
Apabetalone Downregulates Fibrotic, Inflammatory and Calcific Processes in Renal Mesangial Cells and Patients with Renal Impairment

 ,
, 
Abstract
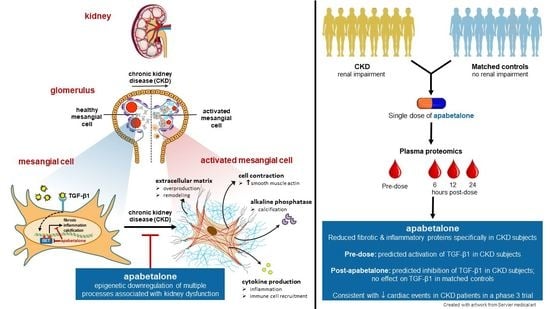
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Apabetalone Suppresses TGF-β1-Mediated HRMC Activation
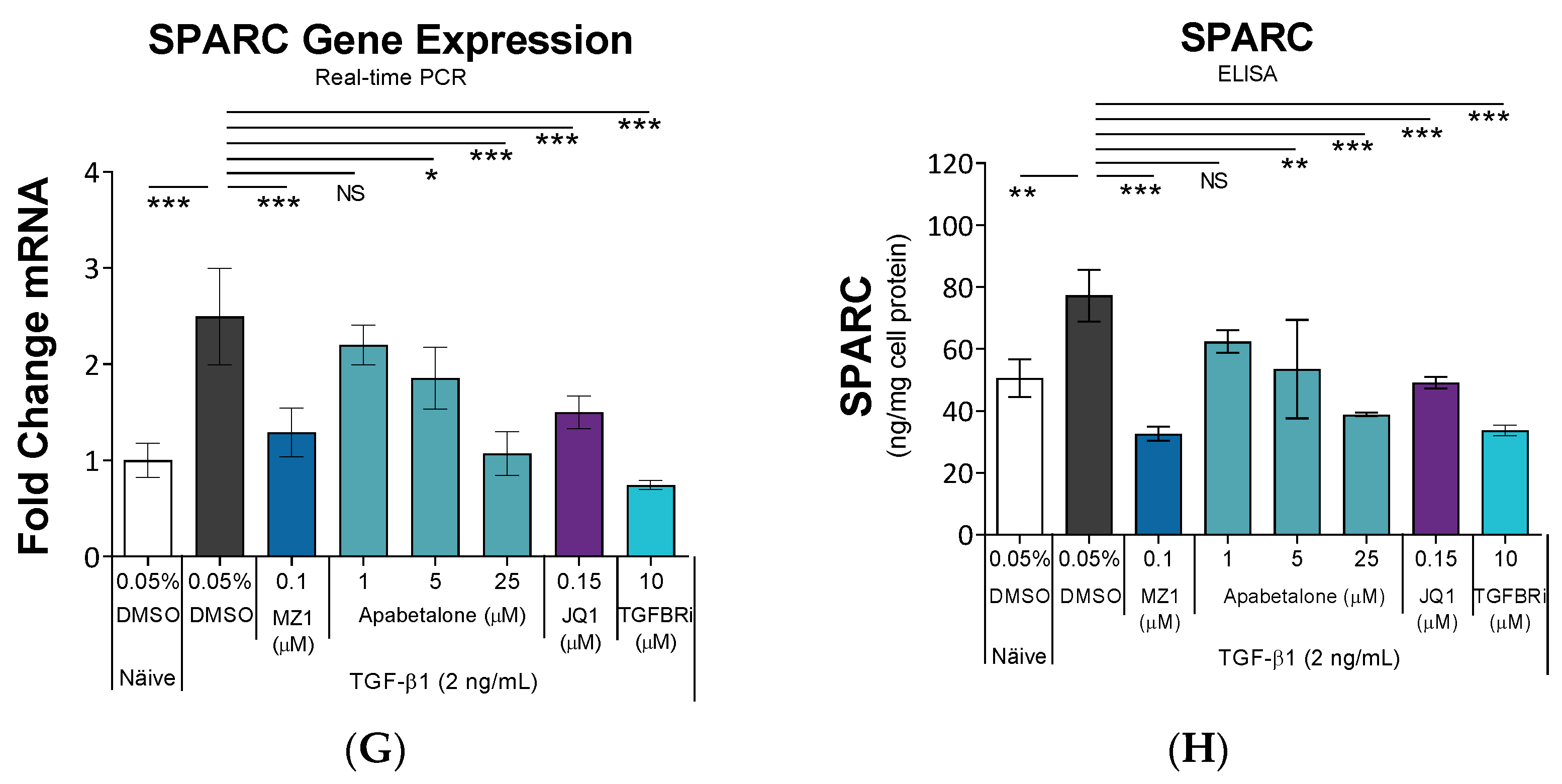
3.2. Apabetalone Blocks TGF-β1 Mediated Expression of Key Drivers of Fibrosis
3.3. Apabetalone Downregulates Inflammatory Mediators in HRMCs
3.4. Apabetalone Downregulates TGF-β1 Mediated Expression of Alkaline Phosphatase
3.5. Apabetalone Alters Transcriptional Response of HRMCs to TGF-β1
3.6. Apabetalone Reduces Levels of Pro-Fibrotic Factors in Human Plasma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int. 2019, 96, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Schlondorff, D.; Banas, B. The mesangial cell revisited: No cell is an island. J. Am. Soc. Nephrol. JASN 2009, 20, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Scindia, Y.M.; Deshmukh, U.S.; Bagavant, H. Mesangial pathology in glomerular disease: Targets for therapeutic intervention. Adv. Drug Deliv. Rev. 2010, 62, 1337–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Hong, S.W.; Iglesias-de la Cruz, M.C.; Isono, M.; Casaretto, A.; Ziyadeh, F.N. The key role of the transforming growth factor-beta system in the pathogenesis of diabetic nephropathy. Ren. Fail. 2001, 23, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Schnaper, H.W.; Hayashida, T.; Hubchak, S.C.; Poncelet, A.C. TGF-beta signal transduction and mesangial cell fibrogenesis. Am. J. Physiol. Ren. Physiol. 2003, 284, F243–F252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottinger, E.P.; Bitzer, M. TGF-beta signaling in renal disease. J. Am. Soc. Nephrol. JASN 2002, 13, 2600–2610. [Google Scholar] [PubMed] [Green Version]
- Gewin, L.S. TGF-beta and Diabetic Nephropathy: Lessons Learned Over the Past 20 Years. Am. J. Med. Sci. 2020, 359, 70–72. [Google Scholar] [CrossRef]
- Tsujino, K.; Reed, N.I.; Atakilit, A.; Ren, X.; Sheppard, D. Transforming growth factor-beta plays divergent roles in modulating vascular remodeling, inflammation, and pulmonary fibrosis in a murine model of scleroderma. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L22–L31. [Google Scholar] [CrossRef] [Green Version]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-beta1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. JASN 2017, 28, 953–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chen, H.Y.; Zhong, X.; Chung, A.C.; Lan, H.Y. Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 2012, 227, 175–188. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharm. 2019, 10, 1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilham, D.; Tsujikawa, L.M.; Sarsons, C.D.; Halliday, C.; Wasiak, S.; Stotz, S.C.; Jahagirdar, R.; Sweeney, M.; Johansson, J.O.; Wong, N.C.W.; et al. Apabetalone downregulates factors and pathways associated with vascular calcification. Atherosclerosis 2019, 280, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin. Epigenet. 2019, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Wasiak, S.; Gilham, D.; Daze, E.; Tsujikawa, L.M.; Halliday, C.; Stotz, S.C.; Rakai, B.D.; Fu, L.; Jahagirdar, R.; Sweeney, M.; et al. Epigenetic Modulation by Apabetalone Counters Cytokine-Driven Acute Phase Response In Vitro, in Mice and in Patients with Cardiovascular Disease. Cardiovasc. Ther. 2020, 2020, 9397109. [Google Scholar] [CrossRef] [PubMed]
- Van der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.L.; Szulcek, R.; Bourgeois, A.; Lampron, M.C.; Habbout, K.; Martineau, S.; et al. Multicenter Preclinical Validation of BET Inhibition for the Treatment of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef]
- Wasiak, S.; Dzobo, K.E.; Rakai, B.D.; Kaiser, Y.; Versloot, M.; Bahjat, M.; Stotz, S.C.; Fu, L.; Sweeney, M.; Johansson, J.O.; et al. BET protein inhibitor apabetalone (RVX-208) suppresses pro-inflammatory hyper-activation of monocytes from patients with cardiovascular disease and type 2 diabetes. Clin. Epigenet. 2020, 12, 166. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhou, Y.; Peng, Y.; Huang, T.; Xia, F.; Yang, T.; Duan, Q.; Zhang, W. Bromodomain-containing protein 4 contributes to renal fibrosis through the induction of epithelial-mesenchymal transition. Exp. Cell Res. 2019, 383, 111507. [Google Scholar] [CrossRef]
- Xiong, C.; Masucci, M.V.; Zhou, X.; Liu, N.; Zang, X.; Tolbert, E.; Zhao, T.C.; Zhuang, S. Pharmacological targeting of BET proteins inhibits renal fibroblast activation and alleviates renal fibrosis. Oncotarget 2016, 7, 69291–69308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgado-Pascual, J.L.; Suarez-Alvarez, B.; Marchant, V.; Basantes, P.; Tharaux, P.L.; Ortiz, A.; Lopez-Larrea, C.; Ruiz-Ortega, M.; Rayego-Mateos, S. Type IV Collagen and SOX9 Are Molecular Targets of BET Inhibition in Experimental Glomerulosclerosis. Int. J. Mol. Sci. 2022, 24, 486. [Google Scholar] [CrossRef]
- Zhou, B.; Mu, J.; Gong, Y.; Lu, C.; Zhao, Y.; He, T.; Qin, Z. Brd4 inhibition attenuates unilateral ureteral obstruction-induced fibrosis by blocking TGF-beta-mediated Nox4 expression. Redox Biol. 2017, 11, 390–402. [Google Scholar] [CrossRef]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, L.M.; Kharenko, O.A.; Stotz, S.C.; Rakai, B.D.; Sarsons, C.D.; Gilham, D.; Wasiak, S.; Fu, L.; Sweeney, M.; Johansson, J.O.; et al. Breaking boundaries: Pan BETi disrupt 3D chromatin structure, BD2-selective BETi are strictly epigenetic transcriptional regulators. Biomed. Pharm. 2022, 152, 113230. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Schwartz, G.G.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 2021, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients With Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2020, 323, 1565–1573. [Google Scholar] [CrossRef]
- Jahagirdar, R.; Zhang, H.; Azhar, S.; Tobin, J.; Attwell, S.; Yu, R.; Wu, J.; McLure, K.G.; Hansen, H.C.; Wagner, G.S.; et al. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis 2014, 236, 91–100. [Google Scholar] [CrossRef]
- Kulikowski, E.; Halliday, C.; Johansson, J.; Sweeney, M.; Lebioda, K.; Wong, N.; Haarhaus, M.; Brandenburg, V.; Beddhu, S.; Tonelli, M.; et al. Apabetalone Mediated Epigenetic Modulation is Associated with Favorable Kidney Function and Alkaline Phosphatase Profile in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2018, 43, 449–457. [Google Scholar] [CrossRef]
- Wasiak, S.; Tsujikawa, L.M.; Halliday, C.; Stotz, S.C.; Gilham, D.; Jahagirdar, R.; Kalantar-Zadeh, K.; Robson, R.; Sweeney, M.; Johansson, J.O.; et al. Benefit of Apabetalone on Plasma Proteins in Renal Disease. Kidney Int. Rep. 2018, 3, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantar-Zadeh, K.; Schwartz, G.G.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kulikowski, E.; Lebioda, K.; Toth, P.P.; Wong, N.; et al. Effect of Apabetalone on Cardiovascular Events in Diabetes, CKD, and Recent Acute Coronary Syndrome: Results from the BETonMACE Randomized Controlled Trial. Clin. J. Am. Soc. Nephrol. 2021, 16, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Gilham, D.; Wasiak, S.; Tsujikawa, L.M.; Halliday, C.; Norek, K.; Patel, R.G.; Kulikowski, E.; Johansson, J.; Sweeney, M.; Wong, N.C. RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 2016, 247, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.F.; Liu, H.; Lv, L.L.; Ma, K.L.; Wen, Y.; Chen, L.; Liu, B.C. CCN3 suppresses TGF-beta1-induced extracellular matrix accumulation in human mesangial cells in vitro. Acta Pharmacol. Sin. 2018, 39, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhang, W.; Li, L.; Zheng, F.; Shao, X.; Zhou, J.; Li, H. Inhibitory effects of honokiol on lipopolysaccharide-induced cellular responses and signaling events in human renal mesangial cells. Eur. J. Pharmacol. 2011, 654, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Gilham, D.; Smith, A.L.; Fu, L.; Moore, D.Y.; Muralidharan, A.; Reid, S.P.M.; Stotz, S.C.; Johansson, J.O.; Sweeney, M.; Wong, N.C.W.; et al. Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-CoV-2 Infection In Vitro. Biomedicines 2021, 9, 437. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Horie, M.; Mihira, H.; Saito, A. Molecular Analysis of Endothelial-mesenchymal Transition Induced by Transforming Growth Factor-beta Signaling. J. Vis. Exp. JoVE 2018, 138, e57577. [Google Scholar]
- Fernley, H.N.; Walker, P.G. Kinetic behaviour of calf-intestinal alkaline phosphatase with 4-methylumbelliferyl phosphate. Biochem. J. 1965, 97, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haneda, M.; Koya, D.; Isono, M.; Kikkawa, R. Overview of glucose signaling in mesangial cells in diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2003, 14, 1374–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, M.; Maruyama, N.; Mitarai, T.; Nagasawa, R.; Yoshida, H.; Sakai, O. Extracellular matrix contraction by cultured mesangial cells: Modulation by transforming growth factor-beta and matrix components. Exp. Mol. Pathol. 1992, 56, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, C.; Takabatake, Y.; Mizui, M.; Isaka, Y.; Kawashi, H.; Rupprecht, H.; Imai, E.; Hugo, C. Antisense oligonucleotides against thrombospondin-1 inhibit activation of tgf-beta in fibrotic renal disease in the rat in vivo. Am. J. Pathol. 2003, 163, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Wiede, J.; Krutzsch, H.C.; Ribeiro, S.M.; Roberts, D.D.; Murphy-Ullrich, J.E.; Hugo, C. Thrombospondin-1 is a major activator of TGF-beta in fibrotic renal disease in the rat in vivo. Kidney Int. 2004, 65, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.H.; Yang, S.H.; Kim, Y.C.; Kim, J.H.; An, J.N.; Moon, K.C.; Oh, Y.K.; Park, J.Y.; Kim, D.K.; Kim, Y.S.; et al. Experimental Inhibition of Periostin Attenuates Kidney Fibrosis. Am. J. Nephrol. 2017, 46, 501–517. [Google Scholar] [CrossRef]
- Francki, A.; Bradshaw, A.D.; Bassuk, J.A.; Howe, C.C.; Couser, W.G.; Sage, E.H. SPARC regulates the expression of collagen type I and transforming growth factor-beta1 in mesangial cells. J. Biol. Chem. 1999, 274, 32145–32152. [Google Scholar] [CrossRef] [Green Version]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, E.Y.; Brezgunova, A.A.; Pevzner, I.B.; Zorova, L.D.; Manskikh, V.N.; Popkov, V.A.; Silachev, D.N.; Zorov, D.B. Mechanisms of LPS-Induced Acute Kidney Injury in Neonatal and Adult Rats. Antioxidants 2018, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shui, H.A.; Ka, S.M.; Wu, W.M.; Lin, Y.F.; Hou, Y.C.; Su, L.C.; Chen, A. LPS-evoked IL-18 expression in mesangial cells plays a role in accelerating lupus nephritis. Rheumatology 2007, 46, 1277–1284. [Google Scholar] [CrossRef] [Green Version]
- Haarhaus, M.; Brandenburg, V.; Kalantar-Zadeh, K.; Stenvinkel, P.; Magnusson, P. Alkaline phosphatase: A novel treatment target for cardiovascular disease in CKD, Nature reviews. Nephrology 2017, 13, 429–442. [Google Scholar]
- Cheung, K.L.; Kim, C.; Zhou, M.M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 728777. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Sheppard, D. Integrin-mediated regulation of TGFbeta in fibrosis. Biochim. Biophys. Acta 2013, 1832, 891–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.H. Mesangial Cells and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 165–194. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Y.; Liu, X.S.; Huang, X.R.; Yu, X.Q.; Lan, H.Y. Diverse Role of TGF-beta in Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef]
- Hohenstein, B.; Daniel, C.; Hausknecht, B.; Boehmer, K.; Riess, R.; Amann, K.U.; Hugo, C.P. Correlation of enhanced thrombospondin-1 expression, TGF-beta signalling and proteinuria in human type-2 diabetic nephropathy. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2008, 23, 3880–3887. [Google Scholar] [CrossRef] [Green Version]
- Wahab, N.A.; Schaefer, L.; Weston, B.S.; Yiannikouris, O.; Wright, A.; Babelova, A.; Schaefer, R.; Mason, R.M. Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia 2005, 48, 2650–2660. [Google Scholar] [CrossRef] [Green Version]
- Bige, N.; Shweke, N.; Benhassine, S.; Jouanneau, C.; Vandermeersch, S.; Dussaule, J.C.; Chatziantoniou, C.; Ronco, P.; Boffa, J.J. Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction. Kidney Int. 2012, 81, 1226–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-beta activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef]
- Bowers, S.L.K.; Davis-Rodriguez, S.; Thomas, Z.M.; Rudomanova, V.; Bacon, W.C.; Beiersdorfer, A.; Ma, Q.; Devarajan, P.; Blaxall, B.C. Inhibition of fibronectin polymerization alleviates kidney injury due to ischemia-reperfusion. Am. J. Physiol. Ren. Physiol. 2019, 316, F1293–F1298. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef] [PubMed]
- Turczyn, A.; Panczyk-Tomaszewska, M. The role of periostin in kidney diseases. Cent. Eur. J. Immunol. 2021, 46, 494–501. [Google Scholar] [CrossRef]
- Wallace, D.P. Periostin in the Kidney. Adv. Exp. Med. Biol. 2019, 1132, 99–112. [Google Scholar] [CrossRef]
- Mael-Ainin, M.; Abed, A.; Conway, S.J.; Dussaule, J.C.; Chatziantoniou, C. Inhibition of periostin expression protects against the development of renal inflammation and fibrosis. J. Am. Soc. Nephrol. JASN 2014, 25, 1724–1736. [Google Scholar] [CrossRef] [Green Version]
- Um, J.E.; Park, J.T.; Nam, B.Y.; Lee, J.P.; Jung, J.H.; Kim, Y.; Kim, S.; Park, J.; Wu, M.; Han, S.H.; et al. Periostin-binding DNA aptamer treatment attenuates renal fibrosis under diabetic conditions. Sci. Rep. 2017, 7, 8490. [Google Scholar] [CrossRef] [Green Version]
- Taneda, S.; Pippin, J.W.; Sage, E.H.; Hudkins, K.L.; Takeuchi, Y.; Couser, W.G.; Alpers, C.E. Amelioration of diabetic nephropathy in SPARC-null mice. J. Am. Soc. Nephrol. JASN 2003, 14, 968–980. [Google Scholar] [CrossRef] [Green Version]
- Kiberd, B.A. Interleukin-6 receptor blockage ameliorates murine lupus nephritis. J. Am. Soc. Nephrol. JASN 1993, 4, 58–61. [Google Scholar] [CrossRef]
- Yamazoe, M.; Mizuno, A.; Nishi, Y.; Niwa, K.; Isobe, M. Serum alkaline phosphatase as a predictor of worsening renal function in patients with acute decompensated heart failure. J. Cardiol. 2016, 67, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Haarhaus, M.; Ray, K.K.; Nicholls, S.J.; Schwartz, G.G.; Kulikowski, E.; Johansson, J.O.; Sweeney, M.; Halliday, C.; Lebioda, K.; Wong, N.; et al. Apabetalone lowers serum alkaline phosphatase and improves cardiovascular risk in patients with cardiovascular disease. Atherosclerosis 2019, 290, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, L.; Wen, X.; Gao, L.; Li, G.; Chang, G.; Qin, S.; Zhang, D. TNAP is a novel regulator of cardiac fibrosis after myocardial infarction by mediating TGF-beta/Smads and ERK1/2 signaling pathways. EBioMedicine 2021, 67, 103370. [Google Scholar] [CrossRef]
- Haarhaus, M.; Gilham, D.; Kulikowski, E.; Magnusson, P.; Kalantar-Zadeh, K. Pharmacologic epigenetic modulators of alkaline phosphatase in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 4–15. [Google Scholar] [CrossRef]
- Zhang, Y.; Reif, G.; Wallace, D.P. Extracellular matrix, integrins, and focal adhesion signaling in polycystic kidney disease. Cell Signal 2020, 72, 109646. [Google Scholar] [CrossRef] [PubMed]
- Eddington, H.; Sinha, S.; Kalra, P.A. Vascular calcification in chronic kidney disease: A clinical review. J. Ren. Care 2009, 35, 45–50. [Google Scholar] [CrossRef]
- Imig, J.D.; Ryan, M.J. Immune and inflammatory role in renal disease. Compr. Physiol. 2013, 3, 957–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 ng/mL TGF-β1 | |||||
|---|---|---|---|---|---|
| 0.05% DMSO | 5 µM Apa | 25 µM Apa | 0.15 µM JQ1 | 0.1 µM MZ1 | |
| # DEGs in “ECM Organization” Gene Set | 143 | 92 | 151 | 162 | 164 |
| # Genes Upregulated | 84 | 33 | 47 | 52 | 54 |
| % Upregulated | 59% | 36% | 31% | 32% | 33% |
| # Genes Downregulated | 59 | 59 | 104 | 110 | 110 |
| % Downregulated | 41% | 64% | 69% | 68% | 67% |
| TGF-β1 IPA Upstream Regulator Analysis | |||||
|---|---|---|---|---|---|
| Pre-dose: | Time post-apabetalone dose | ||||
| 6 h | 12 h | 24 h | |||
| CKD vs. Controls | 2.01 Activated | CKD vs. pre-dose | 1.15 | −2.66 | −3.49 |
| No prediction | Inhibited | Inhibited | |||
| Controls vs. pre-dose | −0.35 | 0.88 | −0.44 | ||
| No prediction | No prediction | No prediction | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilham, D.; Wasiak, S.; Rakai, B.D.; Fu, L.; Tsujikawa, L.M.; Sarsons, C.D.; Carestia, A.; Lebioda, K.; Johansson, J.O.; Sweeney, M.; et al. Apabetalone Downregulates Fibrotic, Inflammatory and Calcific Processes in Renal Mesangial Cells and Patients with Renal Impairment. Biomedicines 2023, 11, 1663. https://doi.org/10.3390/biomedicines11061663
Gilham D, Wasiak S, Rakai BD, Fu L, Tsujikawa LM, Sarsons CD, Carestia A, Lebioda K, Johansson JO, Sweeney M, et al. Apabetalone Downregulates Fibrotic, Inflammatory and Calcific Processes in Renal Mesangial Cells and Patients with Renal Impairment. Biomedicines. 2023; 11(6):1663. https://doi.org/10.3390/biomedicines11061663
Chicago/Turabian StyleGilham, Dean, Sylwia Wasiak, Brooke D. Rakai, Li Fu, Laura M. Tsujikawa, Christopher D. Sarsons, Agostina Carestia, Kenneth Lebioda, Jan O. Johansson, Michael Sweeney, and et al. 2023. "Apabetalone Downregulates Fibrotic, Inflammatory and Calcific Processes in Renal Mesangial Cells and Patients with Renal Impairment" Biomedicines 11, no. 6: 1663. https://doi.org/10.3390/biomedicines11061663
APA StyleGilham, D., Wasiak, S., Rakai, B. D., Fu, L., Tsujikawa, L. M., Sarsons, C. D., Carestia, A., Lebioda, K., Johansson, J. O., Sweeney, M., Kalantar-Zadeh, K., & Kulikowski, E. (2023). Apabetalone Downregulates Fibrotic, Inflammatory and Calcific Processes in Renal Mesangial Cells and Patients with Renal Impairment. Biomedicines, 11(6), 1663. https://doi.org/10.3390/biomedicines11061663