
Methyl-Thiol-Bridged Oxadiazole and Triazole Heterocycles as Inhibitors of NF-κB in Chronic Myelogenous Leukemia Cells

 ,
,  , , , , ,
, , , , , 
Abstract
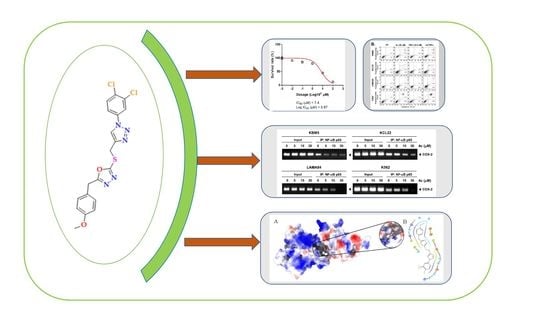
:
1. Introduction
2. Materials and Methods
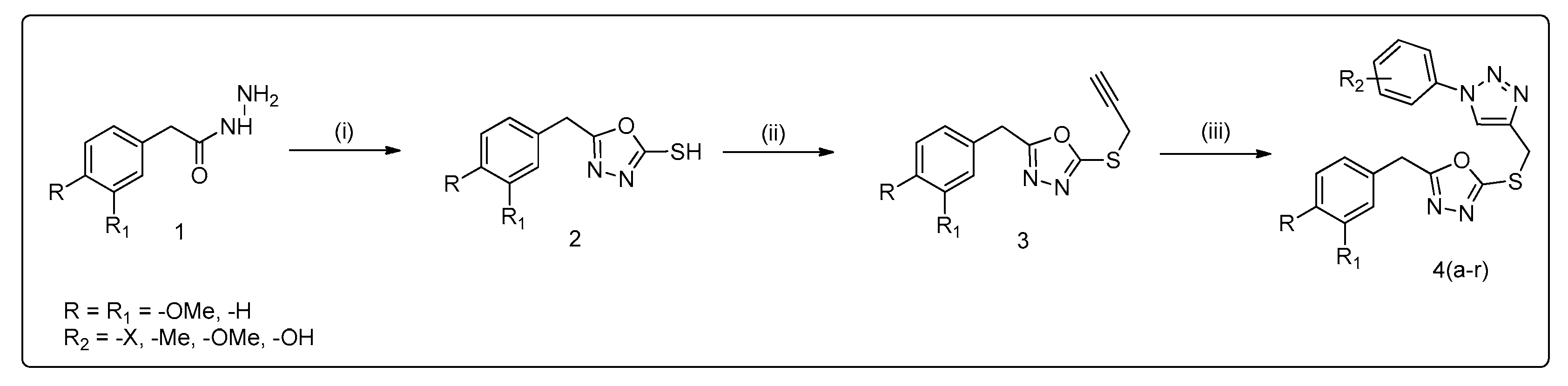
2.1. General Procedure for the Synthesis of 1,2,3-Triazole Derivatives
2.2. 2-(((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5-(4-methoxybenzyl)-1,3,4-oxadiazole (4a)
2.3. 2-(((1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5-(4-methoxybenzyl)-1,3,4-oxadia-zole (4b)
2.4. 2-(((1-(3,4-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5-(4-methoxybenzyl)-1,3,4-oxadiazole (4c)
2.5. 2-(4-Methoxybenzyl)-5-(((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-oxadiazole (4d)
2.6. 2-(((1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5-(4-methoxybenzyl)-1,3,4-oxadiazole (4e)
2.7. 2-(4-Methoxybenzyl)-5-(((1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-oxadiazole (4f)
2.8. 4-(4-(((5-(4-Methoxybenzyl)-1,3,4-oxadiazol-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)phenol (4g)
2.9. 2-(4-Methoxybenzyl)-5-(((1-(o-tolyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-oxadiazole (4h)
2.10. 2-(4-Methoxybenzyl)-5-(((1-(p-tolyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-oxadiazole (4i)
2.11. 2-(((1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5-(3,4-dimethoxybenzyl)-1,3,4-oxadiazole (4j)
2.12. 2-(((1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5-(3,4-dimethoxybenzyl)-1,3,4-oxadiazole (4k)
2.13. 2-(((1-(3,4-Dichlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5-(3,4-dimethoxybenzyl)-1,3,4-oxadiazole (4l)
2.14. 2-(3,4-Dimethoxybenzyl)-5-(((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-oxadiazole (4m)
2.15. 2-(((1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-5-(3,4-dimethoxybenzyl)-1,3,4-oxadiazole (4n)
2.16. 2-(3,4-Dimethoxybenzyl)-5-(((1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-oxadiazole (4o)
2.17. 2-(4-(((5-(3,4-Dimethoxybenzyl)-1,3,4-oxadiazol-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)phenol (4p)
2.18. 2-(3,4-Dimethoxybenzyl)-5-(((1-(p-tolyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-oxadia-zole (4q)
2.19. 2-(3,4-Dimethoxybenzyl)-5-(((1-(o-tolyl)-1H-1,2,3-triazol-4-yl)methyl)thio)-1,3,4-oxadia-zole (4r)
2.20. Reagents
2.21. Cell Lines and Culture Conditions
2.22. MTT Assay
2.23. Electrophoretic Mobility Shift Assay (EMSA)
2.24. Immunocytochemistry
2.25. Western Blot Analysis
2.26. Live and Dead Assay
2.27. Annexin V Assay
2.28. Cell Cycle Analysis
2.29. Chromatin Immunoprecipitation (ChIP) Assay
2.30. In Silico DFT Calculations
2.31. Molecular Docking and Dynamics Analysis
2.32. Statistical Analysis
3. Results
3.1. Method of Synthesis
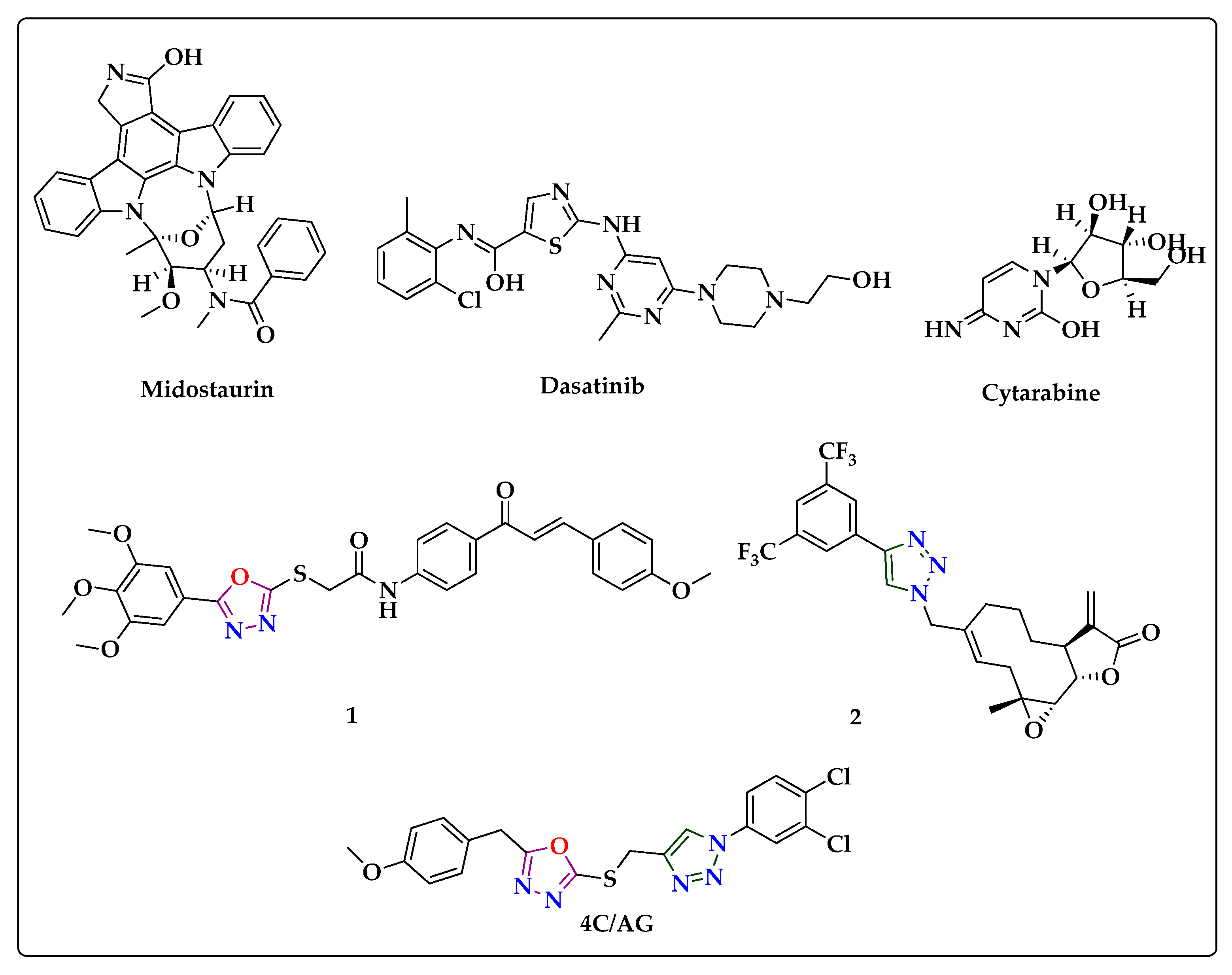
3.2. Efficacy of Title Compounds in Breast Cancer Cells
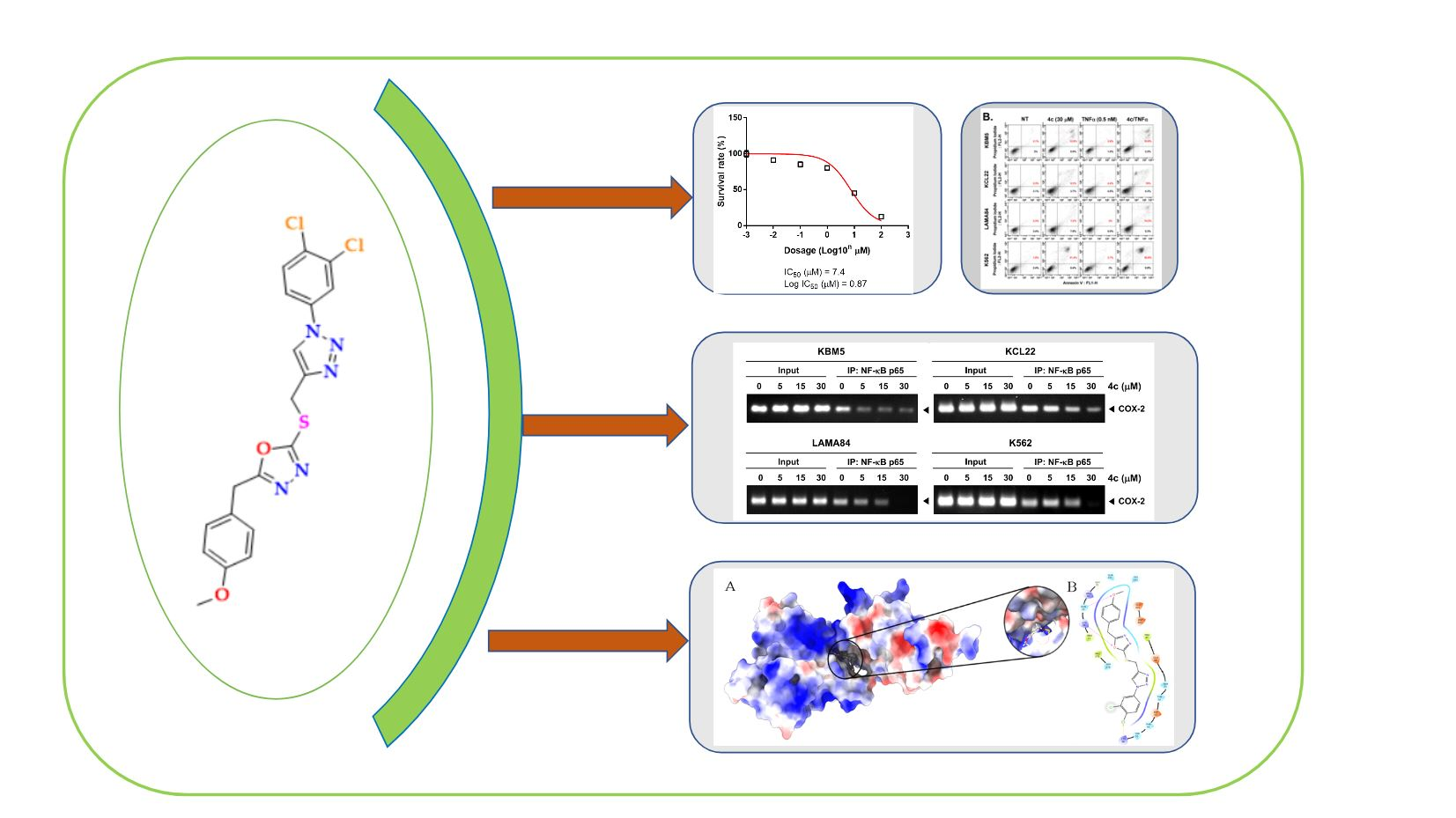
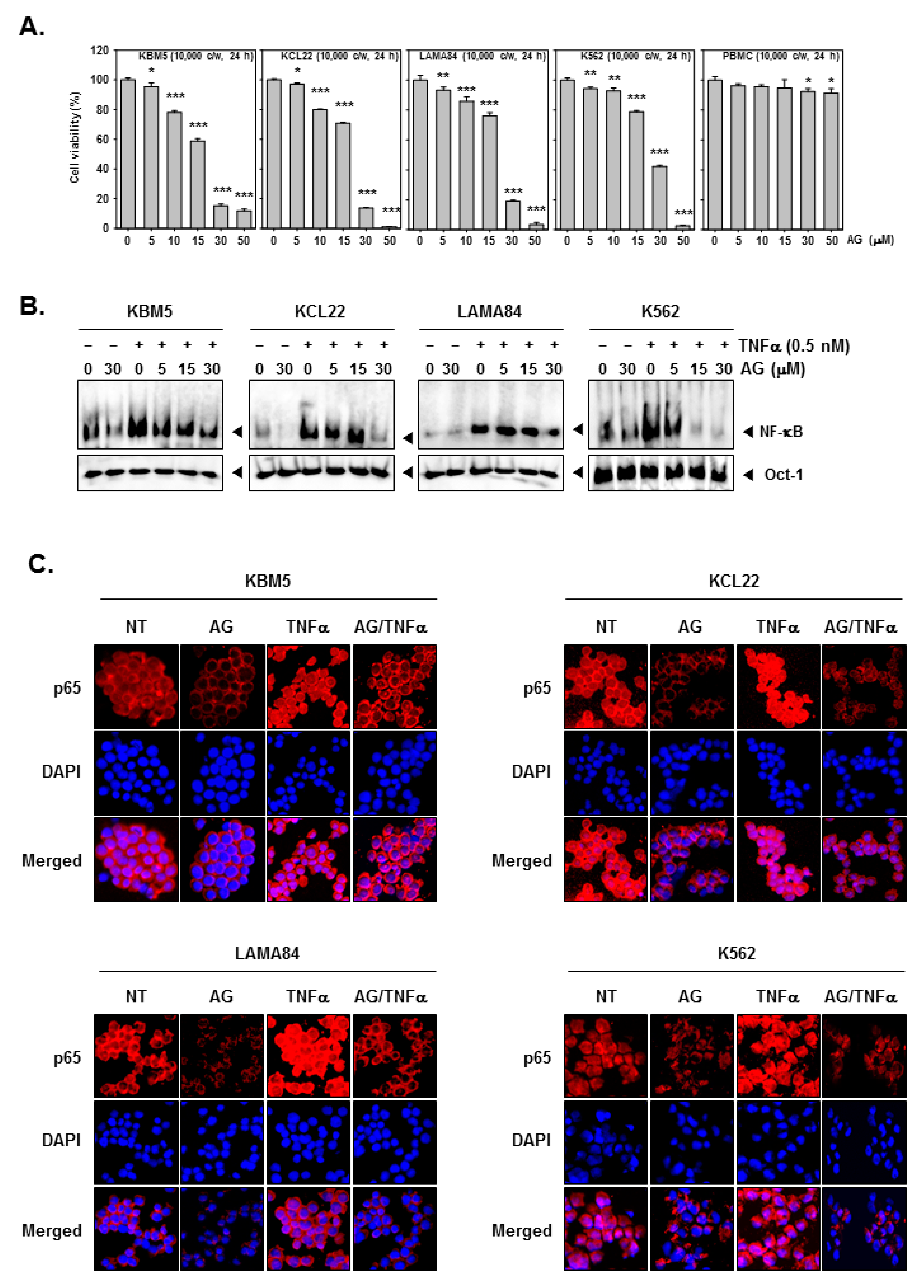
3.3. 4c Suppressed the Viability of CML Cells
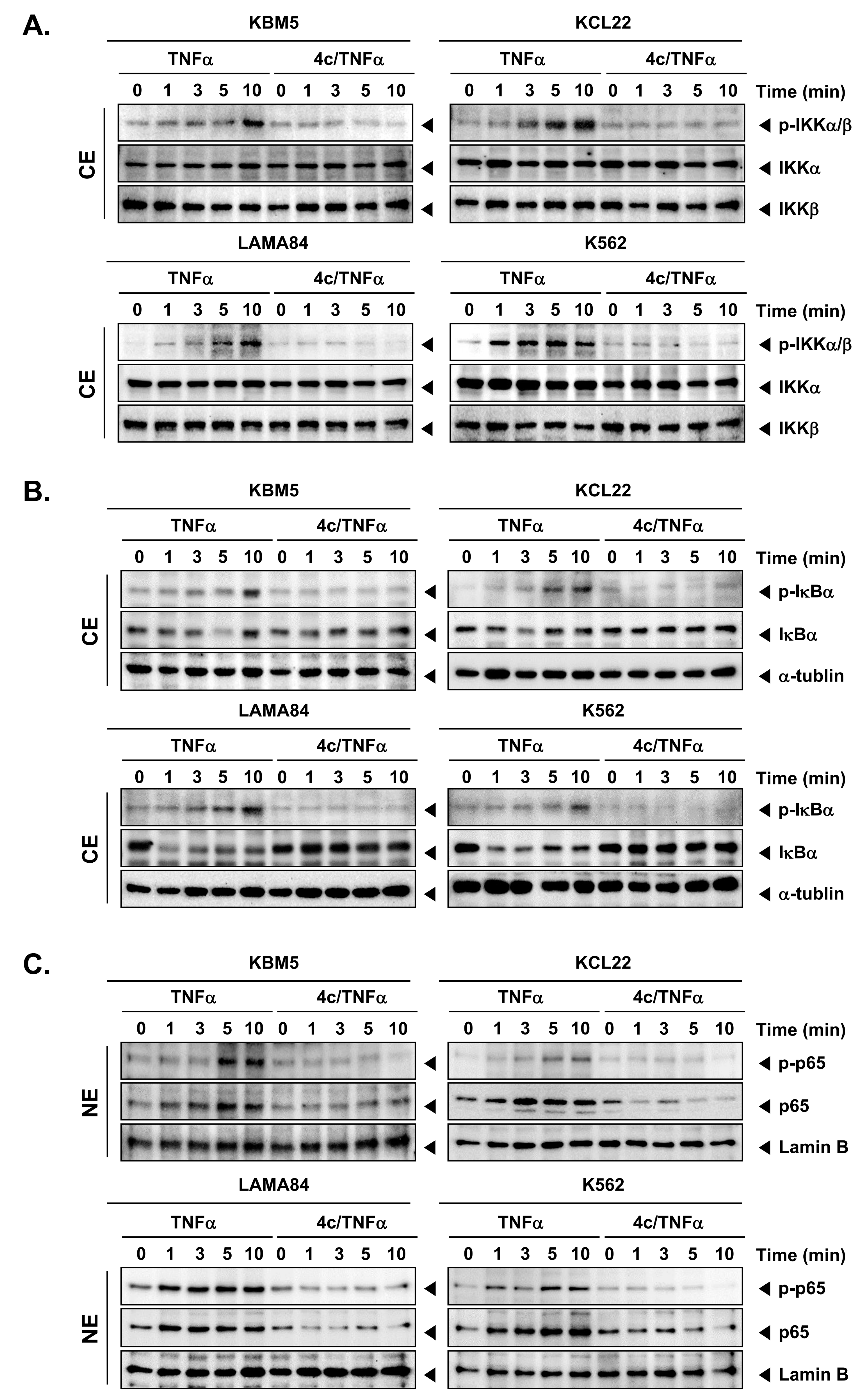
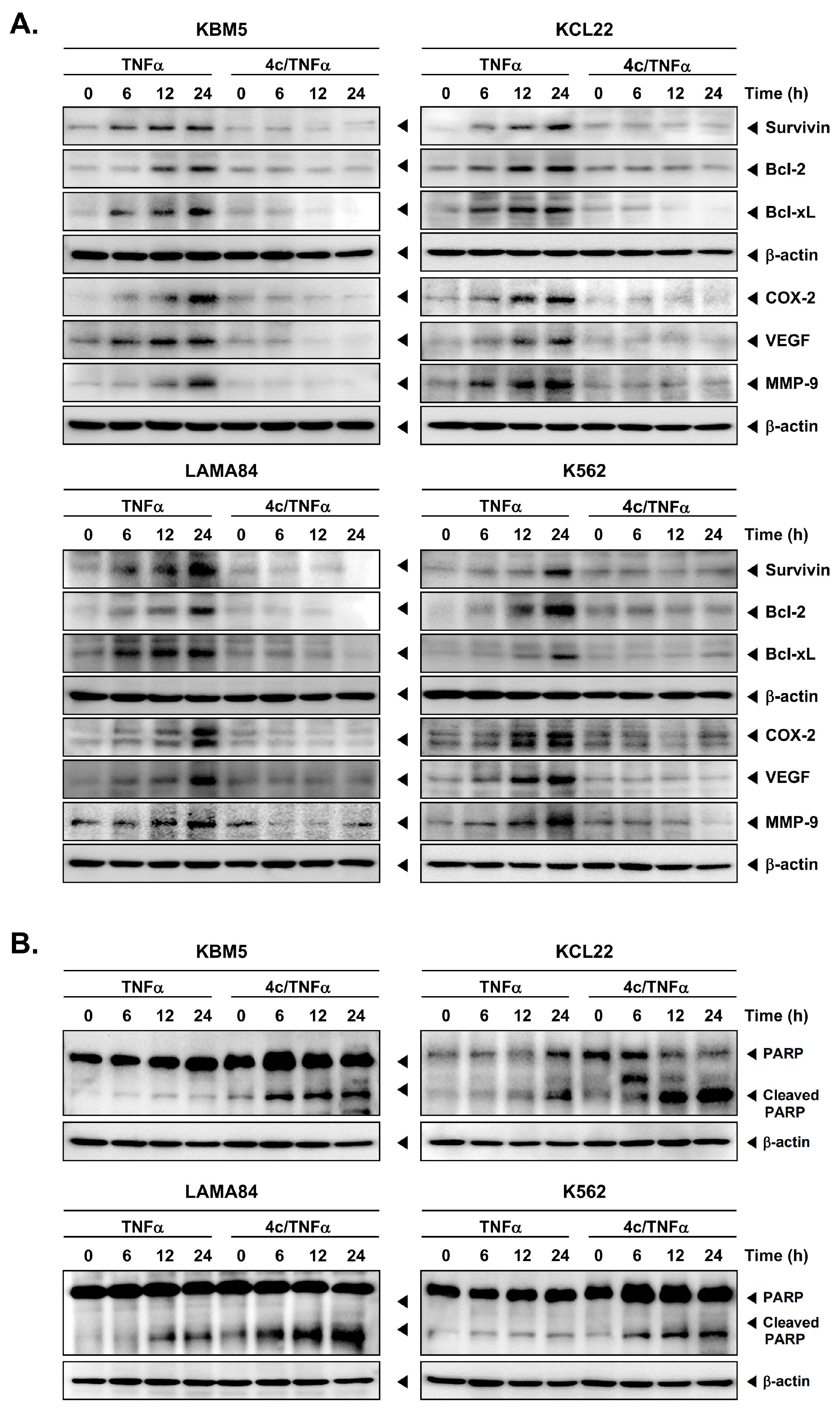
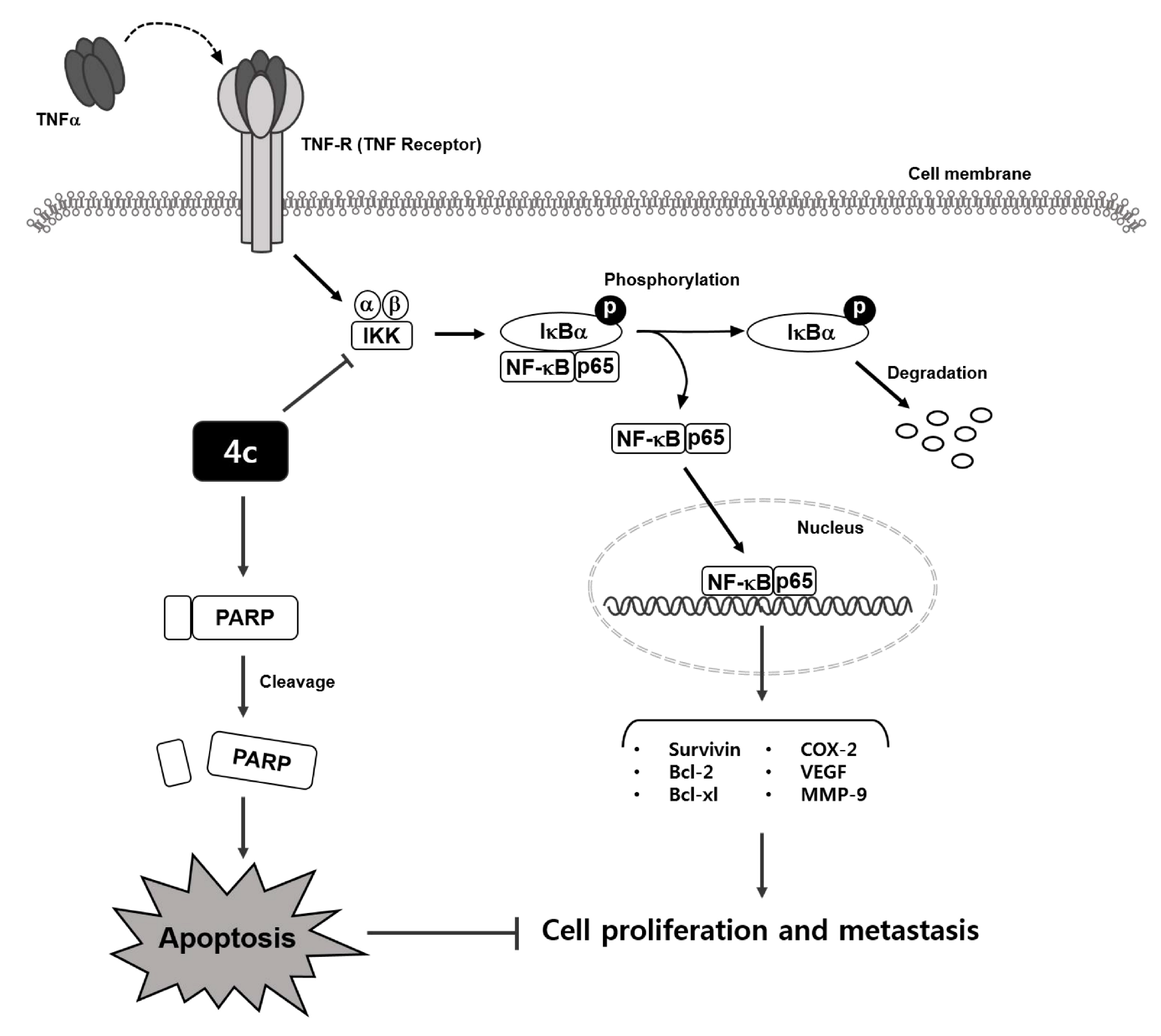
3.4. Compound 4c Inhibited the TNFα Induced NF-κB Activation in CML Cells
3.5. Compound 4c Inhibited NF-κB Complex Formation in CML Cells
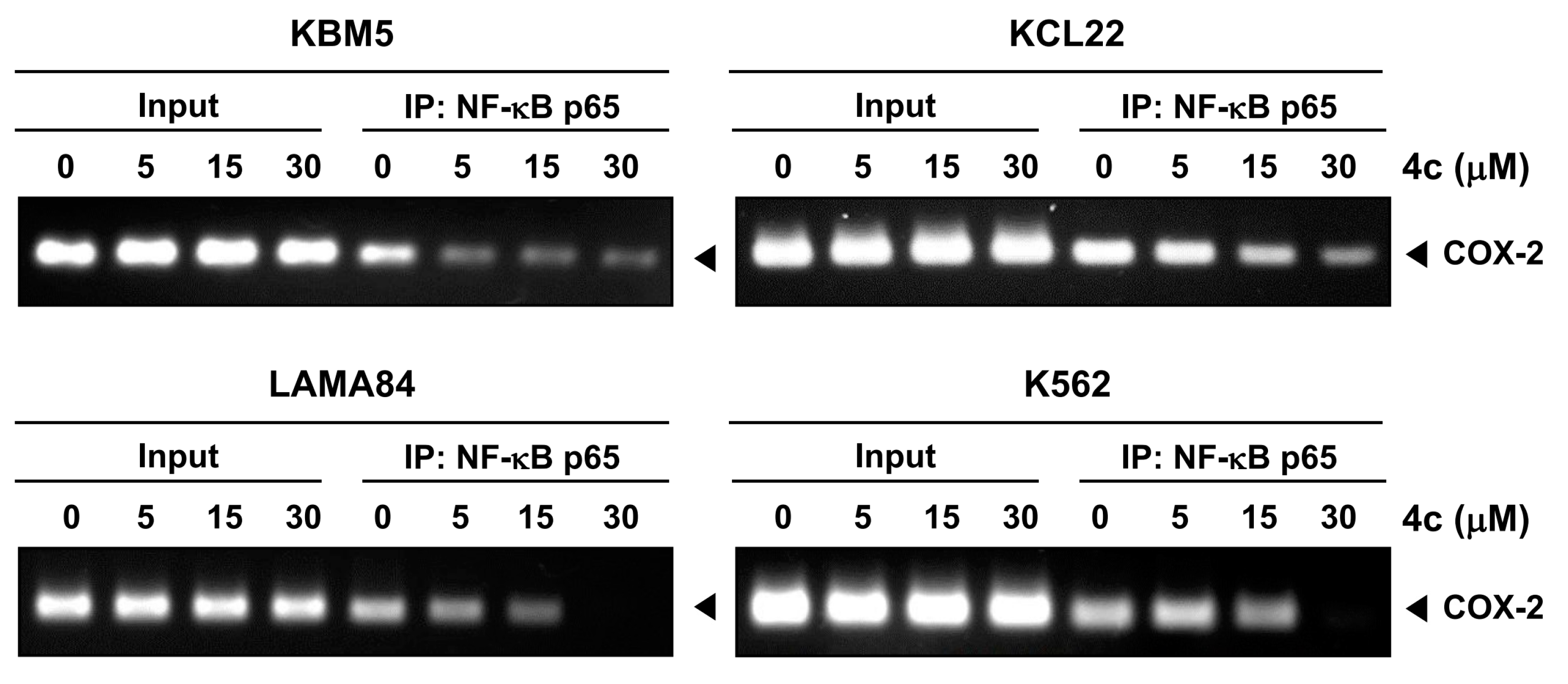
3.6. Compound 4c Inhibits Binding of COX-2 to p65 Promoter in CML Cells
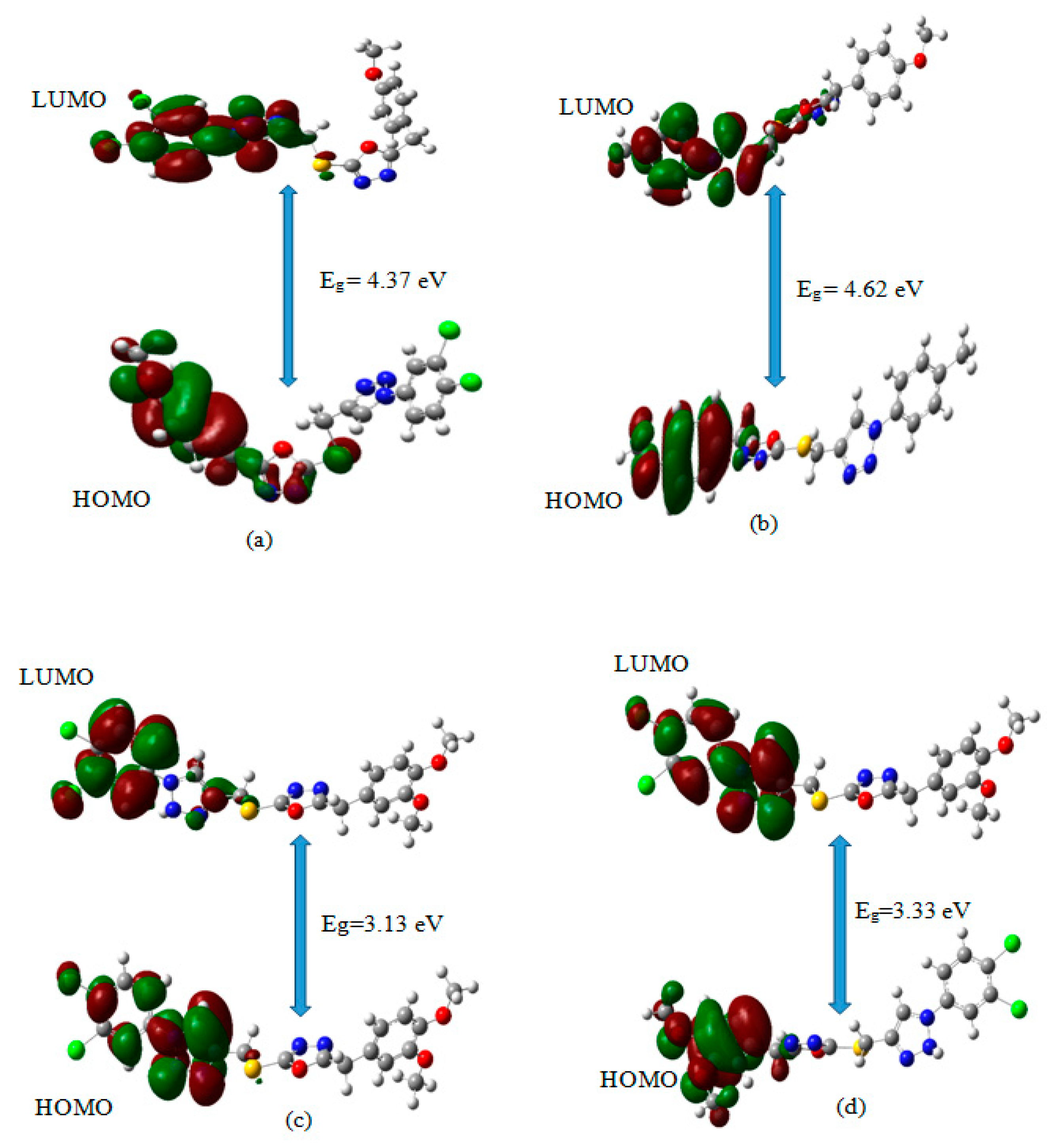
3.7. Frontier Molecular Orbitals (FMO) Calculations for the Title Compounds That Targets NF-κB
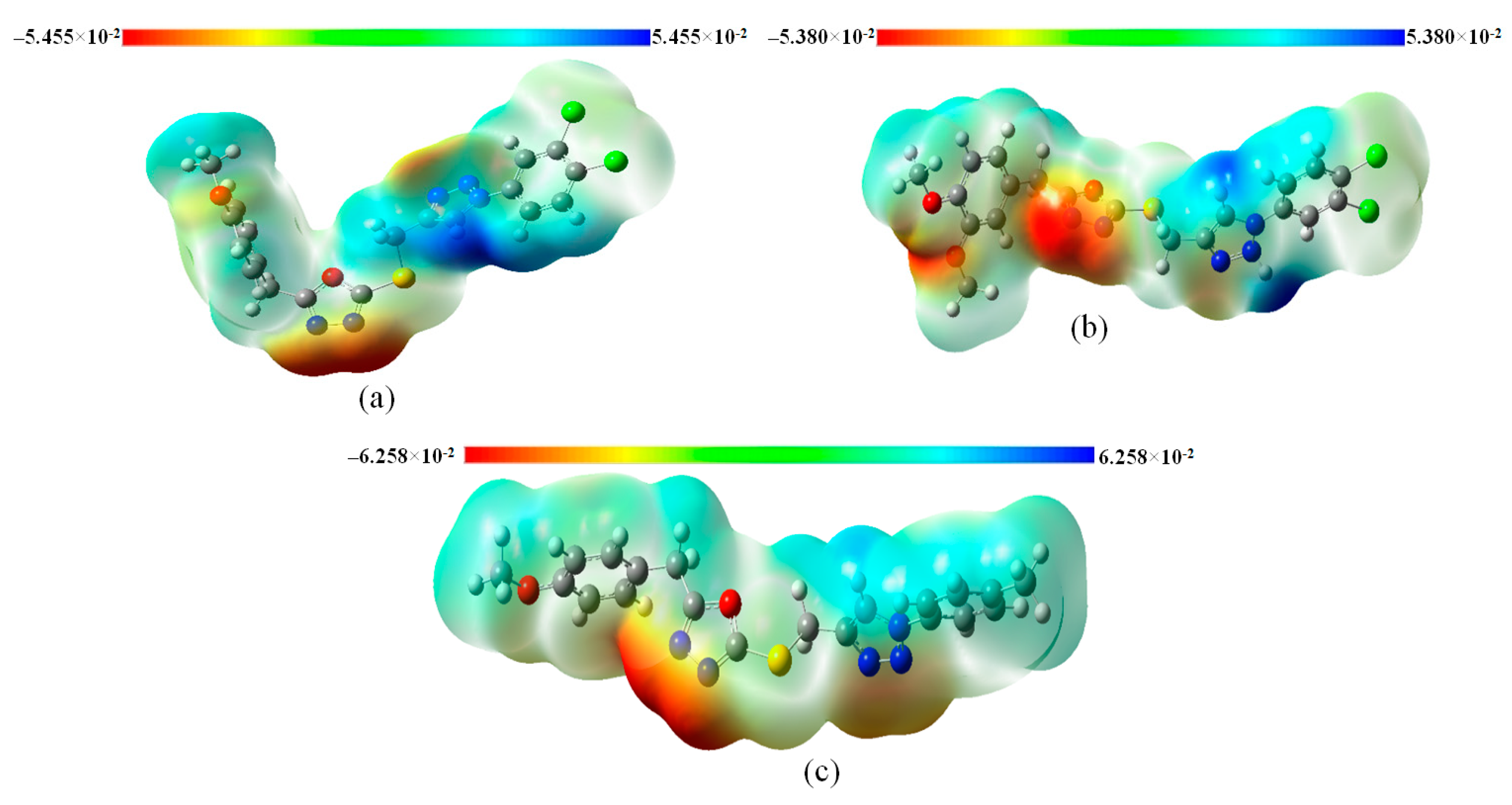
3.8. Molecular Electrostatic Potential (MEP) Analysis of Title Compounds
3.9. Natural Bond Orbital (NBO) Analysis
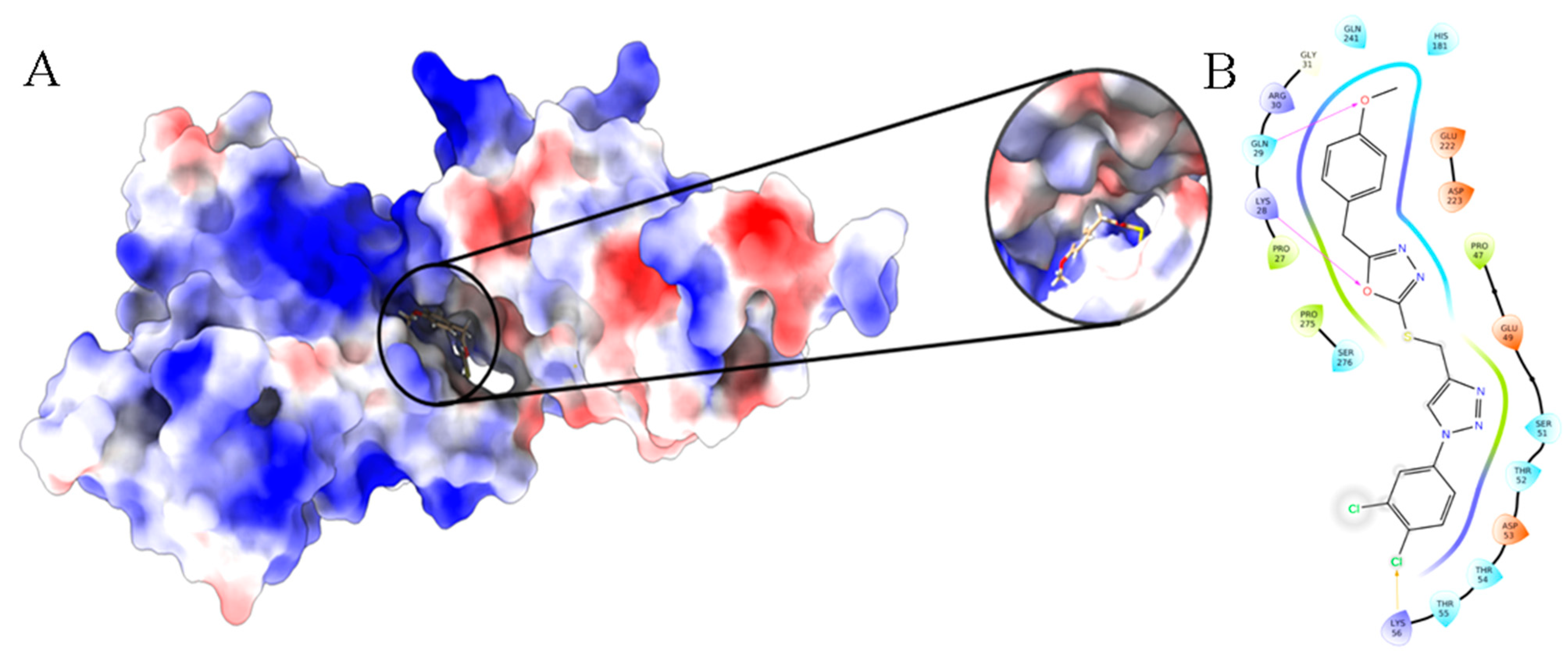
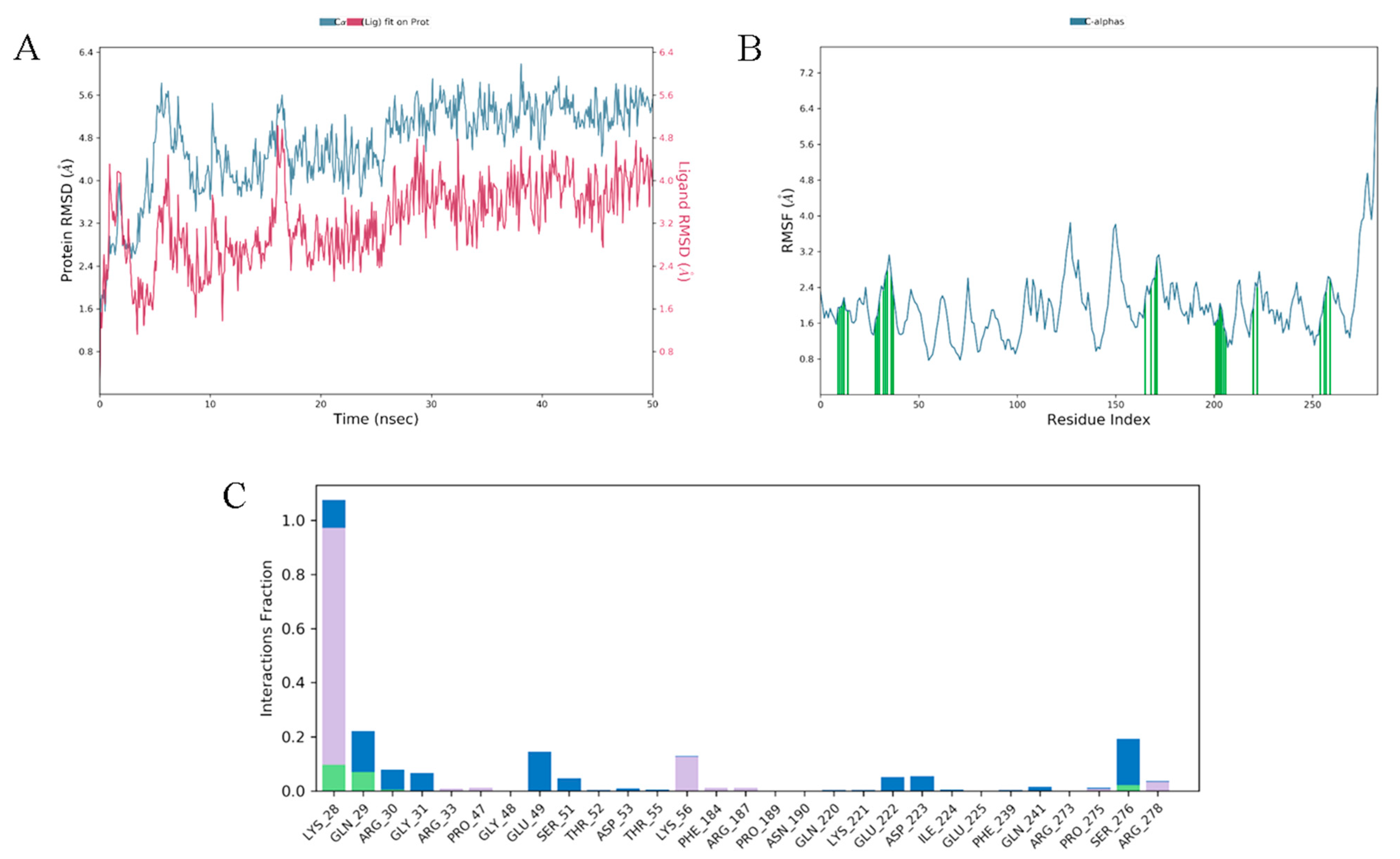
3.10. Molecular Docking and Dynamics Analysis of Compound 4c That Targets NF-κB
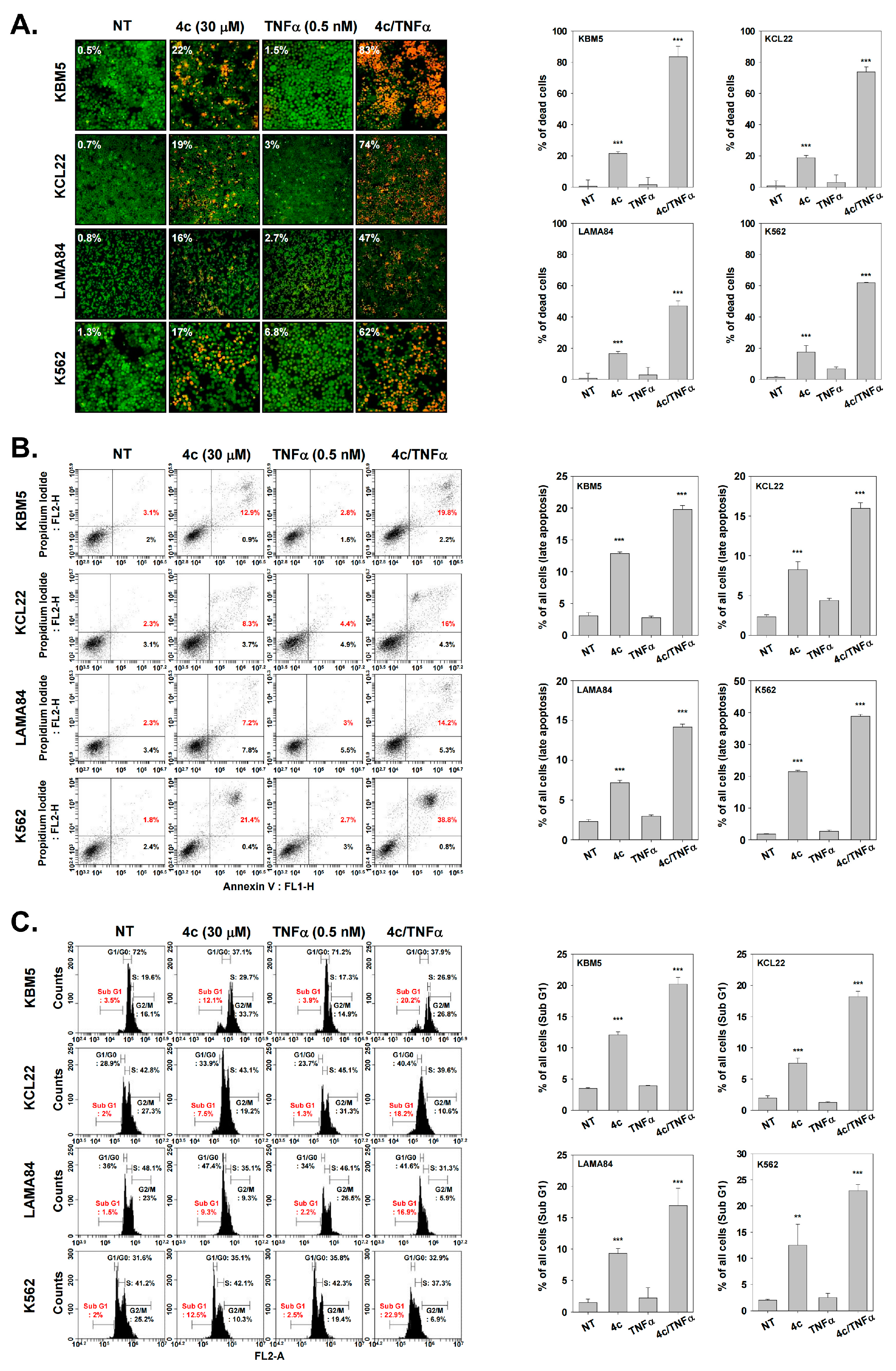
3.11. Compound 4c Induces Apoptosis in CML Cells
3.12. Compound 4c Induced Apoptotic-Cell Death in CML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eden, R.E.; Coviello, J.M. Chronic Myelogenous Leukemia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK531459/ (accessed on 16 January 2023).
- Lin, Q.; Mao, L.; Shao, L.; Zhu, L.; Han, Q.; Zhu, H.; Jin, J.; You, L. Global, Regional, and National Burden of Chronic Myeloid Leukemia, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Front. Oncol. 2020, 10, 580759. [Google Scholar] [CrossRef]
- Yilmaz, M.; Jabbour, E. Tyrosine Kinase Inhibitors Early in the Disease Course: Lessons from Chronic Myelogenous Leukemia. Semin. Oncol. 2015, 42, 876–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, X.; Wang, Z.; Feng, Y.; Jia, Y.; Jiang, L.; Xia, Y.; Cao, J.; Liu, Y. Comparison of Hepatotoxicity Associated with New BCR-ABL Tyrosine Kinase Inhibitors vs. Imatinib among Patients with Chronic Myeloid Leukemia: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e2120165. [Google Scholar] [CrossRef] [PubMed]
- Lindström, H.J.G.; de Wijn, A.S.; Friedman, R. Stochastic modelling of tyrosine kinase inhibitor rotation therapy in chronic myeloid leukaemia. BMC Cancer 2019, 19, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Gutiérrez, V.; Hernández-Boluda, J.C. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front. Oncol. 2019, 9, 603. [Google Scholar] [CrossRef] [Green Version]
- Carrà, G.; Torti, D.; Crivellaro, S.; Panuzzo, C.; Taulli, R.; Cilloni, D.; Guerrasio, A.; Saglio, G.; Morotti, A. The BCR-ABL/NF-κB signal transduction network: A long lasting relationship in Philadelphia positive Leukemias. Oncotarget 2016, 7, 66287–66298. [Google Scholar] [CrossRef] [Green Version]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-κB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Inducibility of κ Immunoglobulin Enhancer-Binding Protein NF-κB by a Posttranslational Mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The Complexity of NF-κB Signaling in Inflammation and Cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Motolani, A.; Martin, M.; Sun, M.; Lu, T. Phosphorylation of the Regulators, a Complex Facet of NF-κB Signaling in Cancer. Biomolecules 2020, 11, 15. [Google Scholar] [CrossRef]
- Ling, J.; Kumar, R. Crosstalk between NFkB and glucocorticoid signaling: A potential target of breast cancer therapy. Cancer Lett. 2012, 322, 119–126. [Google Scholar] [CrossRef]
- Morgan, D.; Garg, M.; Tergaonkar, V.; Tan, S.Y.; Sethi, G. Pharmacological significance of the non-canonical NF-κB pathway in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188449. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sethi, G.; Aggarwal, B.B. Simvastatin potentiates TNF-alpha-induced apoptosis through the down-regulation of NF-kappaB-dependent antiapoptotic gene products: Role of IkappaBalpha kinase and TGF-beta-activated kinase-1. J. Immunol. 2007, 178, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.S.; Sethi, G.; Chaturvedi, M.M.; Aggarwal, B.B. Simvastatin, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, suppresses osteoclastogenesis induced by receptor activator of nuclear factor-kappaB ligand through modulation of NF-kappaB pathway. Int. J. Cancer 2008, 123, 1733–1740. [Google Scholar] [CrossRef]
- Xi, J.B.; Fang, Y.F.; Frett, B.; Zhu, M.L.; Zhu, T.; Kong, Y.N.; Guan, F.J.; Zhao, Y.; Zhang, X.W.; Li, H.Y.; et al. Structure-based design and synthesis of imidazo[1,2-a]pyridine derivatives as novel and potent Nek2 inhibitors with in vitro and in vivo antitumor activities. Eur. J. Med. Chem. 2017, 126, 1083–1106. [Google Scholar] [CrossRef]
- Korunes, K.L.; Liu, J.; Huang, R.; Xia, M.; Houck, K.A.; Corton, J.C. A gene expression biomarker for predictive toxicology to identify chemical modulators of NF-κB. PLoS ONE 2022, 17, e0261854. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.C.; Kim, J.H.; Prasad, S.; Aggarwal, B.B. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010, 29, 405–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Li, Z.; Zhou, J. Tumor Necrosis Factor α in the Onset and Progression of Leukemia. Exp. Hematol. 2017, 45, 17–26. [Google Scholar] [CrossRef]
- Wu, J.T.; Kral, J.G. The NF-kappaB/IkappaB signaling system: A molecular target in breast cancer therapy. J. Surg. Res. 2005, 123, 158–169. [Google Scholar] [CrossRef]
- Chen, Z.J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell. Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solt, L.A.; May, M.J. The IkappaB kinase complex: Master regulator of NF-kappaB signaling. Immunol. Res. 2008, 42, 3–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Ito, K. Leukemia Stem Cells as a Potential Target to Achieve Therapy-Free Remission in Chronic Myeloid Leukemia. Cancers 2021, 13, 5822. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [Green Version]
- Israël, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [Green Version]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.; Elizalde, P.V.; Schillaci, R. Harnessing Tumor Necrosis Factor Alpha to Achieve Effective Cancer Immunotherapy. Cancers 2021, 13, 564. [Google Scholar] [CrossRef]
- Neganova, M.; Liu, J.; Aleksandrova, Y.; Klochkov, S.; Fan, R. Therapeutic Influence on Important Targets Associated with Chronic Inflammation and Oxidative Stress in Cancer Treatment. Cancers 2021, 13, 6062. [Google Scholar] [CrossRef]
- Fathi, M.A.A.; Abd El-Hafeez, A.A.; Abdelhamid, D.; Abbas, S.H.; Montano, M.M.; Abdel-Aziz, M. 1,3,4-Oxadiazole/Chalcone Hybrids: Design, Synthesis, and Inhibition of Leukemia Cell Growth and EGFR, Src, IL-6 and STAT3 Activities. Bioorg. Chem. 2019, 84, 150–163. [Google Scholar] [CrossRef]
- Janganati, V.; Ponder, J.; Balasubramaniam, M.; Bhat-Nakshatri, P.; Bar, E.E.; Nakshatri, H.; Jordan, C.T.; Crooks, P.A. MMB Triazole Analogs Are Potent NF-κB Inhibitors and Anti-Cancer Agents against Both Hematological and Solid Tumor Cells. Eur. J. Med. Chem. 2018, 157, 562–581. [Google Scholar] [CrossRef] [Green Version]
- Bharathkumar, H.; Mohan, C.D.; Rangappa, S.; Kang, T.; Keerthy, H.K.; Fuchs, J.E.; Kwon, N.H.; Bender, A.; Kim, S.; Rangappa, K.S. Screening of quinoline, 1,3-benzoxazine, and 1,3-oxazine-based small molecules against isolated methionyl-tRNA synthetase and A549 and HCT116 cancer cells including an in silico binding mode analysis. Org. Biomol. Chem. 2015, 13, 9381–9387. [Google Scholar] [CrossRef] [Green Version]
- Rüngeler, P.; Castro, V.; Mora, G.; Gören, N.; Vichnewski, W.; Pahl, H.L.; Merfort, I.; Schmidt, T.J. Inhibition of transcription factor NF-kappaB by sesquiterpene lactones: A proposed molecular mechanism of action. Bioorg. Med. Chem. 1999, 7, 2343–2352. [Google Scholar] [CrossRef]
- Mohan, C.D.; Anilkumar, N.C.; Rangappa, S.; Shanmugam, M.K.; Mishra, S.; Chinnathambi, A.; Alharbi, S.A.; Bhattacharjee, A.; Sethi, G.; Kumar, A.P.; et al. Novel 1,3,4-oxadiazole induces anticancer activity by targeting NF-κB in hepatocellular carcinoma cells. Front. Oncol. 2018, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView Version 6 (Shawnee Mission, KS) Semichem Inc. 2016. Available online: http://gaussian.com/uvvisplot (accessed on 26 January 2023).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, W.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- BIOVIA Dassault Systèmes. Discovery Studio Visualizer, 21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2020. [Google Scholar]
- Schrödinger, L.L.C.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol (accessed on 2 January 2022).
- Schrödinger, L.L.C. Schrödinger Release 2020-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2020. [Google Scholar]
- Basappa, K.C.; Rangappa, K.S. Simple and an efficient method for the synthesis of 1-[2-dimethylamino-1-(4-methoxy-phenyl)-ethyl]-cyclohexanol hydrochloride: (+/−) venlafaxine racemic mixtures. Bioorg. Med. Chem. Lett. 2004, 14, 3279–3281. [Google Scholar] [CrossRef] [PubMed]
- Kanchugarakoppal, S.R.; Basappa. New cholinesterase inhibitors: Synthesis and structure-activity relationship studies of 1,2-benzisoxazole series and novel imidazolyl-d 2-isoxazolines. J. Phys. Organ. Chem. 2005, 18, 773–778. [Google Scholar]
- Pavitra, E.; Kancharla, J.; Gupta, V.K.; Prasad, K.; Sung, J.Y.; Kim, J.; Tej, M.B.; Choi, R.; Lee, J.H.; Han, Y.K.; et al. The role of NF-κB in breast cancer initiation, growth, metastasis, and resistance to chemotherapy. Biomed. Pharmacother. 2023, 163, 114822. [Google Scholar] [CrossRef]
- Shono, Y.; Tuckett, A.Z.; Liou, H.C.; Doubrovina, E.; Derenzini, E.; Ouk, S.; Tsai, J.J.; Smith, O.M.; Levy, E.R.; Kreines, F.M.; et al. Characterization of a c-Rel Inhibitor That Mediates Anticancer Properties in Hematologic Malignancies by Blocking NF-κB-Controlled Oxidative Stress Responses. Cancer Res. 2016, 76, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Panicker, C.Y. FT-IR, HOMO–LUMO, NBO, MEP analysis and molecular docking study of 3-Methyl-4-{(E)-[4-(methylsulfanyl)-benzylidene] amino} 1H-1, 2, 4-triazole-5 (4H)-thione. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 151, 198–207. [Google Scholar] [CrossRef]
- Gurupadaswamy, H.D.; Girish, V.; Kavitha, C.V.; Raghavan, S.C.; Khanum, S.A. Synthesis and Evaluation of 2,5-Di(4-Aryloylaryloxymethyl)-1,3,4-Oxadiazoles as Anti-Cancer Agents. Eur. J. Med. Chem. 2013, 63, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Nani, B.D.; Rosalen, P.L.; Lazarini, J.G.; de Cássia Orlandi Sardi, J.; Romário-Silva, D.; de Araújo, L.P.; Dos Reis, M.S.B.; Breseghello, I.; Cunha, T.M.; de Alencar, S.M.; et al. A Study on the Anti-NF-κB, Anti-Candida, and Antioxidant Activities of Two Natural Plant Hormones: Gibberellin A4 and A7. Pharmaceutics 2022, 14, 1347. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Rothman, P. JAK-STAT signaling activated by Abl oncogenes. Oncogene 2000, 19, 2523–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.K.; Langdon, W.Y.; Varticovski, L. Tyrosine phosphorylation of p120cbl in BCR/abl transformed hematopoietic cells mediates enhanced association with phosphatidylinositol 3-kinase. Oncogene 1997, 14, 2217–2228. [Google Scholar] [CrossRef] [Green Version]
- Mizuchi, D.; Kurosu, T.; Kida, A.; Jin, Z.H.; Jin, A.; Arai, A.; Miura, O. BCR/ABL activates Rap1 and B-Raf to stimulate the MEK/Erk signaling pathway in hematopoietic cells. Biochem. Biophys. Res. Commun. 2005, 326, 645–651. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Talpaz, M.; Giles, F.; O’Brien, S.; Cortes, J. New insights into the pathophysiology of chronic myeloid leukemia and imatinib resistance. Ann. Intern. Med. 2006, 145, 913–923. [Google Scholar] [CrossRef]
- Sethi, G.; Ahn, K.S.; Chaturvedi, M.M.; Aggarwal, B.B. Epidermal growth factor (EGF) activates nuclear factor-kappaB through IkappaBalpha kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IkappaBalpha. Oncogene 2007, 26, 7324–7332. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.S.; Sethi, G.; Aggarwal, B.B. Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-κB pathway. Biochem. Pharmacol. 2008, 75, 907–913. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | IC50 (µM) |
|---|---|---|---|
| 4a | 4-methoxy | 4-Cl | 9.58 ± 0.98 |
| 4b | 4-methoxy | 4-Br | 8.44 ± 0.93 |
| 4c | 4-methoxy | 3,4-Cl2 | 7.40 ± 0.87 |
| 4d | 4-methoxy | H | 25.80 ± 1.41 |
| 4e | 4-methoxy | 3-Cl | 16.86 ± 1.23 |
| 4f | 4-methoxy | 4-methoxy | 34.50 ± 1.54 |
| 4g | 4-methoxy | 4-OH | 29.73 ± 1.47 |
| 4h | 4-methoxy | 2-CH3 | 26.22 ± 1.42 |
| 4i | 4-methoxy | 4-CH3 | 45.39 ± 1.66 |
| 4j | 3,4-dimethoxy | 4-Cl | 40.53 ± 1.61 |
| 4k | 3,4-dimethoxy | 4-Br | 31.90 ± 1.50 |
| 4l | 3,4-dimethoxy | 3,4-Cl2 | 23.79 ± 1.38 |
| 4m | 3,4-dimethoxy | H | 28.26 ± 1.45 |
| 4n | 3,4-dimethoxy | 3-Cl | 23.16 ± 1.37 |
| 4o | 3,4-dimethoxy | 4-methoxy | 35.83 ± 1.55 |
| 4p | 3,4-dimethoxy | 4-OH | 65.04 ± 1.81 |
| 4q | 3,4-dimethoxy | 4-CH3 | 19.73 ± 1.30 |
| 4r | 3,4-dimethoxy | 2-CH3 | 35.70 ± 1.55 |
| Tamoxifen | 1.45 ± 0.16 | ||
| Doxorubicin | 0.91 ± 0.04 | ||
| Properties | 4c | (4i) | (4l) | |
|---|---|---|---|---|
| Alpha Orbitals | Beta Orbitals | |||
| EHOMO eV | −6.4199 | −6.2245 | −4.5581 | −5.8096 |
| ELOMO eV | −2.0479 | −1.5989 | −1.4182 | −2.4781 |
| ∆ELUMO−HOMO eV | 4.372 | 4.6256 | 3.1399 | 3.3313 |
| Ionization potential (I) | 6.4199 | 6.2245 | 4.5581 | 5.8096 |
| Electron affinity (A) | 2.0479 | 1.5989 | 1.4182 | 2.4781 |
| Global hardness (ɳ) | 2.1860 | 2.3128 | 1.5699 | 1.6656 |
| Softness (S) | 0.2287 | 0.2161 | 0.3184 | 0.3001 |
| Chemical potential (μ) | −4.2339 | −3.9117 | −2.9881 | −4.1438 |
| Electronegativity (χ) | 4.2339 | 3.9117 | 2.9881 | 4.1438 |
| Electrophilicity (ᴪ) | 4.1001 | 3.3079 | 2.8437 | 5.1546 |
| Donor (i) | ED (i) (e) | Acceptor (j) | ED (j)(e) | E(2) kJ/mol | ∆E a | F (i,j) b a.u. |
|---|---|---|---|---|---|---|
| π (C1–C2) | 0.38305 | π *(C3–C4) | 0.30446 | 1232.816 | 0.01 | 0.083 |
| 0.38305 | π *(C5–C6) | 0.35079 | 1104.492 | 0.01 | 0.08 | |
| π (N17–C20) | 0.90032 | π *(C26–C27) | 0.37632 | 458.943 | 0.07 | 0.1 |
| 0.90032 | π *(C23–C24) | 0.32671 | 359.1546 | 0.08 | 0.097 | |
| 0.90032 | π *(N18–N19) | 0.46421 | 195.0999 | 0.03 | 0.043 | |
| 1.62866 | π *(C15–C16) | 0.33318 | 98.57504 | 0.36 | 0.083 | |
| LP (1) C25 | 1.07521 | π *(C26–C27) | 0.37632 | 254.2198 | 0.16 | 0.103 |
| 1.07521 | π *(C23–C24) | 0.32671 | 248.6133 | 0.17 | 0.107 | |
| π (N10–C11) | 0.30658 | π *(C8–N9) | 0.24318 | 202.4219 | 0.02 | 0.054 |
| π (N18–N19) | 0.46421 | π *(C15–C16) | 0.33318 | 201.9617 | 0.05 | 0.069 |
| π (C23–C24) | 1.67802 | π *(N17–C20) | 0.90032 | 142.0468 | 0.2 | 0.088 |
| LP (2) O12 | 1.71189 | π *(N10–C11) | 0.30658 | 136.231 | 0.35 | 0.095 |
| 1.71189 | π *(C8–N9) | 0.24318 | 132.8838 | 0.37 | 0.097 | |
| LP (2) O21 | 1.84250 | π *(C1–C2) | 0.38305 | 126.0639 | 0.34 | 0.096 |
| π (C26–C27) | 1.67623 | π *(N17–C20) | 0.90032 | 125.5618 | 0.21 | 0.085 |
| 0.37632 | π *(C23–C24) | 0.32671 | 43.97384 | 0.01 | 0.016 | |
| π (C15–C16) | 1.76426 | π *(N18–N19) | 0.46421 | 109.7882 | 0.25 | 0.076 |
| π (N17–C20) | 1.62866 | π *(N18–N19) | 0.46421 | 104.6000 | 0.31 | 0.08 |
| π (N17–C20) | 1.62866 | π *(C15–C16) | 0.33318 | 98.57504 | 0.36 | 0.083 |
| π (C1–C2) | 1.66747 | π *(C5–C6) | 0.35079 | 91.33672 | 0.3 | 0.072 |
| π (C3–C4) | 1.70804 | π *(C1–C2) | 0.38305 | 89.87232 | 0.28 | 0.071 |
| π (C5–C6) | 1.69157 | π *(C3–C4) | 0.30446 | 87.61296 | 0.29 | 0.069 |
| LP (2) S13 | 1.86175 | π *(N10–C11) | 0.30658 | 72.46688 | 0.24 | 0.06 |
| π (C5–C6) | 1.69157 | π *(C1–C2) | 0.38305 | 72.13216 | 0.28 | 0.063 |
| π (C8–N9) | 1.90918 | π *(N10–C11) | 0.30658 | 44.64328 | 0.32 | 0.056 |
| LP (1) N9 | 1.93333 | σ *(C8–O12) | 0.06577 | 43.38808 | 0.72 | 0.077 |
| LP (1) N10 | 1.93465 | σ *(C11–O12) | 0.07854 | 42.92784 | 0.71 | 0.076 |
| σ (N9–N10) | 1.96066 | σ *(C11–S13) | 0.03347 | 29.12064 | 0.96 | 0.073 |
| 1.96066 | σ *(C7–C8) | 0.02796 | 25.68976 | 1.18 | 0.076 | |
| σ (C15–C16) | 1.97644 | σ *(N17–C20) | 0.03907 | 25.3132 | 1.10 | 0.073 |
| σ (C14–H37) | 1.98275 | σ *(C15–C16) | 0.02903 | 21.84048 | 1.09 | 0.068 |
| σ (C24–C25) | 1.97360 | σ *(C26–Cl28) | 0.02784 | 20.0832 | 0.89 | 0.058 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basappa, B.; Jung, Y.Y.; Ravish, A.; Xi, Z.; Swamynayaka, A.; Madegowda, M.; Pandey, V.; Lobie, P.E.; Sethi, G.; Ahn, K.S. Methyl-Thiol-Bridged Oxadiazole and Triazole Heterocycles as Inhibitors of NF-κB in Chronic Myelogenous Leukemia Cells. Biomedicines 2023, 11, 1662. https://doi.org/10.3390/biomedicines11061662
Basappa B, Jung YY, Ravish A, Xi Z, Swamynayaka A, Madegowda M, Pandey V, Lobie PE, Sethi G, Ahn KS. Methyl-Thiol-Bridged Oxadiazole and Triazole Heterocycles as Inhibitors of NF-κB in Chronic Myelogenous Leukemia Cells. Biomedicines. 2023; 11(6):1662. https://doi.org/10.3390/biomedicines11061662
Chicago/Turabian StyleBasappa, Basappa, Young Yun Jung, Akshay Ravish, Zhang Xi, Ananda Swamynayaka, Mahendra Madegowda, Vijay Pandey, Peter E. Lobie, Gautam Sethi, and Kwang Seok Ahn. 2023. "Methyl-Thiol-Bridged Oxadiazole and Triazole Heterocycles as Inhibitors of NF-κB in Chronic Myelogenous Leukemia Cells" Biomedicines 11, no. 6: 1662. https://doi.org/10.3390/biomedicines11061662
APA StyleBasappa, B., Jung, Y. Y., Ravish, A., Xi, Z., Swamynayaka, A., Madegowda, M., Pandey, V., Lobie, P. E., Sethi, G., & Ahn, K. S. (2023). Methyl-Thiol-Bridged Oxadiazole and Triazole Heterocycles as Inhibitors of NF-κB in Chronic Myelogenous Leukemia Cells. Biomedicines, 11(6), 1662. https://doi.org/10.3390/biomedicines11061662