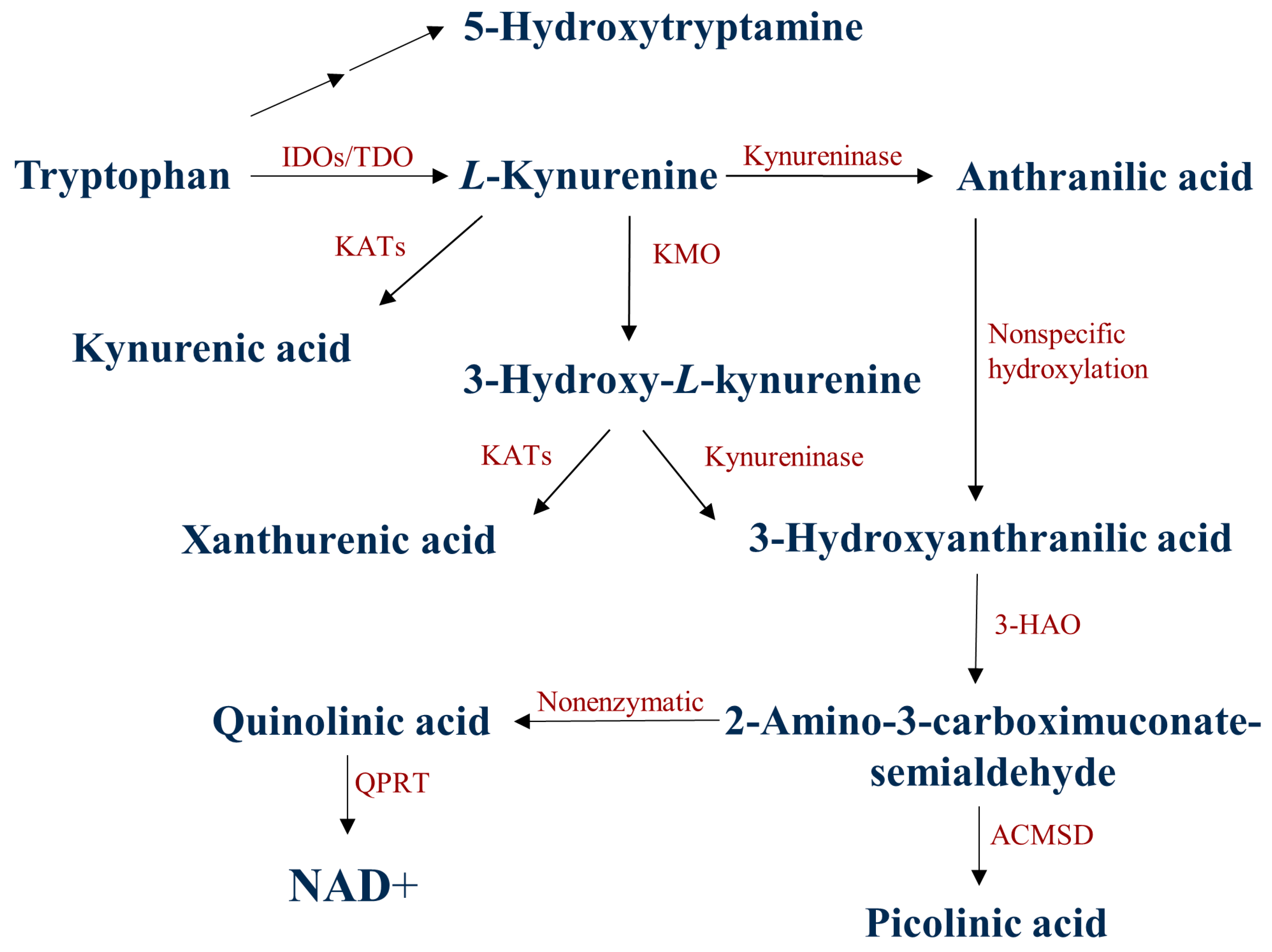
1. Introduction
2. Materials and Methods
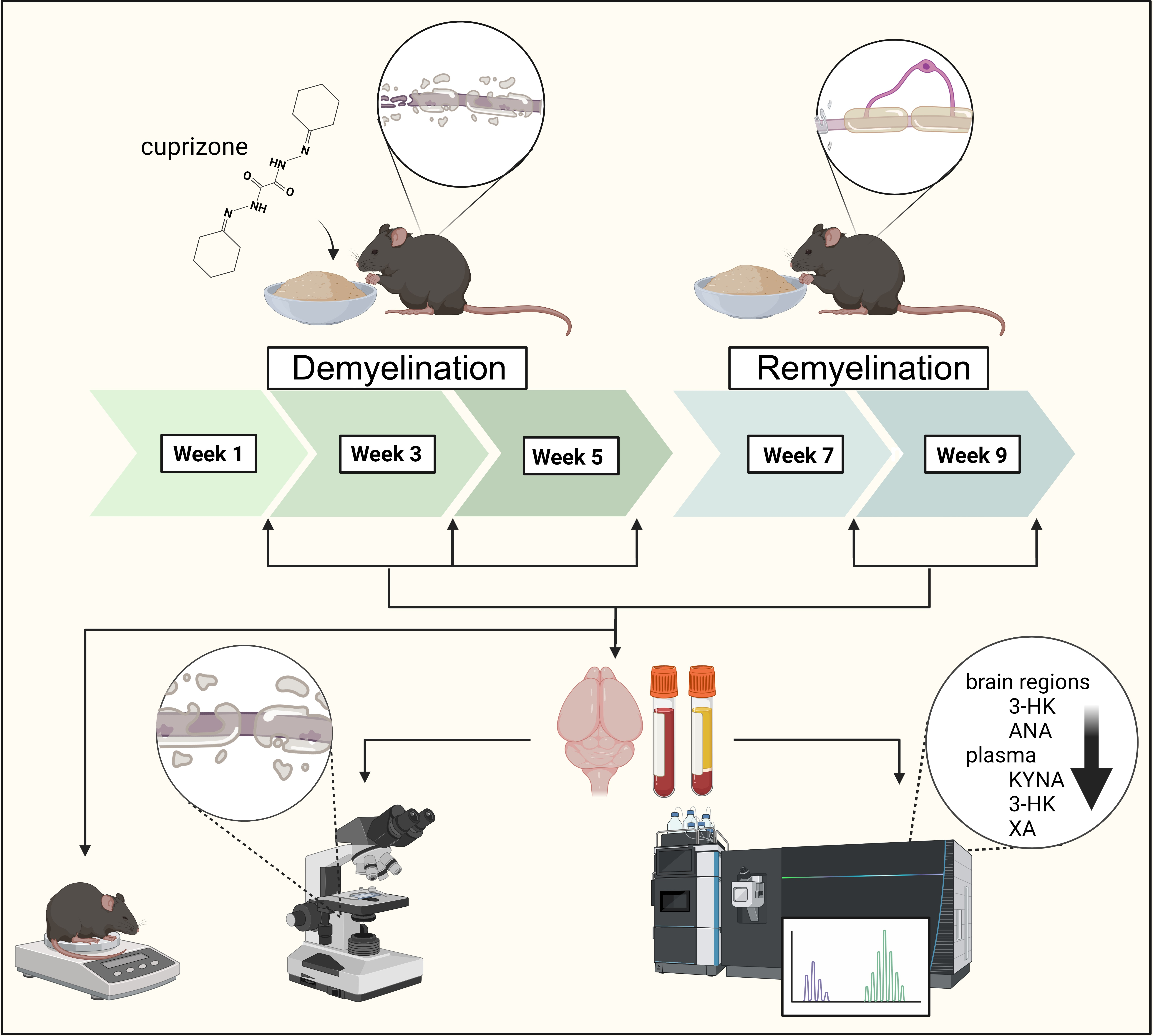
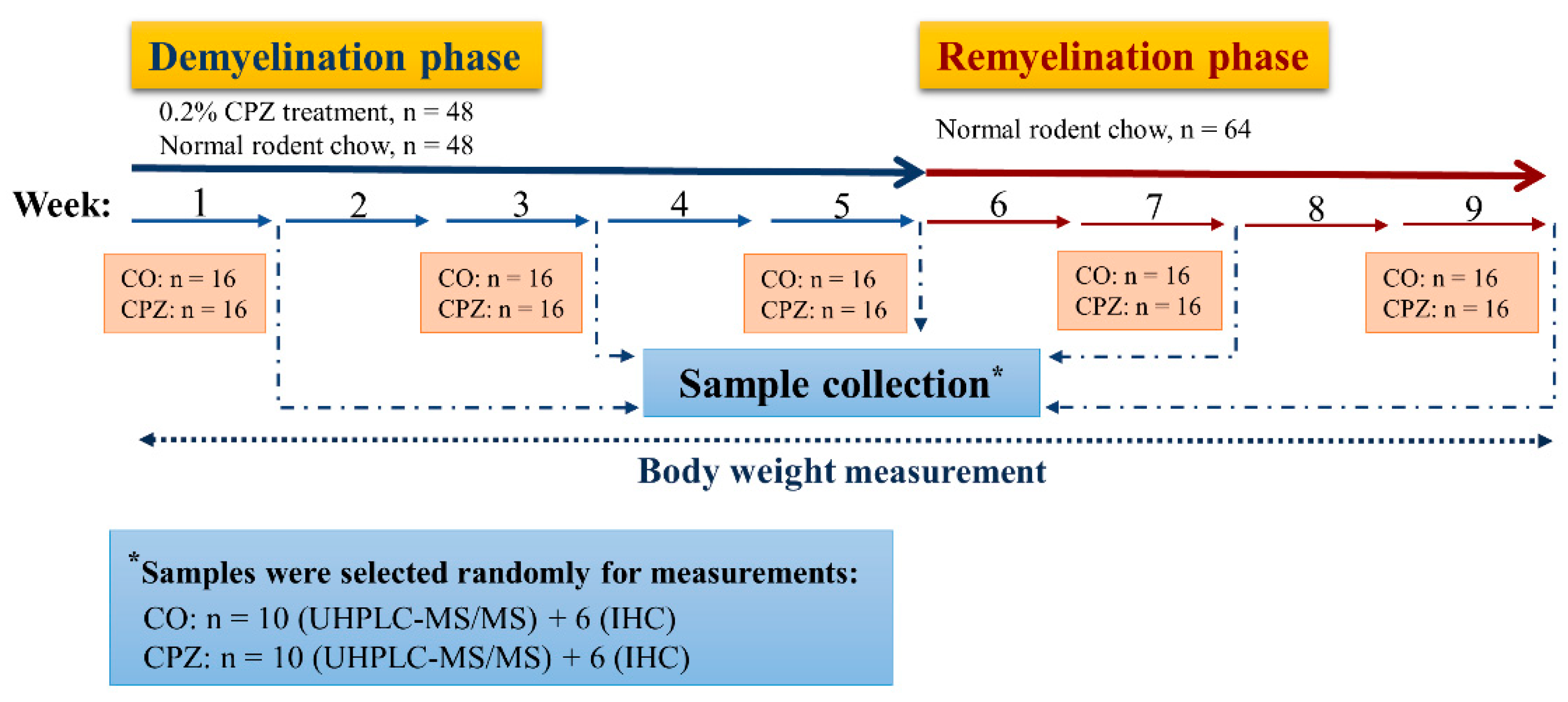
2.1. Animal Experiments and Sample Collection
2.2. Luxol Fast Blue Crystal Violet Staining and Myelin Status Determination by Densitometric Analysis
2.3. Ultra-High-Performance Liquid Chromatography with Tandem Mass Spectrometry Measurement
2.4. Statistical Analysis
3. Results
3.1. Investigation of Body Weight
3.2. Evaluation of Cuprizone Damage in the Demyelination and Remyelination Phases
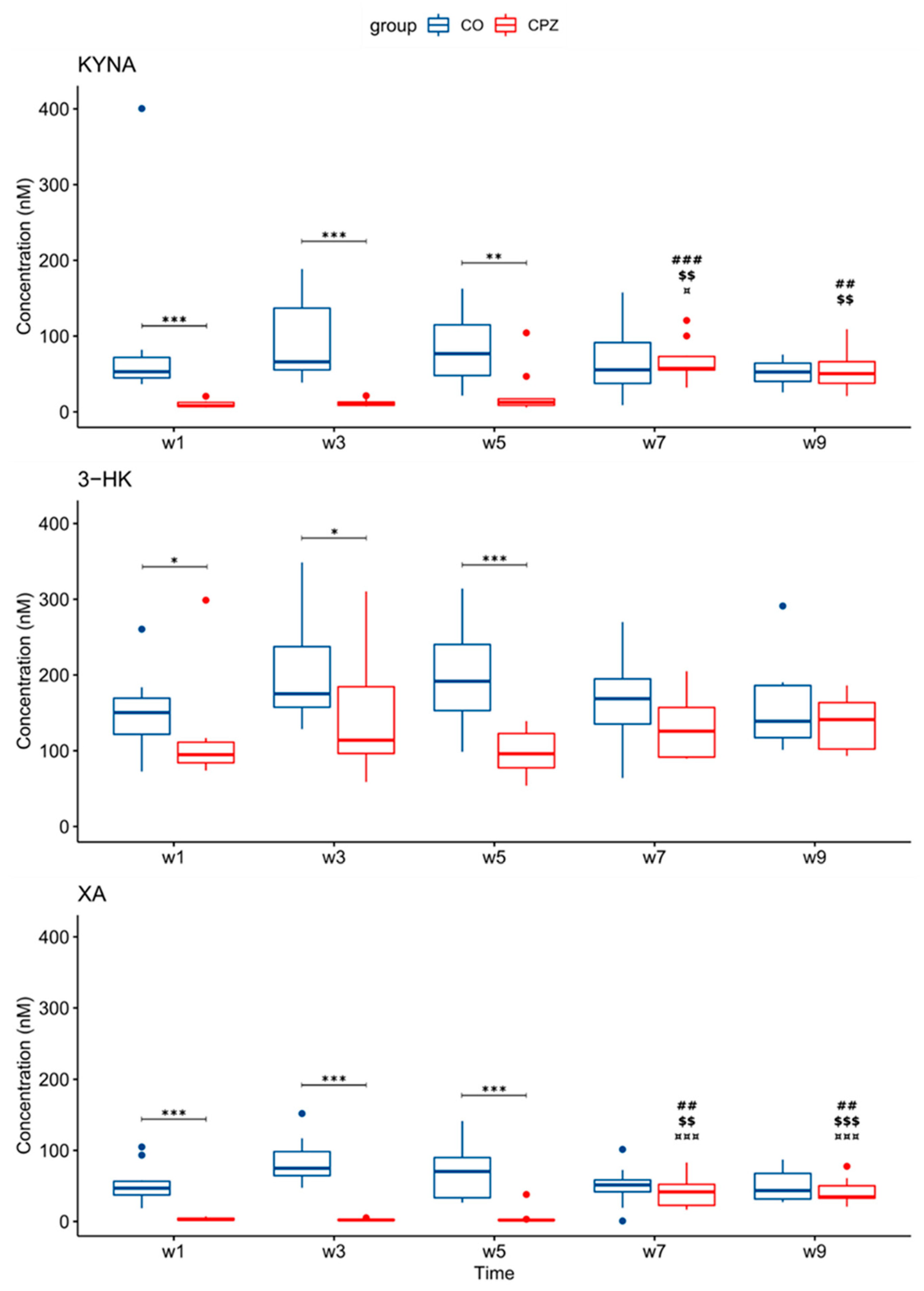
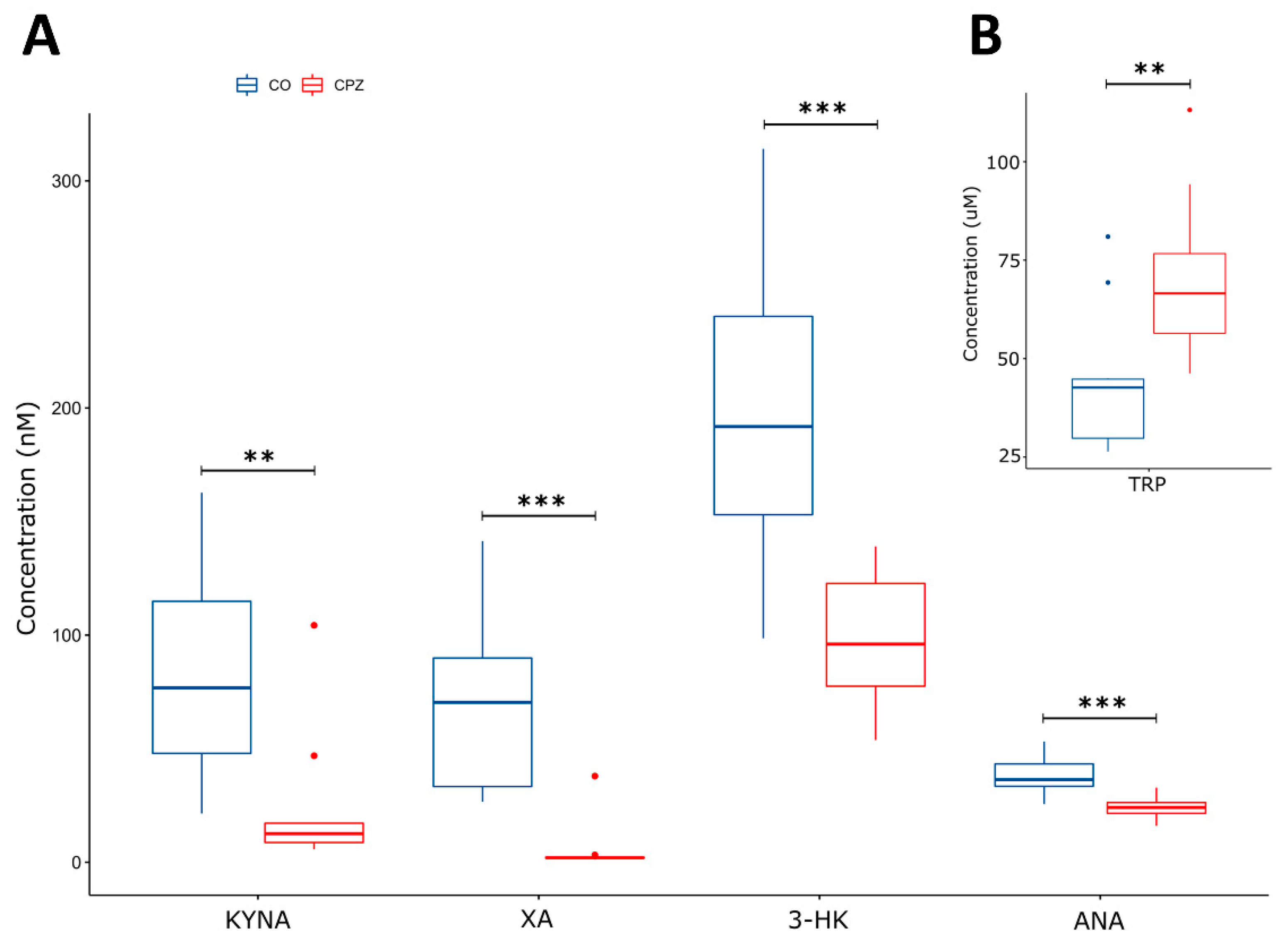
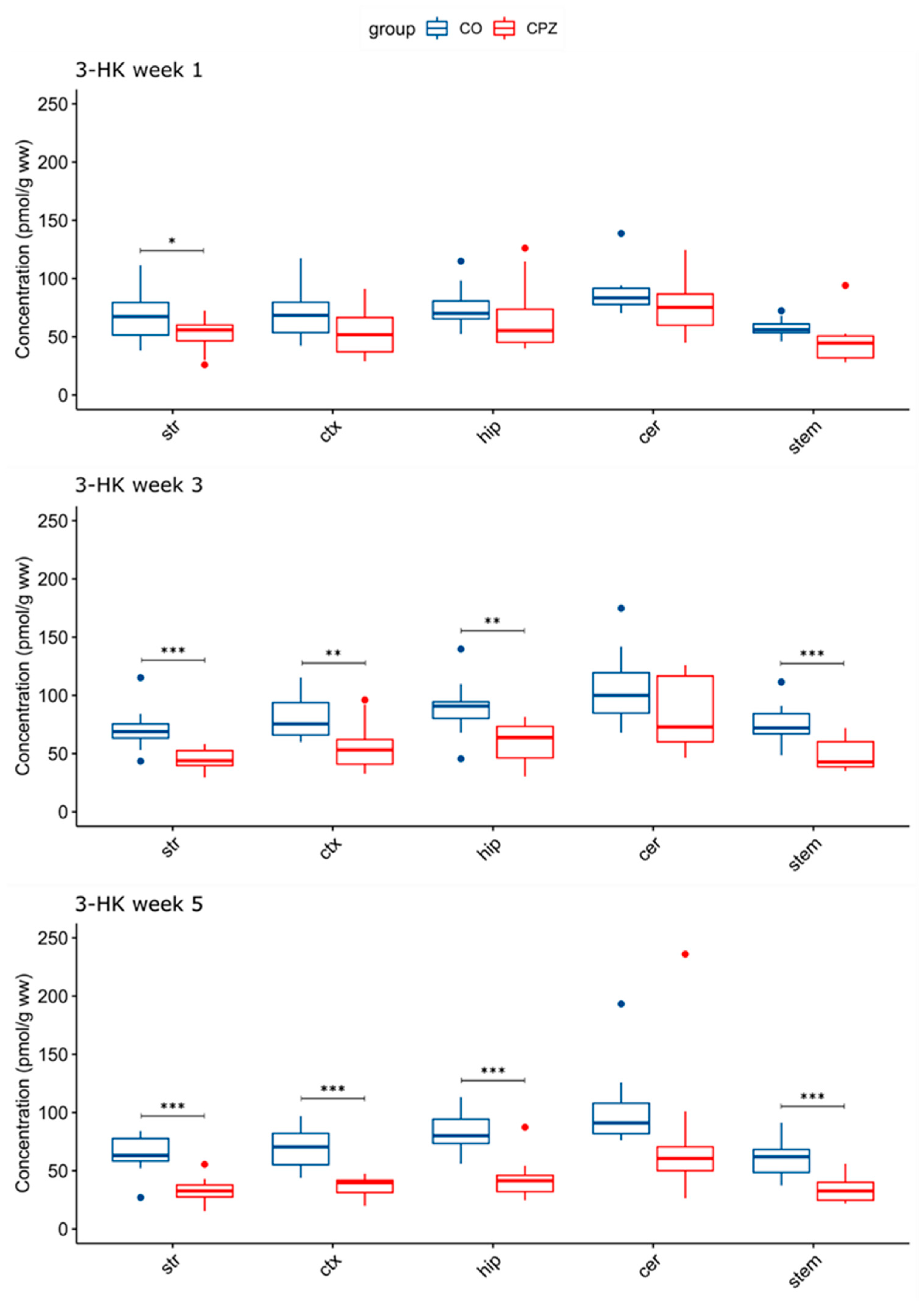
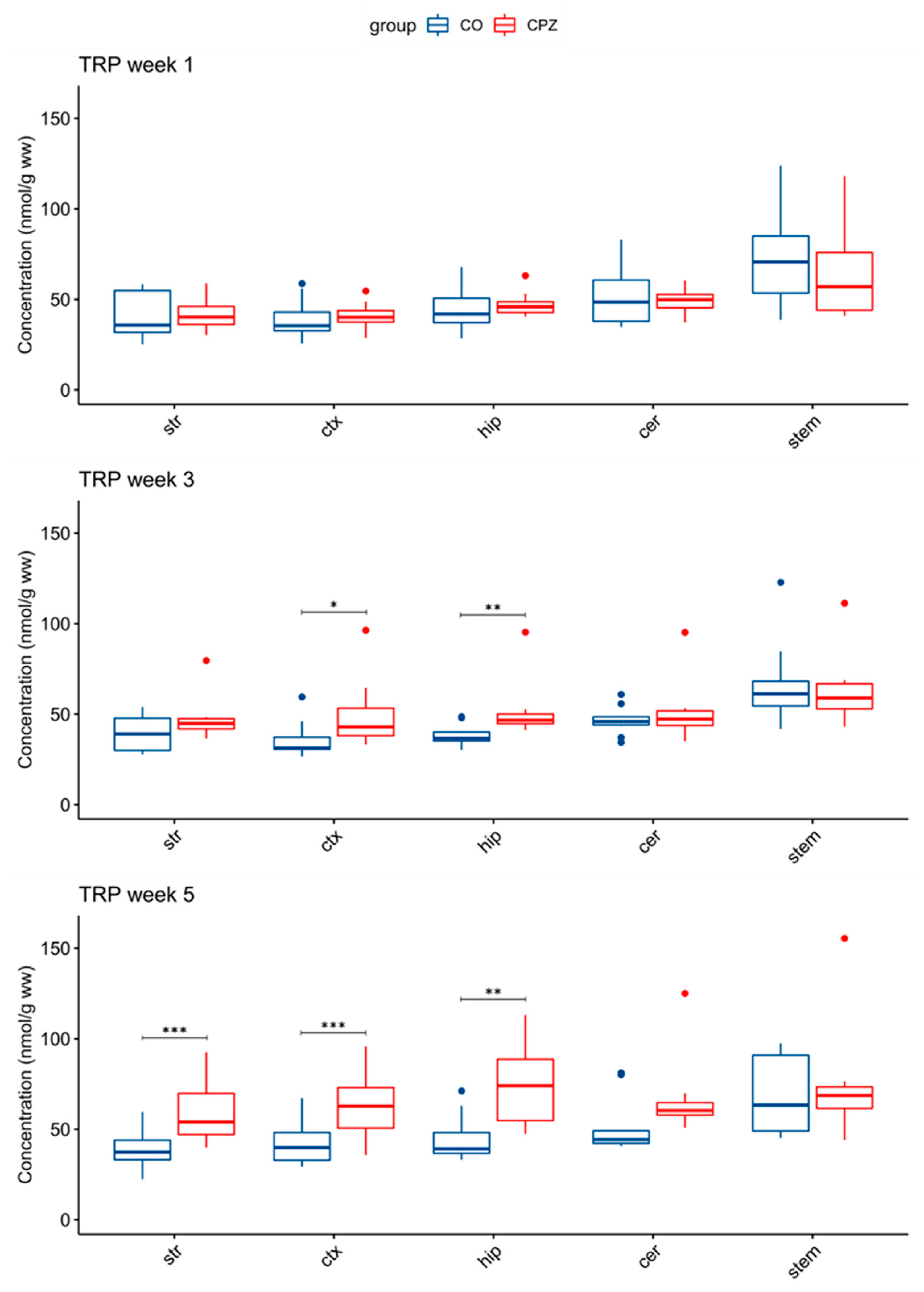
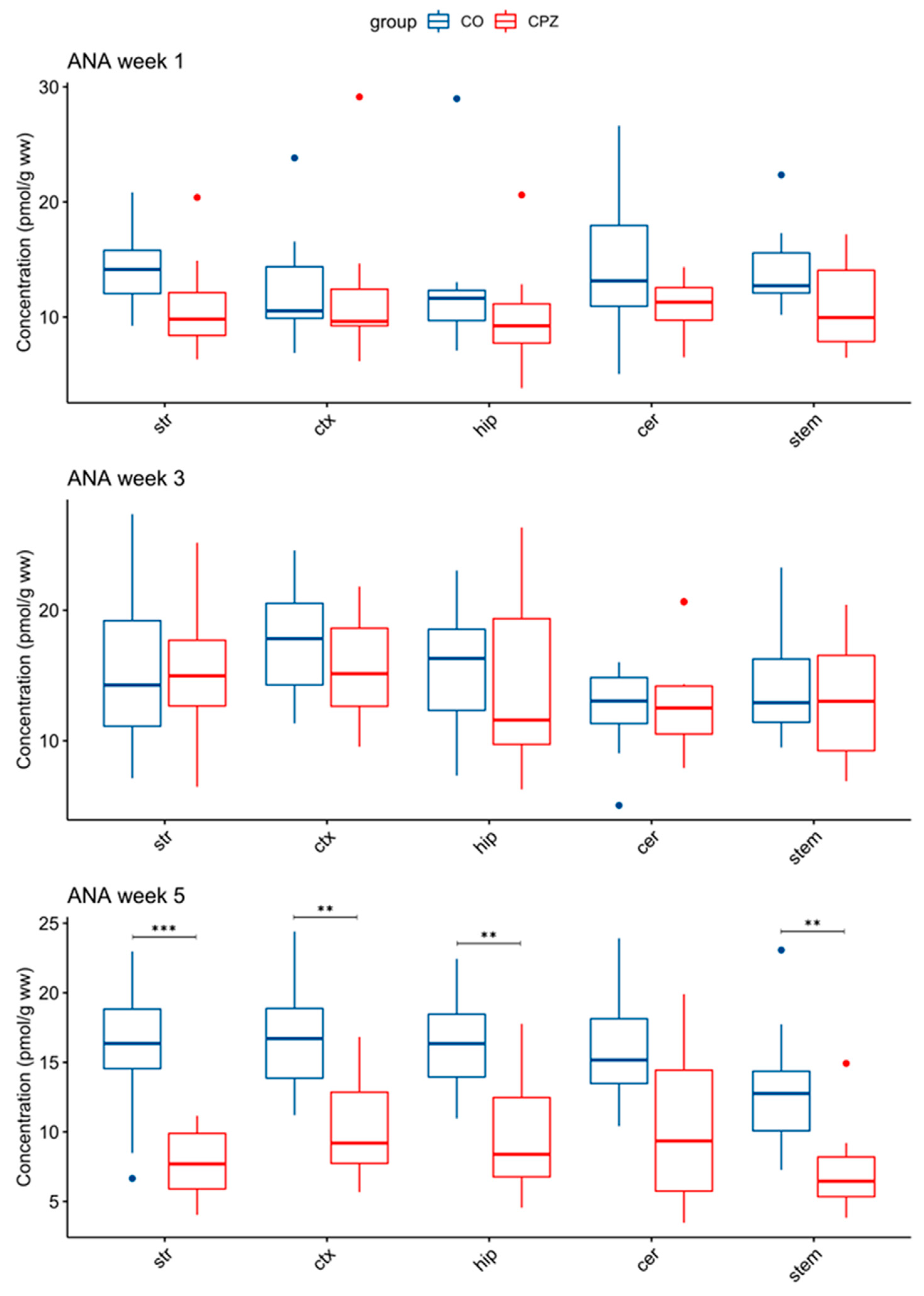
3.3. Ultra-High-Performance Liquid Chromatography with Tandem Mass Spectrometry Measurement of Kynurenine Metabolites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANA | anthranilic acid |
| AMPA | amino-3-hydroxy-5methyl-4 isoxazole propionic acid |
| BBB | blood–brain barrier |
| CO | control |
| CNS | central nervous system |
| CPZ | cuprizone |
| DEM | demyelination |
| 3-HANA | 3-hydroxyanthranilic acid |
| 3-HK | 3-hydroxy-L-kynurenine |
| HPLC | high-performance liquid chromatography |
| IDO | indoleamine 2,3- dioxygenase |
| KAT | kynurenine aminotransferase |
| KMO | kynurenine 3-monooxygenase |
| KP | kynurenine pathway |
| KYN | kynurenine |
| KYNA | kynurenic acid |
| UHPLC-MS/MS | Ultra-high-performance liquid chromatography with tandem mass spectrometry |
| LFB | luxol fast blue |
| MRM | multiple reaction monitoring |
| MS | multiple sclerosis |
| NAD+ | nicotinamide adenine dinucleotide |
| NMDA | N-methyl-D-aspartate |
| ROS | reactive oxygen species |
| PICA | picolinic acid |
| REM | remyelination |
| RRMS | relapsing–remitting multiple sclerosis |
| 5-HT | serotonin |
| TDO | tryptophan 2,3-dioxygenase |
| TRP | tryptophan |
| XA | xanthurenic acid |
| QUIN | quinolinic acid |
References
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Karussis, D.; Karageorgiou, C.; Vaknin-Dembinsky, A.; Gowda-Kurkalli, B.; Gomori, J.M.; Kassis, I.; Bulte, J.W.; Petrou, P.; Ben-Hur, T.; Abramsky, O.; et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Shiri, E.; Pasbakhsh, P.; Borhani-Haghighi, M.; Alizadeh, Z.; Nekoonam, S.; Mojaverrostami, S.; Pirhajati Mahabadi, V.; Mehdi, A.; Zibara, K.; Kashani, I.R. Mesenchymal Stem Cells Ameliorate Cuprizone-Induced Demyelination by Targeting Oxidative Stress and Mitochondrial Dysfunction. Cell Mol. Neurobiol. 2021, 41, 1467–1481. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Calandri, E.; Graziano, F.; Borghi, M.; Bonino, S. Young adults’ adjustment to a recent diagnosis of multiple sclerosis: The role of identity satisfaction and self-efficacy. Disabil. Health J. 2019, 12, 72–78. [Google Scholar] [CrossRef]
- Rajda, C.; Pukoli, D.; Bende, Z.; Majláth, Z.; Vécsei, L. Excitotoxins, Mitochondrial and Redox Disturbances in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 353. [Google Scholar] [CrossRef] [Green Version]
- Cree, B.A.C.; Arnold, D.L.; Chataway, J.; Chitnis, T.; Fox, R.J.; Pozo Ramajo, A.; Murphy, N.; Lassmann, H. Secondary Progressive Multiple Sclerosis. Neurology 2021, 97, 378–388. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Battaglia, S.; Thayer, J.F. Functional interplay between central and autonomic nervous systems in human fear conditioning. Trends Neurosci. 2022, 45, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Orsolini, S.; Borgomaneri, S.; Barbieri, R.; Diciotti, S.; di Pellegrino, G. Characterizing cardiac autonomic dynamics of fear learning in humans. Psychophysiology 2022, 59, e14122. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, F.; La Porta, F.; Petrone, V.; Battaglia, S.; Orlandi, S.; Ippolito, G.; Romei, V.; Piperno, R.; Lullini, G. Accuracy of EEG Biomarkers in the Detection of Clinical Outcome in Disorders of Consciousness after Severe Acquired Brain Injury: Preliminary Results of a Pilot Study Using a Machine Learning Approach. Biomedicines 2022, 10, 1897. [Google Scholar] [CrossRef] [PubMed]
- Nyatega, C.O.; Qiang, L.; Adamu, M.J.; Kawuwa, H.B. Gray matter, white matter and cerebrospinal fluid abnormalities in Parkinson’s disease: A voxel-based morphometry study. Front. Psychiatry 2022, 13, 1027907. [Google Scholar] [CrossRef]
- Nyatega, C.O.; Qiang, L.; Adamu, M.J.; Younis, A.; Kawuwa, H.B. Altered Dynamic Functional Connectivity of Cuneus in Schizophrenia Patients: A Resting-State fMRI Study. Appl. Sci. 2021, 11, 11392. [Google Scholar] [CrossRef]
- Orso, B.; Lorenzini, L.; Arnaldi, D.; Girtler, N.; Brugnolo, A.; Doglione, E.; Mattioli, P.; Biassoni, E.; Massa, F.; Peira, E.; et al. The Role of Hub and Spoke Regions in Theory of Mind in Early Alzheimer’s Disease and Frontotemporal Dementia. Biomedicines 2022, 10, 544. [Google Scholar] [CrossRef]
- Okanda Nyatega, C.; Qiang, L.; Jajere Adamu, M.; Bello Kawuwa, H. Altered striatal functional connectivity and structural dysconnectivity in individuals with bipolar disorder: A resting state magnetic resonance imaging study. Front. Psychiatry 2022, 13, 1054380. [Google Scholar] [CrossRef]
- Battaglia, S.; Fabius, J.H.; Moravkova, K.; Fracasso, A.; Borgomaneri, S. The Neurobiological Correlates of Gaze Perception in Healthy Individuals and Neurologic Patients. Biomedicines 2022, 10, 627. [Google Scholar] [CrossRef]
- Nyatega, C.O.; Qiang, L.; Jajere, M.A.; Kawuwa, H.B. Atypical Functional Connectivity of Limbic Network in Attention Deficit/Hyperactivity Disorder. Clin. Schizophr. Relat. Psychoses 2022, 16, 2. [Google Scholar] [CrossRef]
- Datki, Z.; Sinka, R. Translational biomedicine-oriented exploratory research on bioactive rotifer-specific biopolymers. Adv. Clin. Exp. Med. 2022, 31, 931–935. [Google Scholar] [CrossRef]
- Kwon, K.-M.; Lee, M.-J.; Chung, H.-S.; Pak, J.-H.; Jeon, C.-J. The Organization of Somatostatin-Immunoreactive Cells in the Visual Cortex of the Gerbil. Biomedicines 2022, 10, 92. [Google Scholar] [CrossRef]
- Lieb, A.; Thaler, G.; Fogli, B.; Trovato, O.; Posch, M.A.; Kaserer, T.; Zangrandi, L. Functional Characterization of Spinocerebellar Ataxia Associated Dynorphin A Mutant Peptides. Biomedicines 2021, 9, 1882. [Google Scholar] [CrossRef] [PubMed]
- Palotai, M.; Telegdy, G.; Tanaka, M.; Bagosi, Z.; Jászberényi, M. Neuropeptide AF induces anxiety-like and antidepressant-like behavior in mice. Behav. Brain Res. 2014, 274, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, C.; Soga, T.; Ahemad, N.; Bhuvanendran, S.; Parhar, I. Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from α-Synuclein-Induced Toxicity In Vitro. Int. J. Mol. Sci. 2022, 23, 5193. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Cho, G.-W.; Vijayakumar, K.A.; Moon, C.; Ang, M.J.; Kim, J.; Park, I.; Jang, C.H. Neuroprotective Effect of Valproic Acid on Salicylate-Induced Tinnitus. Int. J. Mol. Sci. 2021, 23, 23. [Google Scholar] [CrossRef] [PubMed]
- Telegdy, G.; Tanaka, M.; Schally, A.V. Effects of the growth hormone-releasing hormone (GH-RH) antagonist on brain functions in mice. Behav. Brain Res. 2011, 224, 155–158. [Google Scholar] [CrossRef]
- Tanaka, M.; Kádár, K.; Tóth, G.; Telegdy, G. Antidepressant-like effects of urocortin 3 fragments. Brain Res. Bull. 2011, 84, 414–418. [Google Scholar] [CrossRef]
- Ibos, K.E.; Bodnár, É.; Bagosi, Z.; Bozsó, Z.; Tóth, G.; Szabó, G.; Csabafi, K. Kisspeptin-8 Induces Anxiety-like Behavior and Hypolocomotion by Activating the HPA Axis and Increasing GABA Release in the Nucleus Accumbens in Rats. Biomedicines 2021, 9, 112. [Google Scholar] [CrossRef]
- Rákosi, K.; Masaru, T.; Zarándi, M.; Telegdy, G.; Tóth, G.K. Short analogs and mimetics of human urocortin 3 display antidepressant effects in vivo. Peptides 2014, 62, 59–66. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabó, Á.; Vécsei, L. Integrating Armchair, Bench, and Bedside Research for Behavioral Neurology and Neuropsychiatry: Editorial. Biomedicines 2022, 10, 2999. [Google Scholar] [CrossRef]
- Younis, A.; Qiang, L.; Nyatega, C.O.; Adamu, M.J.; Kawuwa, H.B. Brain Tumor Analysis Using Deep Learning and VGG-16 Ensembling Learning Approaches. Appl. Sci. 2022, 12, 7282. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue ‘Dissecting Neurological and Neuropsychiatric Diseases: Neurodegeneration and Neuroprotection. Int. J. Mol. Sci. 2022, 23, 6991. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue “Crosstalk between Depression, Anxiety, and Dementia: Comorbidity in Behavioral Neurology and Neuropsychiatry”. Biomedicines 2021, 9, 517. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M. Crosstalk between Depression, Anxiety, and Dementia: Comorbidity in Behavioral Neurology and Neuropsychiatry. Biomedicines 2022. [Google Scholar] [CrossRef]
- Praet, J.; Guglielmetti, C.; Berneman, Z.; Van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 47, 485–505. [Google Scholar] [CrossRef] [Green Version]
- Sen, M.K.; Mahns, D.A.; Coorssen, J.R.; Shortland, P.J. Behavioural phenotypes in the cuprizone model of central nervous system demyelination. Neurosci. Biobehav. Rev. 2019, 107, 23–46. [Google Scholar] [CrossRef]
- Kalman, B.; Laitinen, K.; Komoly, S. The involvement of mitochondria in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2007, 188, 1–12. [Google Scholar] [CrossRef]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial response during cuprizone-induced de- and remyelination in the CNS: Lessons learned. Front. Cell Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Hiremath, M.M.; Saito, Y.; Knapp, G.W.; Ting, J.P.-Y.; Suzuki, K.; Matsushima, G.K. Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J. Neuroimmunol. 1998, 92, 38–49. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.È.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Skripuletz, T.; Hackstette, D.; Bauer, K.; Gudi, V.; Pul, R.; Voss, E.; Berger, K.; Kipp, M.; Baumgärtner, W.; Stangel, M. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain 2013, 136, 147–167. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.L.; Jones, J.J.; Taniike, M.; Morell, P.; Suzuki, K.; Matsushima, G.K. Mature oligodendrocyte apoptosis precedes IGF-1 production and oligodendrocyte progenitor accumulation and differentiation during demyelination/remyelination. J. Neurosci. Res. 2000, 61, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, G.K.; Morell, P. The Neurotoxicant, Cuprizone, as a Model to Study Demyelination and Remyelination in the Central Nervous System. Brain Pathol. 2001, 11, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Nyamoya, S.; Steinle, J.; Chrzanowski, U.; Kaye, J.; Schmitz, C.; Beyer, C.; Kipp, M. Laquinimod Supports Remyelination in Non-Supportive Environments. Cells 2019, 8, 1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azami Tameh, A.; Clarner, T.; Beyer, C.; Atlasi, M.A.; Hassanzadeh, G.; Naderian, H. Regional regulation of glutamate signaling during cuprizone-induced demyelination in the brain. Ann. Anat.—Anat. Anz. 2013, 195, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Chomyk, A.M.; Chang, A.; Ribaudo, M.V.; Deckard, S.A.; Doud, M.K.; Edberg, D.D.; Bai, B.; Li, M.; Baranzini, S.E.; et al. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann. Neurol. 2013, 73, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Dewar, D.; Underhill, S.M.; Goldberg, M.P. Oligodendrocytes and ischemic brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Morales-Puerto, N.; Giménez-Gómez, P.; Pérez-Hernández, M.; Abuin-Martínez, C.; Gil de Biedma-Elduayen, L.; Vidal, R.; Gutiérrez-López, M.D.; O’Shea, E.; Colado, M.I. Addiction and the kynurenine pathway: A new dancing couple? Pharmacol. Ther. 2021, 223, 107807. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Kynurenine pathway and human systems. Exp. Gerontol. 2020, 129, 110770. [Google Scholar] [CrossRef]
- Rajda, C.; Majláth, Z.; Pukoli, D.; Vécsei, L. Kynurenines and Multiple Sclerosis: The Dialogue between the Immune System and the Central Nervous System. Int. J. Mol. Sci. 2015, 16, 18270–18282. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, T.; Sandi, D.; Bencsik, K.; Vécsei, L. Kynurenines in the Pathogenesis of Multiple Sclerosis: Therapeutic Perspectives. Cells 2020, 9, 1564. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-M.; Bao, C.-H.; Wu, Y.; Liang, S.-H.; Wang, D.; Wu, L.-Y.; Huang, Y.; Liu, H.R.; Wu, H.G. Tryptophan-kynurenine metabolism: A link between the gut and brain for depression in inflammatory bowel disease. J. Neuroinflamm. 2021, 18, 135. [Google Scholar] [CrossRef]
- Taleb, O.; Maammar, M.; Klein, C.; Maitre, M.; Mensah-Nyagan, A.G. A Role for Xanthurenic Acid in the Control of Brain Dopaminergic Activity. Int. J. Mol. Sci. 2021, 22, 6974. [Google Scholar] [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenate and FG9041 have both competitive and non-competitive antagonist actions at excitatory amino acid receptors. Eur. J. Pharmacol. 1988, 151, 313–315. [Google Scholar] [CrossRef]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A glycine site associated with N-methyl-D-aspartic acid receptors: Characterization and identification of a new class of antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Klivényi, P.; Plangár, I.; Toldi, J.; Vécsei, L. Endogenous neuroprotection in chronic neurodegenerative disorders: With particular regard to the kynurenines. J. Cell Mol. Med. 2011, 15, 701–717. [Google Scholar] [CrossRef]
- Bohár, Z.; Toldi, J.; Fülöp, F.; Vécsei, L. Changing the Face of Kynurenines and Neurotoxicity: Therapeutic Considerations. Int. J. Mol. Sci. 2015, 16, 9772–9793. [Google Scholar] [CrossRef] [Green Version]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef]
- Sandi, D.; Fricska-Nagy, Z.; Bencsik, K.; Vécsei, L. Neurodegeneration in Multiple Sclerosis: Symptoms of Silent Progression, Biomarkers and Neuroprotective Therapy—Kynurenines Are Important Players. Molecules 2021, 26, 3423. [Google Scholar] [CrossRef]
- Sandi, D.; Biernacki, T.; Szekeres, D.; Füvesi, J.; Kincses, Z.T.; Rózsa, C.; Mátyás, K.; Kása, K.; Matolcsi, J.; Zboznovits, D.; et al. Prevalence of cognitive impairment among Hungarian patients with relapsing-remitting multiple sclerosis and clinically isolated syndrome. Mult. Scler. Relat. Disord. 2017, 17, 57–62. [Google Scholar] [CrossRef]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Spekker, E.; Szabó, Á.; Polyák, H.; Vécsei, L. Modelling the neurodevelopmental pathogenesis in neuropsychiatric disorders. Bioactive kynurenines and their analogues as neuroprotective agents—In celebration of 80th birthday of Professor Peter Riederer. J. Neural. Transm. 2022, 129, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rejdak, K.; Petzold, A.; Kocki, T.; Kurzepa, J.; Grieb, P.; Turski, W.A.; Stelmasiak, Z. Astrocytic activation in relation to inflammatory markers during clinical exacerbation of relapsing-remitting multiple sclerosis. J. Neural Transm. 2007, 114, 1011. [Google Scholar] [CrossRef] [PubMed]
- Rejdak, K.; Bartosik-Psujek, H.; Dobosz, B.; Kocki, T.; Grieb, P.; Giovannoni, G.; Turski, W.A.; Stelmasiak, Z. Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci. Lett. 2002, 331, 63–65. [Google Scholar] [CrossRef]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef] [Green Version]
- Saraste, M.; Matilainen, M.; Rajda, C.; Galla, Z.; Sucksdorff, M.; Vécsei, L.; Airas, L. Association between microglial activation and serum kynurenine pathway metabolites in multiple sclerosis patients. Mult. Scler. Relat. Disord. 2022, 59, 103667. [Google Scholar] [CrossRef]
- Polyák, H.; Cseh, E.K.; Bohár, Z.; Rajda, C.; Zádori, D.; Klivényi, P.; Toldi, J.; Vécsei, L. Cuprizone markedly decreases kynurenic acid levels in the rodent brain tissue and plasma. Heliyon 2021, 7, e06124. [Google Scholar] [CrossRef]
- Acs, P.; Kipp, M.; Norkute, A.; Johann, S.; Clarner, T.; Braun, A.; Berente, Z.; Komoly, S.; Beyer, C. 17β-estradiol and progesterone prevent cuprizone provoked demyelination of corpus callosum in male mice. Glia 2009, 57, 807–814. [Google Scholar] [CrossRef]
- Tömösi, F.; Kecskeméti, G.; Cseh, E.K.; Szabó, E.; Rajda, C.; Kormány, R.; Szabó, Z.; Vécsei, L.; Janáky, T. A validated UHPLC-MS method for tryptophan metabolites: Application in the diagnosis of multiple sclerosis. J. Pharm. Biomed. Anal. 2020, 185, 113246. [Google Scholar] [CrossRef] [PubMed]
- Galla, Z.; Rajda, C.; Rácz, G.; Grecsó, N.; Baráth, Á.; Vécsei, L.; Bereczki, C.; Monostori, P. Simultaneous determination of 30 neurologically and metabolically important molecules: A sensitive and selective way to measure tyrosine and tryptophan pathway metabolites and other biomarkers in human serum and cerebrospinal fluid. J. Chromatogr. A 2021, 1635, 461775. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Mann, T.; Joost, S.; Behrangi, N.; Frank, M.; Kipp, M. The Cuprizone Model: Dos and Do Nots. Cells 2020, 9, 843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, J.; Daniel, M.; van Heuvel, Y.; Victor, M.; Beyer, C.; Clarner, T.; Kipp, M. Short-Term Cuprizone Feeding Induces Selective Amino Acid Deprivation with Concomitant Activation of an Integrated Stress Response in Oligodendrocytes. Cell Mol. Neurobiol. 2013, 33, 1087–1098. [Google Scholar] [CrossRef]
- Capucciati, A.; Galliano, M.; Bubacco, L.; Zecca, L.; Casella, L.; Monzani, E.; Nicolis, S. Neuronal Proteins as Targets of 3-Hydroxykynurenine: Implications in Neurodegenerative Diseases. ACS Chem. Neurosci. 2019, 10, 3731–3739. [Google Scholar] [CrossRef]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Lennon, M.J.; Lim, C.K.; Jacobs, K.; Guillemin, G.J.; Brew, B.J. Recent evidence for an expanded role of the kynurenine pathway of tryptophan metabolism in neurological diseases. Neuropharmacology 2017, 112, 373–388. [Google Scholar] [CrossRef]
- Zinger, A.; Barcia, C.; Herrero, M.T.; Guillemin, G.J. The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinsons. Dis. 2011, 2011, 716859. [Google Scholar] [CrossRef] [Green Version]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood–Brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Kita, T.; Morrison, P.F.; Heyes, M.P.; Markey, S.P. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J. Neurochem. 2002, 82, 258–268. [Google Scholar] [CrossRef]
- Cathomas, F.; Guetter, K.; Seifritz, E.; Klaus, F.; Kaiser, S. Quinolinic acid is associated with cognitive deficits in schizophrenia but not major depressive disorder. Sci. Rep. 2021, 11, 9992. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.; van der Hart, M.; Roeser, J.; Summergrad, P. Anthranilic Acid: A Potential Biomarker and Treatment Target for Schizophrenia. Ann. Psychiatry Ment. Health 2016, 4, 1059. [Google Scholar]
- Bloomfield, P.S.; Selvaraj, S.; Veronese, M.; Rizzo, G.; Bertoldo, A.; Owen, D.R.; Bloomfield, M.A.; Bonoldi, I.; Kalk, N.; Turkheimer, F.; et al. Microglial Activity in People at Ultra High Risk of Psychosis and in Schizophrenia: An [(11)C]PBR28 PET Brain Imaging Study. Am. J. Psychiatry 2016, 173, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Brisch, R.; Wojtylak, S.; Saniotis, A.; Steiner, J.; Gos, T.; Kumaratilake, J.; Henneberg, M.; Wolf, R. The role of microglia in neuropsychiatric disorders and suicide. Eur. Arch. Psychiatry Clin. Neurosci. 2022, 272, 929–945. [Google Scholar] [CrossRef]
- Cakmak, J.D.; Liu, L.; Poirier, S.E.; Schaefer, B.; Poolacherla, R.; Burhan, A.M.; Sabesan, P.; St Lawrence, K.; Théberge, J.; Hicks, J.W.; et al. The functional and structural associations of aberrant microglial activity in major depressive disorder. J. Psychiatry Neurosci. 2022, 47, E197–E208. [Google Scholar] [CrossRef] [PubMed]
- El-Sewedy, S.M.; Abdel-Tawab, G.A.; El-Zoghby, S.M.; Zeitoun, R.; Mostafa, M.H.; Shalaby, S.M. Studies with tryptophan metabolites in vitro. effect of zinc, manganese, copper and cobalt ions on kynurenine hydrolase and kynurenine aminotransferase in normal mouse liver. Biochem. Pharmacol. 1974, 23, 2557–2565. [Google Scholar] [CrossRef]
- Acs, P.; Selak, M.A.; Komoly, S.; Kalman, B. Distribution of oligodendrocyte loss and mitochondrial toxicity in the cuprizone-induced experimental demyelination model. J. Neuroimmunol. 2013, 262, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, H.; Jiang, W.; Xiao, L.; Yan, B.; He, J.; Wang, Y.; Bi, X.; Li, X.; Kong, J.; et al. Quetiapine alleviates the cuprizone-induced white matter pathology in the brain of C57BL/6 mouse. Schizophr. Res. 2008, 106, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Ljutakova, S.G.; Russanov, E.M. Differences in the in vivo effects of cuprizone on superoxide dismutase activity in rat liver cytosol and mitochondrial intermembrane space. Acta Physiol. Pharmacol. Bulg. 1985, 11, 56–61. [Google Scholar]
- Fridovich, I. Superoxide Dismutases. Annu. Rev. Biochem. 1975, 44, 147–159. [Google Scholar] [CrossRef]
- Blackburn, N.J.; Mason, H.S.; Knowles, P.F. Dopamine-β-hydroxylase: Evidence for binuclear copper sites. Biochem. Biophys. Res. Commun. 1980, 95, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; McIntire, W.S. Cloning and sequencing of a copper-containing, topa quinone-containing monoamine oxidase from human placenta. Gene 1996, 179, 279–286. [Google Scholar] [CrossRef]
- Horn, D.; Barrientos, A. Mitochondrial Copper Metabolism and Delivery to Cytochrome c Oxidase. IUBMB Life 2008, 60, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Kako, K.; Kashiwabara, S.; Takehara, A.; Inada, Y.; Arai, H.; Nakada, K.; Kodama, H.; Hayashi, J.; Baba, T.; et al. Mammalian Copper Chaperone Cox17p Has an Essential Role in Activation of Cytochrome c Oxidase and Embryonic Development. Mol. Cell Biol. 2002, 22, 7614–7621. [Google Scholar] [CrossRef] [Green Version]
- Herring, N.R.; Konradi, C. Myelin, copper, and the cuprizone model of schizophrenia. Front. Biosci. 2011, 3, 23–40. [Google Scholar]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic Acid: An Endogenous Neurotoxin with Multiple Targets. Oxid. Med. Cell Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [Green Version]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef]
- Chobot, V.; Hadacek, F.; Weckwerth, W.; Kubicova, L. Iron chelation and redox chemistry of anthranilic acid and 3-hydroxyanthranilic acid: A comparison of two structurally related kynurenine pathway metabolites to obtain improved insights into their potential role in neurological disease development. J. Organomet. Chem. 2015, 782, 103–110. [Google Scholar] [CrossRef] [Green Version]
- González Esquivel, D.; Ramírez-Ortega, D.; Pineda, B.; Castro, N.; Ríos, C.; Pérez de la Cruz, V. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 2017, 112, 331–345. [Google Scholar] [CrossRef]
- Lima, V.L.A.; Dias, F.; Nunes, R.D.; Pereira, L.O.; Santos, T.S.R.; Chiarini, L.B.; Ramos, T.D.; Silva-Mendes, B.J.; Perales, J.; Valente, R.H.; et al. The Antioxidant Role of Xanthurenic Acid in the Aedes aegypti Midgut during Digestion of a Blood Meal. PLoS ONE 2012, 7, e38349. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K.; Ito, M.; Yoshino, M. Xanthurenic acid inhibits metal ion-induced lipid peroxidation and protects NADP-isocitrate dehydrogenase from oxidative inactivation. J. Nutr. Sci. Vitaminol. 2001, 47, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Pérez-González, A.; Alvarez-Idaboy, J.R.; Galano, A. Free-radical scavenging by tryptophan and its metabolites through electron transfer based processes. J. Mol. Model 2015, 21, 213. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Staniek, K.; Kepplinger, B.; Stur, J.; Draxler, M.; Nohl, H. Kynurenines and the respiratory parameters on rat heart mitochondria. Life Sci. 2003, 72, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Hypothesis kynurenic and quinolinic acids: The main players of the kynurenine pathway and opponents in inflammatory disease. Med. Hypotheses 2018, 118, 129–138. [Google Scholar] [CrossRef]
- Negrotto, L.; Correale, J. Amino Acid Catabolism in Multiple Sclerosis Affects Immune Homeostasis. J. Immunol. 2017, 198, 1900–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturini, G. Enzymic Activities and Sodium, Potassium and Copper Concentrations in Mouse Brain and Liver after Cuprizone Treatment in Vivo. J. Neurochem. 1973, 21, 1147–1151. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Asano, M.; Kurono, C. Mechanism of the formation of megamitochondria induced by copper-chelating agents. II. Isolation and some properties of megamitochondria from the cuprizone-treated mouse liver. Acta Pathol. Jpn. 1975, 25, 39–49. [Google Scholar]
- Song, S.-K.; Yoshino, J.; Le, T.Q.; Lin, S.-J.; Sun, S.-W.; Cross, A.H.; Armstrong, R.C. Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage 2005, 26, 132–140. [Google Scholar] [CrossRef]
- Fairless, A.H.; Dow, H.C.; Toledo, M.M.; Malkus, K.A.; Edelmann, M.; Li, H.; Talbot, K.; Arnold, S.E.; Abel, T.; Brodkin, E.S. Low Sociability is Associated with Reduced Size of the Corpus Callosum in the BALB/cJ Inbred Mouse Strain. Brain Res. 2008, 1230, 211–217. [Google Scholar] [CrossRef]
- Parenti, R.; Cicirata, F.; Zappalà, A.; Catania, A.; La Delia, F.; Cicirata, V.; Tress, O.; Willecke, K. Dynamic expression of Cx47 in mouse brain development and in the cuprizone model of myelin plasticity. Glia 2010, 58, 1594–1609. [Google Scholar] [CrossRef]
- Kesterson, J.W.; Carlton, W.W. Cuprizone toxicosis in mice—Attempts to antidote the toxicity. Toxicol. Appl. Pharmacol. 1972, 22, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Biancotti, J.C.; Kumar, S.; de Vellis, J. Activation of inflammatory response by a combination of growth factors in cuprizone-induced demyelinated brain leads to myelin repair. Neurochem. Res. 2008, 33, 2615–2628. [Google Scholar] [CrossRef] [PubMed]
- Sadok, I.; Gamian, A.; Staniszewska, M.M. Chromatographic analysis of tryptophan metabolites. J. Sep. Sci. 2017, 40, 3020–3045. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. 1500 scientists lift the lid on reproducibility. Nature 2016, 533, 452–454. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polyák, H.; Galla, Z.; Nánási, N.; Cseh, E.K.; Rajda, C.; Veres, G.; Spekker, E.; Szabó, Á.; Klivényi, P.; Tanaka, M.; et al. The Tryptophan-Kynurenine Metabolic System Is Suppressed in Cuprizone-Induced Model of Demyelination Simulating Progressive Multiple Sclerosis. Biomedicines 2023, 11, 945. https://doi.org/10.3390/biomedicines11030945
Polyák H, Galla Z, Nánási N, Cseh EK, Rajda C, Veres G, Spekker E, Szabó Á, Klivényi P, Tanaka M, et al. The Tryptophan-Kynurenine Metabolic System Is Suppressed in Cuprizone-Induced Model of Demyelination Simulating Progressive Multiple Sclerosis. Biomedicines. 2023; 11(3):945. https://doi.org/10.3390/biomedicines11030945
Chicago/Turabian StylePolyák, Helga, Zsolt Galla, Nikolett Nánási, Edina Katalin Cseh, Cecília Rajda, Gábor Veres, Eleonóra Spekker, Ágnes Szabó, Péter Klivényi, Masaru Tanaka, and et al. 2023. "The Tryptophan-Kynurenine Metabolic System Is Suppressed in Cuprizone-Induced Model of Demyelination Simulating Progressive Multiple Sclerosis" Biomedicines 11, no. 3: 945. https://doi.org/10.3390/biomedicines11030945
APA StylePolyák, H., Galla, Z., Nánási, N., Cseh, E. K., Rajda, C., Veres, G., Spekker, E., Szabó, Á., Klivényi, P., Tanaka, M., & Vécsei, L. (2023). The Tryptophan-Kynurenine Metabolic System Is Suppressed in Cuprizone-Induced Model of Demyelination Simulating Progressive Multiple Sclerosis. Biomedicines, 11(3), 945. https://doi.org/10.3390/biomedicines11030945