
Current Therapies for Cholestatic Diseases



{kind=link}
{kind=link}
Abstract
:1. Introduction
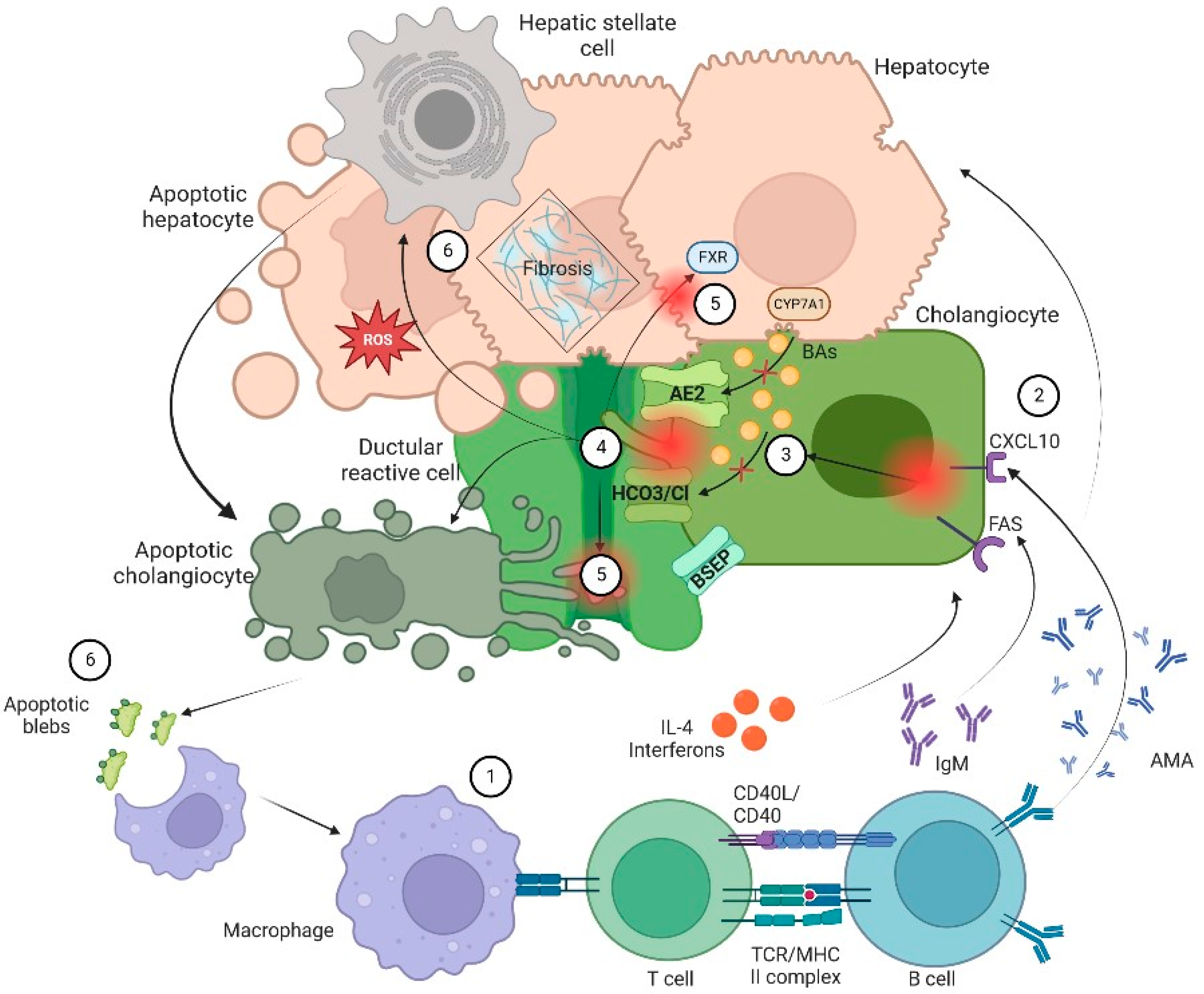
2. Pathophysiology of Cholangiopathies
2.1. Cholangiocytes Biology
2.2. Ductular Reaction
2.3. Biliary Stasis
2.4. Citotoxic Profile of Biliary Acids
2.5. Profibrotic State
3. Therapeutic Options
3.1. Hydrophilic Bile Acids (BA): UDCA and Nor-UDCA
3.2. FXR Agonists: Obeticholic Acid, Tropifexor (LJN-452), Cilofexor (GS-9674) and EDP-305
3.3. Fibroblast Growth Factor 19 (FGF-19) Analogs
3.4. PPAR Agonists: Fibrates, Seladelpar, Elafibranor
3.5. ASBT Inhibitors
3.6. Immune-Modulation Drugs: Corticosteroids and Biological Therapies
3.7. Other Therapies: Bile Acid Sequestrant and Antibiotics
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sanjel, B.; Shim, W.S. Recent advances in understanding the molecular mechanisms of cholestatic pruritus: A review. Biochim. Biophys. Acta Mol. Basis 2020, 1866, 165958. [Google Scholar] [CrossRef] [PubMed]
- Gazda, J.; Drazilova, S.; Janicko, M.; Jarcuska, P. The Epidemiology of Primary Biliary Cholangitis in European Countries: A Systematic Review and Meta-Analysis. Can. J. Gastroenterol. Hepatol. 2021, 2021, 9151525. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Ali, A.H.; Lindor, K.D. Primary Sclerosing Cholangitis, Part 1: Epidemiology, Etiopathogenesis, Clinical Features, and Treatment. Gastroenterol. Hepatol. 2018, 14, 293–304. [Google Scholar]
- Malik, A.; Kardashian, A.A.; Zakharia, K.; Bowlus, C.L.; Tabibian, J.H. Preventative care in cholestatic liver disease: Pearls for the specialist and subspecialist. Liver Res. 2019, 3, 118–127. [Google Scholar] [CrossRef]
- Gerussi, A.; Restelli, U.; Croce, D.; Bonfanti, M.; Invernizzi, P.; Carbone, M. Cost of illness of Primary Biliary Cholangitis—A population-based study. Dig. Liver Dis. 2021, 53, 1167–1170. [Google Scholar] [CrossRef]
- Yokoda, R.T.; Rodriguez, E.A. Review: Pathogenesis of cholestatic liver diseases. World J. Hepatol. 2020, 12, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Sánchez, N. Bile Acids in Health and Disease Foreword. Ann. Hepatol. 2017, 16 (Suppl. S1), s3. [Google Scholar] [CrossRef] [PubMed]
- Desmet, V.J. Ductal plates in hepatic ductular reactions. Hypothesis and implications. I. Types of ductular reaction reconsidered. Virchows Arch. 2011, 458, 251–259. [Google Scholar] [CrossRef]
- Goldstein, J.; Levy, C. Novel and emerging therapies for cholestatic liver diseases. Liver Int. 2018, 38, 1520–1535. [Google Scholar] [CrossRef] [Green Version]
- Hirschfield, G.M.; Chazouillères, O.; Drenth, J.P.; Thorburn, D.; Harrison, S.A.; Landis, C.S.; Mayo, M.J.; Muir, A.J.; Trotter, J.F.; Leeming, D.J.; et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase 2 trial. J. Hepatol. 2019, 70, 483–493. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N. Management of primary biliary cholangitis: The importance to identify patients’ non-responders to standard treatment. Minerva Med. 2018, 109, 407–409. [Google Scholar] [CrossRef]
- Cabrera, D.; Arab, J.P.; Arrese, M. UDCA, NorUDCA, and TUDCA in Liver Diseases: A Review of Their Mechanisms of Action and Clinical Applications. Handb. Exp. Pharmacol. 2019, 256, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Fiorotto, R.; Spirli, C.; Cadamuro, M.; Mariotti, V.; Perugorria, M.J.; Banales, J.M.; Strazzabosco, M. Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Hartl, L.; Haslinger, K.; Angerer, M.; Semmler, G.; Schneeweiss-Gleixner, M.; Jachs, M.; Simbrunner, B.; Bauer, D.J.M.; Eigenbauer, E.; Strassl, R.; et al. Progressive cholestasis and associated sclerosing cholangitis are frequent complications of COVID-19 in patients with chronic liver disease. Hepatology 2022, 76, 1563–1575. [Google Scholar] [CrossRef]
- Beuers, U.; Spengler, U.; Kruis, W.; Aydemir, U.; Wiebecke, B.; Heldwein, W.; Weinzierl, M.; Pape, G.R.; Sauerbruch, T.; Paumgartner, G. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: A placebo-controlled trial. Hepatology 1992, 16, 707–714. [Google Scholar] [CrossRef]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62 (Suppl. S1), S25–S37. [Google Scholar] [CrossRef] [Green Version]
- Poupon, R.; Chrétien, Y.; Poupon, R.E.; Ballet, F.; Calmus, Y.; Darnis, F. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet 1987, 1, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Poupon, R.E.; Balkau, B.; Eschwège, E.; Poupon, R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA-PBC Study Group. N. Engl. J. Med. 1991, 324, 1548–1554. [Google Scholar] [CrossRef]
- Poupon, R.E.; Balkau, B.; Guéchot, J.; Heintzmann, F. Predictive factors in ursodeoxycholic acid-treated patients with primary biliary cirrhosis: Role of serum markers of connective tissue. Hepatology 1994, 19, 635–640. [Google Scholar] [CrossRef]
- Poupon, R.E.; Bonnand, A.M.; Chrétien, Y.; Poupon, R. Ten-year survival in ursodeoxycholic acid-treated patients with primary biliary cirrhosis. UDCA-PBC Study Group Hepatol. 1999, 29, 1668–1671. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Shi, J.; Li, Z.; Zeng, X.; Lin, Y.; Xie, W.F. Ursodeoxycholic acid in primary sclerosing cholangitis: Meta-analysis of randomized controlled trials. Hepatol. Res. 2009, 39, 865–873. [Google Scholar] [CrossRef]
- Triantos, C.K.; Koukias, N.M.; Nikolopoulou, V.N.; Burroughs, A.K. Meta-analysis: Ursodeoxycholic acid for primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2011, 34, 901–910. [Google Scholar] [CrossRef]
- Olsson, R.; Boberg, K.M.; de Muckadell, O.S.; Lindgren, S.; Hultcrantz, R.; Folvik, G.; Bell, H.; Gangsøy-Kristiansen, M.; Matre, J.; Rydning, A.; et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: A 5-year multicenter, randomized, controlled study. Gastroenterology 2005, 129, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; Jorgensen, R.; Petz, J.; Keach, J.; et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009, 50, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Black, D.D.; Mack, C.; Kerkar, N.; Miloh, T.; Sundaram, S.S.; Anand, R.; Gupta, A.; Alonso, E.; Arnon, R.; Bulut, P.; et al. A Prospective Trial of Withdrawal and Reinstitution of Ursodeoxycholic Acid in Pediatric Primary Sclerosing Cholangitis. Hepatol. Commun. 2019, 3, 1482–1495. [Google Scholar] [CrossRef] [Green Version]
- Pardi, D.S.; Loftus, E.V., Jr.; Kremers, W.K.; Keach, J.; Lindor, K.D. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 2003, 124, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Tung, B.Y.; Emond, M.J.; Haggitt, R.C.; Bronner, M.P.; Kimmey, M.B.; Kowdley, K.V.; Brentnall, T.A. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann. Intern. Med. 2001, 134, 89–95. [Google Scholar] [CrossRef]
- Lindström, L.; Boberg, K.M.; Wikman, O.; Friis-Liby, I.; Hultcrantz, R.; Prytz, H.; Sandberg-Gertzén, H.; Sangfelt, P.; Rydning, A.; Folvik, G.; et al. High dose ursodeoxycholic acid in primary sclerosing cholangitis does not prevent colorectal neoplasia. Aliment. Pharmacol. Ther. 2012, 35, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, J.E.; Silveira, M.G.; Pardi, D.S.; Sinakosm, E.; Kowdley, K.V.; Luketic, V.A.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am. J. Gastroenterol. 2011, 106, 1638–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Khanna, S.; Pardi, D.S.; Loftus, E.V., Jr.; Talwalkar, J.A. Effect of ursodeoxycholic acid use on the risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: A systematic review and meta-analysis. Inflamm. Bowel Dis. 2013, 19, 1631–1638. [Google Scholar] [CrossRef]
- Hansen, J.D.; Kumar, S.; Lo, W.K.; Poulsen, D.M.; Halai, U.A.; Tater, K.C. Ursodiol and colorectal cancer or dysplasia risk in primary sclerosing cholangitis and inflammatory bowel disease: A meta-analysis. Dig. Dis. Sci. 2013, 58, 3079–3087. [Google Scholar] [CrossRef]
- Bowlus, C.L.; Arrivé, L.; Bergquist, A.; Deneau, M.; Forman, L.; Ilyas, S.I.; Lunsford, K.E.; Martinez, M.; Sapisochin, G.; Shroff, R. AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 2023, 77, 659–702. [Google Scholar] [CrossRef]
- Fickert, P.; Hirschfield, G.M.; Denk, G.; Marschall, H.U.; Altorjay, I.; Färkkilä, M.; Schramm, C.; Spengler, U.; Chapman, R.; Bergquist, A. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J. Hepatol. 2017, 67, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Floreani, A.; Gabbia, D.; de Martin, S. Update on the Pharmacological Treatment of Primary Biliary Cholangitis. Biomedicines 2022, 10, 2033. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Dunkelberg, J.; Roy, P.K. Obeticholic acid for the treatment of primary sclerosing cholangitis: A systematic review and meta-analysis. Arab. J. Gastroenterol. 2012, 13, 103–110. [Google Scholar] [CrossRef]
- Schramm, C.; Wedemeyer, H.; Mason, A.; Hirschfield, G.M.; Levy, C.; Kowdley, K.V.; Milkiewicz, P.; Janczewska, E.; Malova, E.S.; Sanni, J.; et al. Farnesoid X receptor agonist tropifexor attenuates cholestasis in a randomised trial in patients with primary biliary cholangitis. JHEP Rep. 2022, 4, 100544. [Google Scholar] [CrossRef] [PubMed]
- Schramm, C.; Hirschfield, G.M.; Mason, A.; Wedemeyer, H.; Klickstein, L.; Neelakantham, S.; Koo, P.; Sanni, J.; Badman, M.; Jones, D. Early assessment of safety and efficacy of tropifexor, a potent non bile-acid FXR agonist, in patients with primary biliary cholangitis: An interim analysis of an ongoing phase 2 study. J. Hepatol. 2018, 68, S103. [Google Scholar] [CrossRef]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients with Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M.; Bowlus, C.L.; Gulamhusein, A.; Hameed, B.; Caldwell, S.H.; Shiffman, M.L.; Landis, C.; Muir, A.J.; Billin, A.; Xu, J.; et al. Safety and sustained efficacy of the farnesoid X receptor (FXR) agonist cilofexor over a 96-week open-label extension in patients with PSC. Clin. Gastroenterol. Hepatol. 2023, 21, 1552–1560.e2. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Bonder, A.; Heneghan, M.A.; Hodge, A.D.; Ryder, S.D.; Sanchez, A.J.; Vargas, V.; Zeuzem, S.; Ahmad, A.; Larson, K.; et al. Final data of the phase 2a intrepid study with EDP-305, a non-bile acid farnesoid x receptor (FXR) agonist. Hepatology 2020, 72 (Suppl. S1), 746A–747A. [Google Scholar] [CrossRef]
- Mayo, M.J.; Wigg, A.J.; Leggett, B.A.; Arnold, H.; Thompson, A.J.; Weltman, M.; Carey, E.J.; Muir, A.J.; Ling, L.; Rossi, S.J.; et al. NGM282 for Treatment of Patients with Primary Biliary Cholangitis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Hepatol. Commun. 2018, 2, 1037–1050. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients with Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231.e1. [Google Scholar] [CrossRef] [PubMed]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [Green Version]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Ghonem, N.S.; Assis, D.N.; Boyer, J.L. On Fibrates and Cholestasis: A review. Hepatology 2015, 62, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, R.; Takamatsu, S.; Kimura, T.; Kokuryu, H.; Osaki, Y.; Tomono, N. Bezafibrate may attenuate biliary damage associated with chronic liver diseases accompanied by high serum biliary enzyme levels. J. Gastroenterol. 2006, 41, 686–692. [Google Scholar] [CrossRef]
- Hazzan, R.; Tur-Kaspa, R. Bezafibrate treatment of primary biliary cirrhosis following incomplete response to ursodeoxycholic acid. J. Clin. Gastroenterol. 2010, 44, 371–373. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Ikeda, F.; Fujioka, S.; Takaki, T.; Osawa, T.; Yasunaka, T.; Miyake, Y.; Takaki, A.; Iwasaki, Y.; Kobashi, H.; et al. Additive improvement induced by bezafibrate in patients with primary biliary cirrhosis showing refractory response to ursodeoxycholic acid. J. Gastroenterol. Hepatol. 2011, 26, 1395–1401. [Google Scholar] [CrossRef]
- Iwasaki, S.; Ohira, H.; Nishiguchi, S.; Zeniya, M.; Kaneko, S.; Onji, M.; Ishibashi, H.; Sakaida, I.; Kuriyama, S.; Ichida, T.; et al. The efficacy of ursodeoxycholic acid and bezafibrate combination therapy for primary biliary cirrhosis: A prospective, multicenter study. Hepatol. Res. 2008, 38, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Ohmoto, K.; Yoshioka, N.; Yamamoto, S. Long-term effect of bezafibrate on parameters of hepatic fibrosis in primary biliary cirrhosis. J. Gastroenterol. 2006, 41, 502–503. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Sato, Y.; Ueno, T.; Sata, M. Fenofibrate treatment in patients with primary biliary cirrhosis. Am. J. Gastroenterol. 2002, 97, 2147–2149. [Google Scholar] [CrossRef]
- Dohmen, K.; Mizuta, T.; Nakamuta, M.; Shimohashi, N.; Ishibashi, H.; Yamamoto, K. Fenofibrate for patients with asymptomatic primary biliary cirrhosis. World J. Gastroenterol. 2004, 10, 894–898. [Google Scholar] [CrossRef] [Green Version]
- Levy, C.; Peter, J.A.; Nelson, D.R.; Keach, J.; Petz, J.; Cabrera, R.; Clark, V.; Firpi, R.J.; Morelli, G.; Soldevila-Pico, C.; et al. Pilot study: Fenofibrate for patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Aliment. Pharmacol. Ther. 2011, 33, 235–242. [Google Scholar] [CrossRef]
- Han, X.F.; Wang, Q.X.; Liu, Y.; You, Z.R.; Bian, Z.L.; Qiu, D.K.; Ma, X. Efficacy of fenofibrate in Chinese patients with primary biliary cirrhosis partially responding to ursodeoxycholic acid therapy. J. Dig. Dis. 2012, 13, 219–224. [Google Scholar] [CrossRef]
- Liberopoulos, E.N.; Florentin, M.; Elisaf, M.S.; Mikhailidis, D.; Tsianos, E. Fenofibrate in primary biliary cirrhosis: A pilot study. Open Cardiovasc. Med. J. 2010, 4, 120–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, S.; Feng, Y.; Zhang, Q.; Xie, B. Efficacy of fibrates in the treatment of primary biliary cholangitis: A meta-analysis. Clin. Exp. Med. 2022. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; He, L.; Wang, F.; Chen, K.; Li, J.; Liu, T.; Zheng, Y.; Lu, W.; Zhou, Y.; et al. Combination therapy of fenofibrate and ursodeoxycholic acid in patients with pri mary biliary cirrhosis who respond incompletely to UDCA monotherapy: A meta-analysis. Drug Des. Devel Ther. 2015, 9, 2757–2766. [Google Scholar] [CrossRef] [Green Version]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Tanaka, A.; Hirohara, J.; Nakano, T.; Matsumoto, K.; Chazouillères, O.; Takikawa, H.; Hansen, B.E.; Carrat, F.; Corpechot, C. Association of bezafibrate with transplant-free survival in patients with primary biliary cholangitis. J. Hepatol. 2021, 75, 565–571. [Google Scholar] [CrossRef]
- Jones, D.; Boudes, P.F.; Sawin, M.G.; Bowlus, C.L.; Galambos, M.R.; Bacon, B.R.; Doerffel, Y.; Gitlin, N.; Gordon, S.C.; Odin, J.A.; et al. Seladelpar (MBX-8025), a selective PPAR-δ agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: A double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol. Hepatol. 2017, 2, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Bowlus, C.L.; Galambos, M.R.; Aspinall, R.J.; Hirschfield, G.M.; Jones, D.E.J.; Dörffel, Y.; Gordon, S.C.; Harrison, S.A.; Kremer, A.E.; Mayo, M.J.; et al. A phase 2, randomized, open-label, 52-week study of seladelpar in patients with primary biliary cholangitis. J. Hepatol. 2022, 77, 353–364. [Google Scholar] [CrossRef]
- Kremer, A.E.; Mayo, M.J.; Hirschfield, G.; Levy, C.; Bowlus, C.L.; Jones, D.E.; Steinberg, A.; McWherter, C.A.; Choi, Y.J. Seladelpar improved measures of pruritus, sleep, and fatigue and decreased serum bile acids in patients with primary biliary cholangitis. Liver Int. 2022, 42, 112–123. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, X.; Meng, Z.; Dong, B.; Shiah, S.; Moore, D.D.; Huang, W. Significance and mechanism of CYP7a1 gene regulation during the acute phase of liver regeneration. Mol. Endocrinol. 2009, 23, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colapietro, F.; Gershwin, M.E.; Lleo, A. PPAR agonists for the treatment of primary biliary cholangitis: Old and new tales. J. Transl. Autoimmun. 2023, 6, 100188. [Google Scholar] [CrossRef] [PubMed]
- Westerouen Van Meeteren, M.J.; Drenth, J.P.H.; Tjwa, E.T.T.L. Elafibranor: A potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Pares, A.; Kowdley, K.V.; Heneghan, M.A.; Caldwell, S.A.; Pratt, D.; Bonder, A.; Hirschfield, G.M.; Levy, C.; Vierling, J.; et al. randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J. Hepatol. 2021, 74, 1344–1354. [Google Scholar] [CrossRef]
- Tao, L.; Ren, X.; Zhai, W.; Chen, Z. Progress and Prospects of Non-Canonical NF-κB Signaling Pathway in the Regulation of Liver Diseases. Molecules 2022, 27, 4275. [Google Scholar] [CrossRef]
- Li, F.; Patterson, A.D.; Krausz, K.W.; Jiang, C.; Bi, H.; Sowers, A.L.; Cook, J.A.; Mitchell, J.B.; Gonzalez, F.J. Metabolomics reveals an essential role for peroxisome proliferator-activated receptor α in bile acid homeostasis. J. Lipid Res. 2012, 53, 1625–1635. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhang, T.; Han, H. PPARα: A potential therapeutic target of cholestasis. Front. Pharmacol. 2022, 13, 916866. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Dong, Y.Q.; Jia, G.X.; Fan, S.M.; Li, S.Z.; Yang, S.S.; Li, Y.B. ASBT(SLC10A2): A promising target for treatment of diseases and drug discovery. Biomed. Pharmacother. 2020, 132, 110835. [Google Scholar] [CrossRef]
- Salic, K.; Kleemann, R.; Wilkins-Port, C.; McNulty, J.; Verschuren, L.; Palmer, M. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of non-alcoholic steatohepatitis in Ldlr-/-.Leiden mice. PLoS ONE 2019, 14, e0218459. [Google Scholar] [CrossRef]
- Kunst, R.F.; de Waart, D.R.; Wolters, F.; Duijst, S.; Vogels, E.W.; Bolt, I.; Verheij, J.; Beuers, U.; Elferink, R.P.O.; van de Graaf, S.F. Systemic ASBT inactivation protects against liver damage in obstructive cholestasis in mice. JHEP Rep. 2022, 4, 100573. [Google Scholar] [CrossRef]
- Gonzales, E.; Hardikar, W.; Stormon, M.; Baker, A.; Hierro, L.; Gliwicz, D.; Lacaille, F.; Lachaux, A.; Sturm, E.; Setchell, K.D.R.; et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): A randomised phase 2 study. Lancet 2021, 398, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Hegade, V.S.; Kendrick, S.F.; Dobbins, R.L.; Miller, S.R.; Thompson, D.; Richards, D.; Storey, J.; Dukes, G.E.; Corrigan, M.; Elferink, R.P.O.; et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: A double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet 2017, 389, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Ino, H.; Endo, A.; Wakamatsu, A.; Ogura, H.; Numachi, Y.; Kendrick, S. Safety, Tolerability, Pharmacokinetic and Pharmacodynamic Evaluations Following Single Oral Doses of GSK2330672 in Healthy Japanese Volunteer. Clin. Pharmacol. Drug. Dev. 2019, 8, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Levy, C.; Kendrick, S.; Bowlus, C.L.; Tanaka, A.; Jones, D.; Kremer, A.E.; Mayo, M.J.; Haque, N.; von Maltzahn, R.; Allinder, M.; et al. GLIMMER: A Randomized Phase 2b Dose-Ranging Trial of Linerixibat in Primary Biliary Cholangitis Patients with Pruritus. Clin. Gastroenterol. Hepatol. 2022, 4, S1542-3565(22)01021-7. [Google Scholar] [CrossRef]
- Thompson, R.; Arnell, H.; Artan, R.; Baumann, U.; Calvo, P.L.C.; Czubkowski, P.; Dalgic, B.; D’Antiga, L.; Durmaz, Ö.; Fischler, B.; et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: A randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol. Hepatol. 2022, 7, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.M. Odevixibat: First Approval. Drugs 2021, 81, 1781–1786. [Google Scholar] [CrossRef]
- Blendis, L. Steroids in the management of PBC: Why do we need them? Gastroenterology 2005, 129, 1350–1352. [Google Scholar] [CrossRef] [PubMed]
- Al-Aqil, F.A.; Monte, M.J.; Peleteiro-Vigil, A.; . Briz, O.; Rosales, R.; González, R.; Aranda, C.J.; Ocón, B.; Uriarte, I.; de Medina, F.S.; et al. Interaction of glucocorticoids with FXR/FGF19/FGF21-mediated ileum-liver crosstalk. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2927–2937. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, J.; Dai, W.; Wang, F.; Shen, M.; Yang, J.; Zhu, R.; Zhang, H.; Chen, K.; Cheng, P.; et al. Combination Therapy of Ursodeoxycholic Acid and Corticosteroids for Primary Biliary Cirrhosis with Features of Autoimmune Hepatitis: A Meta-Analysis. Gastroenterol. Res. Pract. 2013, 2013, 490731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, S.; Yang, J.; Zheng, Y.; Wang, J.; Lu, W.; Zhou, Y.; Yin, Q.; Zhu, R.; Guo, C. A meta-analysis of ursodeoxycholic acid therapy versus combination therapy with corticosteroids for PBC-AIH-overlap syndrome: Evidence from 97 monotherapy and 117 combinations. Prz. Gastroenterol. 2015, 10, 148–155. [Google Scholar] [CrossRef]
- Silveira, M.G.; Lindor, K.D. Obeticholic acid and budesonide for the treatment of primary biliary cirrhosis. Expert. Opin. Pharmacother. 2014, 15, 365–372. [Google Scholar] [CrossRef]
- Hempfling, W.; Grunhage, F.; Dilger, K.; Reichel, C.; Beuers, U.; Sauerbruch, T. Pharmacokinetics and pharmacodynamic action of budesonide in early and late-stage primary biliary cirrhosis. Hepatology 2003, 38, 196–202. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Beuers, U.; Kupcinskas, L.; Ott, P.; Bergquist, A.; Färkkilä, M.; Manns, M.P.; Parés, A.; Spengler, U.; Stiess, M.; et al. A placebo-controlled randomised trial of budesonide for PBC following an insufficient response to UDCA. J. Hepatol. 2021, 74, 321–329. [Google Scholar] [CrossRef]
- Geier, A.; Gartung, C.; Dietrich, C.G.; Wasmuth, H.E.; Reinartz, P.; Matern, S. Side effects of budesonide in liver cirrhosis due to chronic autoimmune hepatitis: Influence of hepatic metabolism versus portosystemic shunts on a patients complicated with HCC. World J. Gastroenterol. 2003, 9, 2681–2685. [Google Scholar] [CrossRef]
- Myers, R.P.; Swain, M.G.; Lee, S.S.; Shaheen, A.A.; Burak, K.W. B-cell depletion with rituximab in patients with primary biliary cirrhosis refractory to ursodeoxycholic acid. Am. J. Gastroenterol. 2013, 108, 933–941. [Google Scholar] [CrossRef]
- Tsuda, M.; Moritoki, Y.; Lian, Z.X.; Zhang, W.; Yoshida, K.; Wakabayashi, K.; Yang, G.X.; Nakatani, T.; Vierling, J.; Lindor, K.; et al. Biochemical and immunologic effects of rituximab in patients with primary biliary cirrhosis and incomplete response to ursodeoxycholic acid. Hepatology 2012, 55, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.A.; Topazian, M.D.; Witzig, T.E.; Clain, J.E.; Gleeson, F.C.; Klebig, R.R.; Levy, M.J.; Pearson, R.K.; Petersen, B.T.; Smyrk, T.C.; et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: The Mayo Clinic experience. Gut 2012, 62, 1607–1615. [Google Scholar] [CrossRef]
- Yamada, Y.; Hoshino, K.; Fuchimoto, Y.; Matsubara, K.; Hibi, T.; Yagi, H.; Abe, Y.; Shinoda, M.; Kitago, M.; Obara, H.; et al. Rituximab induction to prevent the recurrence of PSC after liver transplantation—The lessons learned from ABO-Incompatible living donor liver transplantation. Transplant. Direct. 2018, 4, e342. [Google Scholar] [CrossRef] [PubMed]
- Bowlus, C.L.; Yang, G.-X.; Liu, C.H.; Johnson, C.R.; Dhaliwal, S.S.; Frank, D.; Levy, C.; Peters, M.G.; Vierling, J.M.; Gershwin, M.E. Therapeutic trials of biologics in primary biliary cholangitis: An open label study of abatacept and review of the literature. J. Autoimmun. 2019, 101, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.C.; Trudeau, S.; Regev, A.; Uhas, J.M.; Chakladar, S.; Pinto-Correia, A.; Gottlieb, K.; Schlichting, D. Baricitinib and primary biliary cholangitis. J. Transl. Autoimmun. 2021, 4, 100107. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Gershwin, M.E.; Strauss, R.; Mayo, M.J.; Levy, C.; Zou, B.; Johanns, J.; Nnane, I.P.; Dasgupta, B.; Li, K.; et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: A proof-of-concept study. Hepatology 2016, 64, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.D.; Chapman, R.W.; Keshav, S.; Montano-Loza, A.L.; Mason, A.L.; Kremer, A.E.; Vetter, M.; de Krijger, M.; Ponsioen, C.Y.; Trivedi, P.; et al. Effects of vedolizumab in patients with primary sclerosing cholangitis and inflammatory bowel diseases. Clin. Gastroenterol. Hepatol. 2019, 18, 179–187.e6. [Google Scholar] [CrossRef] [Green Version]
- De Graaf, K.L.; Lapeyre, G.; Guilhot, F.; Ferlin, W.; Curbishley, S.M.; Carbone, M.; Richardson, P.; Moreea, S.; McCune, C.A.; Ryder, S.D.; et al. NI-0801, an anti-chemokine (C-X-C motif) ligand 10 antibody, in patients with primary biliary cholangitis and an incomplete response to ursodeoxycholic acid. Hepatol. Commun. 2018, 2, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Rowe, B.H.; Vandermeer, B.; Bain, V.G. The Efficacy and Safety of Bile Acid Binding Agents, Opioid Antagonists, or Rifampin in the Treatment of Cholestasis-Associated Pruritus. Am. J. Gastroenterol. 2007, 102, 1528–1536. [Google Scholar] [CrossRef]
- Kuiper, E.M.; van Erpecum, K.J.; Beuers, U.; Hansen, B.E.; Thio, H.B.; de Man, R.A.; Janssen, H.L.; van Buuren, H.R. The potent bile acid sequestrant colesevelam is not effective in cholestatic pruritus: Results of a double-blind, randomized, placebo-controlled trial. Hepatology 2010, 52, 1334–1340. [Google Scholar] [CrossRef]
- Shah, A.; Crawford, D.; Burger, D.; Martin, N.; Walker, M.; Talley, N.J.; Tallis, C.; Jones, M.; Stuart, K.; Keely, S.; et al. Effects of Antibiotic Therapy in Primary Sclerosing Cholangitis with and without Inflammatory Bowe Disease: A Systematic Review and Meta-Analysis. Semin. Liver Dis. 2019, 39, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Elfaki, D.A.; Lindor, K.D. Antibiotics for the Treatment of Primary Sclerosing Cholangitis. Am. J. Ther. 2011, 18, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Damman, J.L.; Rodriguez, E.A.; Ali, A.H.; Buness, C.W.; Cox, K.L.; Carey, E.J.; Lindor, K.D. Review article: The evidence that vancomycin is a therapeutic option for primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2018, 47, 886–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abarbanel, D.N.; Seki, S.M.; Davies, Y.; Marlen, N.; Benavides, J.A.; Cox, K.; Nadeau, K.C.; Cox, K.L. Immunomodulatory effect of vancomycin on Treg in pediatric inflammatory bowel disease and primary sclerosing cholangitis. J. Clin. Immunol. 2013, 33, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, Y.K.; Tsay, C.J.; Caccamo, D.V.; Cox, K.M.; Castillo, R.O.; Cox, K.L. Successful treatment of recurrent primary sclerosing cholangitis after orthotopic liver transplantation with oral vancomycin. Case Rep. Transplant. 2013, 2013, 314292. [Google Scholar] [CrossRef] [Green Version]
- Khurana, S.; Singh, P. Rifampin is safe for treatment of pruritus due to chronic cholestasis: A meta-analysis of prospective randomized-controlled trials. Liver Int. 2006, 26, 943–948. [Google Scholar] [CrossRef]
- Hague, W.M.; Callaway, L.; Chambers, J.; Chappel, L.; Coat, S.; de Haan-Jebbink, J.; Dekker, M.A.; Dixon, P.; Dodd, J.; Fuller, M.; et al. A multi-centre, open label, randomised, parallel-group, superiority Trial to compare the efficacy of URsodeoxycholic acid with RIFampicin in the management of women with severe early onset Intrahepatic Cholestasis of pregnancy: The TURRIFIC randomised trial. BMC Pregnancy Childbirth 2021, 21, 51. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méndez-Sánchez, N.; Coronel-Castillo, C.E.; Ordoñez-Vázquez, A.L. Current Therapies for Cholestatic Diseases. Biomedicines 2023, 11, 1713. https://doi.org/10.3390/biomedicines11061713
Méndez-Sánchez N, Coronel-Castillo CE, Ordoñez-Vázquez AL. Current Therapies for Cholestatic Diseases. Biomedicines. 2023; 11(6):1713. https://doi.org/10.3390/biomedicines11061713
Chicago/Turabian StyleMéndez-Sánchez, Nahum, Carlos E. Coronel-Castillo, and Ana L. Ordoñez-Vázquez. 2023. "Current Therapies for Cholestatic Diseases" Biomedicines 11, no. 6: 1713. https://doi.org/10.3390/biomedicines11061713
APA StyleMéndez-Sánchez, N., Coronel-Castillo, C. E., & Ordoñez-Vázquez, A. L. (2023). Current Therapies for Cholestatic Diseases. Biomedicines, 11(6), 1713. https://doi.org/10.3390/biomedicines11061713