

Activation and Regulation of Pancreatic Stellate Cells in Chronic Pancreatic Fibrosis: A Potential Therapeutic Approach for Chronic Pancreatitis

Abstract
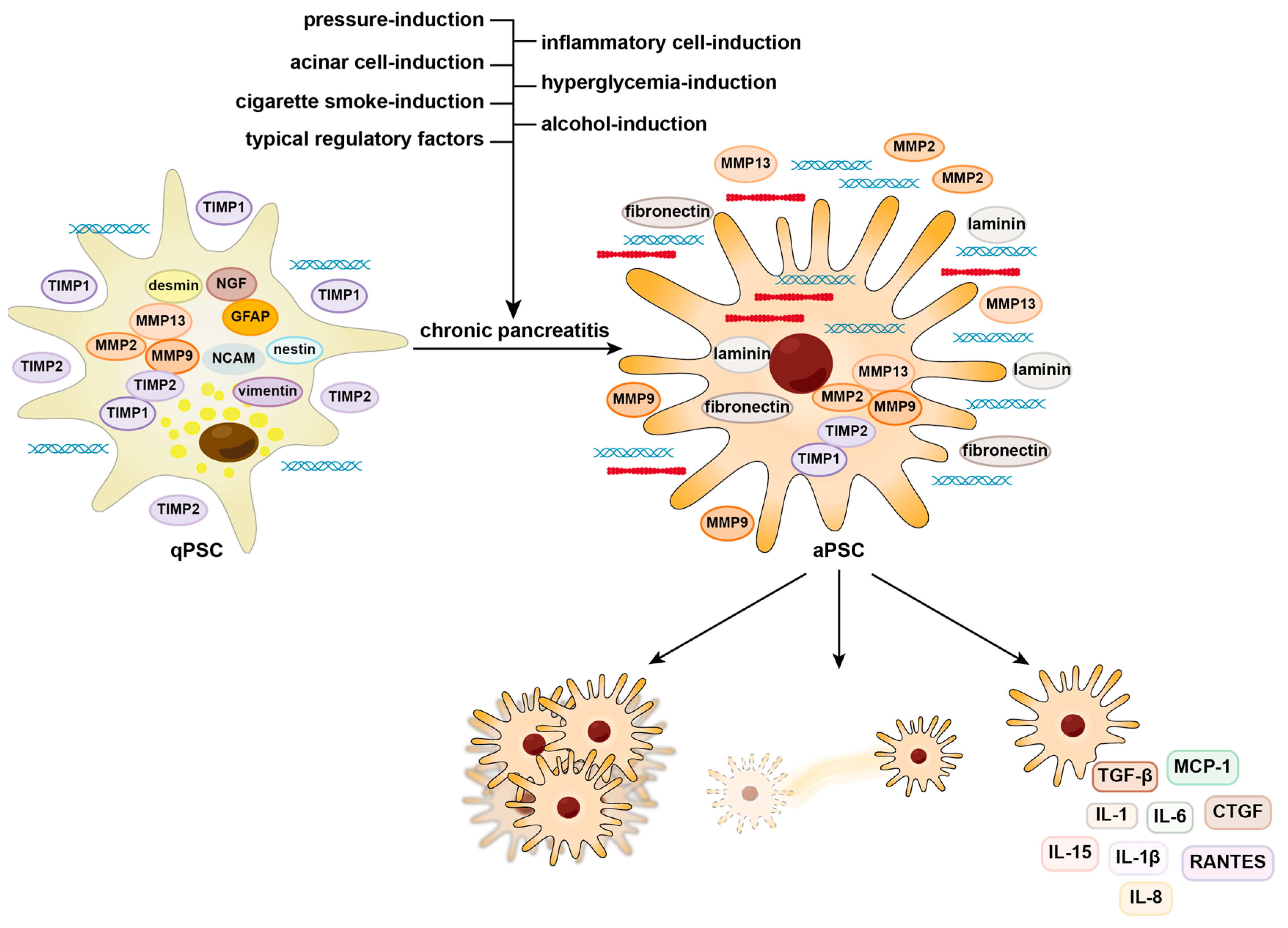
:1. Introduction
2. Activation and Contribution of PSCs to Fibrosis
2.1. Alcohol-Induced Activation of PSCs
2.2. Cigarette Smoke-Induced Activation of PSCs
2.3. Hyperglycemia-Induced Activation of PSCs
2.4. Pressure-Induced Activation of PSCs
2.5. Acinar-Cell-Induced Activation of PSCs
2.6. Inflammatory-Cell-Induced Activation of PSCs
2.7. Typical Regulatory Factors of PSC Activation
3. Prospects for PSC Therapy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Aoki, H.; Ohnishi, H.; Hama, K.; Ishijima, T.; Satoh, Y.; Hanatsuka, K.; Ohashi, A.; Wada, S.; Miyata, T.; Kita, H.; et al. Autocrine loop between TGF-beta1 and IL-1beta through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am. J. Physiol. Cell Physiol. 2006, 290, C1100–C1108. [Google Scholar] [CrossRef]
- Andoh, A.; Takaya, H.; Saotome, T.; Shimada, M.; Hata, K.; Araki, Y.; Nakamura, F.; Shintani, Y.; Fujiyama, Y.; Bamba, T. Cytokine regulation of chemokine (IL-8, MCP-1, and RANTES) gene expression in human pancreatic periacinar myofibroblasts. Gastroenterology 2000, 119, 211–219. [Google Scholar] [CrossRef]
- Apte, M.V.; Wilson, J.S. Stellate cell activation in alcoholic pancreatitis. Pancreas 2003, 27, 316–320. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells induce angiogenesis. Pancreas 2009, 38, 483. [Google Scholar] [CrossRef]
- Whitcomb, D.C. Inflammation and Cancer V. Chronic pancreatitis and pancreatic cancer. Am. J. Physiol.-Gastrointest. Liver Physiol. 2004, 287, G315–G319. [Google Scholar] [CrossRef]
- Vege, S.S.; Chari, S.T. Chronic Pancreatitis. N. Engl. J. Med. 2022, 386, 869–878. [Google Scholar] [CrossRef]
- Braganza, J.M.; Lee, S.H.; McCloy, R.F.; McMahon, M.J. Chronic pancreatitis. Lancet 2011, 377, 1184–1197. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Kodama, T.; Sato, K.; Murai, K.; Yoshioka, T.; Shigekawa, M.; Yamada, R.; Hikita, H.; Sakamori, R.; Akita, H.; et al. Dysregulation of PI3K and Hippo signaling pathways synergistically induces chronic pancreatitis via CTGF upregulation. J. Clin. Investig. 2021, 131, e143414. [Google Scholar] [CrossRef] [PubMed]
- Ferdek, P.E.; Jakubowska, M.A.; Gerasimenko, J.V.; Gerasimenko, O.V.; Petersen, O.H. Bile acids induce necrosis in pancreatic stellate cells dependent on calcium entry and sodium-driven bile uptake. J. Physiol. 2016, 594, 6147–6164. [Google Scholar] [CrossRef] [PubMed]
- Spanier, B.W.; Dijkgraaf, M.G.; Bruno, M.J. Epidemiology, aetiology and outcome of acute and chronic pancreatitis: An update. Best Pract Res. Clin. Gastroenterol. 2008, 22, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Norton, I.D.; Wilson, J.S. Ethanol induced acinar cell injury. Alcohol. Alcohol. Suppl. 1994, 2, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.S.; Apte, M.V. Role of alcohol metabolism in alcoholic pancreatitis. Pancreas 2003, 27, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.S.; Apte, M.V.; Applegate, T.L.; Norton, I.D.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Metabolism of ethanol by rat pancreatic acinar cells. J. Lab. Clin. Med. 1998, 132, 294–302. [Google Scholar] [CrossRef]
- Apte, M.; Pirola, R.; Wilson, J. The fibrosis of chronic pancreatitis: New insights into the role of pancreatic stellate cells. Antioxid Redox Signal 2011, 15, 2711–2722. [Google Scholar] [CrossRef]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Battle-scarred pancreas: Role of alcohol and pancreatic stellate cells in pancreatic fibrosis. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. S3), S97–S101. [Google Scholar] [CrossRef]
- Petersen, O.H.; Gerasimenko, J.V.; Gerasimenko, O.V.; Gryshchenko, O.; Peng, S. The roles of calcium and ATP in the physiology and pathology of the exocrine pancreas. Physiol. Rev. 2021, 101, 1691–1744. [Google Scholar] [CrossRef]
- Detlefsen, S.; Sipos, B.; Feyerabend, B.; Kloppel, G. Fibrogenesis in alcoholic chronic pancreatitis: The role of tissue necrosis, macrophages, myofibroblasts and cytokines. Mod. Pathol. 2006, 19, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Phillips, P.A.; Fahmy, R.G.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Naidoo, D.; Wilson, J.S. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology 2000, 118, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, J.; Wang, J.; Ma, S.; Feng, W.; Wu, Z.; Guo, Y.; Zhou, H.; Mi, W.; Chen, W.; et al. Very-low-density lipoprotein receptor-enhanced lipid metabolism in pancreatic stellate cells promotes pancreatic fibrosis. Immunity 2022, 55, 1185–1199.e8. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, J.A.; Phillips, P.A.; Park, S.; Doherty, E.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cell activation by ethanol and acetaldehyde: Is it mediated by the mitogen-activated protein kinase signaling pathway? Pancreas 2003, 27, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Galli, A.; Pignalosa, P.; Frulloni, L.; Grappone, C.; Milani, S.; Pederzoli, P.; Cavallini, G.; Surrenti, C. Collagen type I synthesized by pancreatic periacinar stellate cells (PSC) co-localizes with lipid peroxidation-derived aldehydes in chronic alcoholic pancreatitis. J. Pathol. 2000, 192, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Satoh, A.; Watanabe, T.; Kikuta, K.; Satoh, M.; Suzuki, N.; Satoh, K.; Shimosegawa, T. Effects of ethanol and its metabolites on human pancreatic stellate cells. Dig. Dis. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef]
- Gao, R.; Brigstock, D.R. Connective tissue growth factor (CCN2) in rat pancreatic stellate cell function: Integrin alpha5beta1 as a novel CCN2 receptor. Gastroenterology 2005, 129, 1019–1030. [Google Scholar] [CrossRef]
- Hu, R.; Wang, Y.L.; Edderkaoui, M.; Lugea, A.; Apte, M.V.; Pandol, S.J. Ethanol augments PDGF-induced NADPH oxidase activity and proliferation in rat pancreatic stellate cells. Pancreatology 2007, 7, 332–340. [Google Scholar] [CrossRef]
- Lawrencia, C.; Charrier, A.; Huang, G.; Brigstock, D.R. Ethanol-mediated expression of connective tissue growth factor (CCN2) in mouse pancreatic stellate cells. Growth Factors 2009, 27, 91–99. [Google Scholar] [CrossRef]
- Charrier, A.; Chen, R.; Chen, L.; Kemper, S.; Hattori, T.; Takigawa, M.; Brigstock, D.R. Connective tissue growth factor (CCN2) and microRNA-21 are components of a positive feedback loop in pancreatic stellate cells (PSC) during chronic pancreatitis and are exported in PSC-derived exosomes. J. Cell. Commun. Signal. 2014, 8, 147–156. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Satoh, M.; Satoh, A.; Shimosegawa, T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J. Pharmacol. Exp. Ther. 2002, 302, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Ito, T.; Nakamura, T.; Igarashi, H.; Oono, T.; Fujimori, N.; Kawabe, K.; Suzuki, K.; Jensen, R.T.; Takayanagi, R. ERK pathway and sheddases play an essential role in ethanol-induced CX3CL1 release in pancreatic stellate cells. Lab. Investg. 2013, 93, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Ito, T.; Nakamura, T.; Hijioka, M.; Igarashi, H.; Oono, T.; Kato, M.; Nakamura, K.; Suzuki, K.; Takayanagi, R.; et al. Pancreatic stellate cells and CX3CR1: Occurrence in normal pancreas and acute and chronic pancreatitis and effect of their activation by a CX3CR1 agonist. Pancreas 2014, 43, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Dolai, S.; Liang, T.; Lam, P.P.L.; Fernandez, N.A.; Chidambaram, S.; Gaisano, H.Y. Effects of ethanol metabolites on exocytosis of pancreatic acinar cells in rats. Gastroenterology 2012, 143, 832–843.e7. [Google Scholar] [CrossRef] [PubMed]
- Gryshchenko, O.; Gerasimenko, J.V.; Peng, S.; Gerasimenko, O.V.; Petersen, O.H. Calcium signalling in the acinar environment of the exocrine pancreas: Physiology and pathophysiology. J. Physiol. 2018, 596, 2663–2678. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H.; Tepikin, A.V.; Gerasimenko, J.V.; Gerasimenko, O.V.; Sutton, R.; Criddle, D.N. Fatty acids, alcohol and fatty acid ethyl esters: Toxic Ca2+ signal generation and pancreatitis. Cell. Calcium. 2009, 45, 634–642. [Google Scholar] [CrossRef]
- Kusiak, A.A.; Jakubowska, M.A.; Stopa, K.B.; Zhang, X.; Huang, W.; Gerasimenko, J.V.; Gerasimenko, O.V.; Sutton, R.; Petersen, O.H.; Ferdek, P.E. Activation of pancreatic stellate cells attenuates intracellular Ca2+ signals due to downregulation of TRPA1 and protects against cell death induced by alcohol metabolites. Cell Death Dis. 2022, 13, 744. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Zhang, X.; Yang, L.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Withdrawal of alcohol promotes regression while continued alcohol intake promotes persistence of LPS-induced pancreatic injury in alcohol-fed rats. Gut 2011, 60, 238–246. [Google Scholar] [CrossRef]
- Bhatia, R.; Thompson, C.M.; Clement, E.J.; Ganguly, K.; Cox, J.L.; Rauth, S.; Siddiqui, J.A.; Mashiana, S.S.; Jain, M.; Wyatt, T.A.; et al. Malondialdehyde-Acetaldehyde Extracellular Matrix Protein Adducts Attenuate Unfolded Protein Response During Alcohol and Smoking-Induced Pancreatitis. Gastroenterology 2022, 163, 1064–1078.e10. [Google Scholar] [CrossRef]
- Coté, G.A.; Yadav, D.; Slivka, A.; Hawes, R.H.; Anderson, M.A.; Burton, F.R.; Brand, R.E.; Banks, P.A.; Lewis, M.D.; Disario, J.A.; et al. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin. Gastroenterol. Hepatol. 2011, 9, 266–273, quiz e227. [Google Scholar] [CrossRef]
- Law, R.; Parsi, M.; Lopez, R.; Zuccaro, G.; Stevens, T. Cigarette smoking is independently associated with chronic pancreatitis. Pancreatology 2010, 10, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Alsamarrai, A.; Das, S.L.; Windsor, J.A.; Petrov, M.S. Factors that affect risk for pancreatic disease in the general population: A systematic review and meta-analysis of prospective cohort studies. Clin. Gastroenterol. Hepatol. 2014, 12, 1635–1644.e5. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Hawes, R.H.; Brand, R.E.; Anderson, M.A.; Money, M.E.; Banks, P.A.; Bishop, M.D.; Baillie, J.; Sherman, S.; DiSario, J.; et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch. Intern. Med. 2009, 169, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Harris, R.A. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat. Neurosci. 2005, 8, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Lowenfels, A.B.; Müllhaupt, B.; Cavallini, G.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellöf, L.; Frulloni, L.; et al. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut 2005, 54, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Lugea, A.; Gerloff, A.; Su, H.Y.; Xu, Z.; Go, A.; Hu, C.; French, S.W.; Wilson, J.S.; Apte, M.V.; Waldron, R.T.; et al. The Combination of Alcohol and Cigarette Smoke Induces Endoplasmic Reticulum Stress and Cell Death in Pancreatic Acinar Cells. Gastroenterology 2017, 153, 1674–1686. [Google Scholar] [CrossRef]
- Lee, A.T.; Xu, Z.; Pothula, S.P.; Patel, M.B.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Alcohol and cigarette smoke components activate human pancreatic stellate cells: Implications for the progression of chronic pancreatitis. Alcohol. Clin. Exp. Res. 2015, 39, 2123–2133. [Google Scholar] [CrossRef]
- Yadav, D.; Whitcomb, D.C. The role of alcohol and smoking in pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 131–145. [Google Scholar] [CrossRef]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Primers 2017, 3, 17060. [Google Scholar] [CrossRef]
- Xue, J.; Zhao, Q.; Sharma, V.; Nguyen, L.P.; Lee, Y.N.; Pham, K.L.; Edderkaoui, M.; Pandol, S.J.; Park, W.; Habtezion, A. Aryl Hydrocarbon Receptor Ligands in Cigarette Smoke Induce Production of Interleukin-22 to Promote Pancreatic Fibrosis in Models of Chronic Pancreatitis. Gastroenterology 2016, 151, 1206–1217. [Google Scholar] [CrossRef]
- Rioux, N.; Castonguay, A. 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone modulation of cytokine release in U937 human macrophages. Cancer Immunol. Immunother. 2001, 49, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Li, Z.; Li, M.; Jin, T.; Zhang, X.; Liu, X.; Hao, J. Mitochondria oxidative stress mediated nicotine-promoted activation of pancreatic stellate cells by regulating mitochondrial dynamics. Toxicol In Vitro 2022, 84, 105436. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ. Health Perspect. 1997, 105 (Suppl. S4), 875–882. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, W.J.; Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef]
- Nordman, J.C.; Kabbani, N. Microtubule dynamics at the growth cone are mediated by α7 nicotinic receptor activation of a Gαq and IP3 receptor pathway. FASEB J. 2014, 28, 2995–3006. [Google Scholar] [CrossRef]
- Li, Z.; Lu, D.; Jin, T.; Liu, X.; Hao, J. Nicotine facilitates pancreatic fibrosis by promoting activation of pancreatic stellate cells via α7nAChR-mediated JAK2/STAT3 signaling pathway in rats. Toxicol. Lett. 2021, 349, 84–91. [Google Scholar] [CrossRef]
- Hong, O.-K.; Lee, S.-H.; Rhee, M.; Ko, S.-H.; Cho, J.-H.; Choi, Y.-H.; Song, K.-H.; Son, H.-Y.; Yoon, K.-H. Hyperglycemia and hyperinsulinemia have additive effects on activation and proliferation of pancreatic stellate cells: Possible explanation of islet-specific fibrosis in type 2 diabetes mellitus. J. Cell. Biochem. 2007, 101, 665–675. [Google Scholar] [CrossRef]
- Ko, S.-H.; Hong, O.-K.; Kim, J.-W.; Ahn, Y.-B.; Song, K.-H.; Cha, B.-Y.; Son, H.-Y.; Kim, M.-J.; Jeong, I.-K.; Yoon, K.-H. High glucose increases extracellular matrix production in pancreatic stellate cells by activating the renin-angiotensin system. J. Cell. Biochem. 2006, 98, 343–355. [Google Scholar] [CrossRef]
- Hama, K.; Ohnishi, H.; Yasuda, H.; Ueda, N.; Mashima, H.; Satoh, Y.; Hanatsuka, K.; Kita, H.; Ohashi, A.; Tamada, K.; et al. Angiotensin II stimulates DNA synthesis of rat pancreatic stellate cells by activating ERK through EGF receptor transactivation. Biochem. Biophys. Res. Commun. 2004, 315, 905–911. [Google Scholar] [CrossRef]
- Nomiyama, Y.; Tashiro, M.; Yamaguchi, T.; Watanabe, S.; Taguchi, M.; Asaumi, H.; Nakamura, H.; Otsuki, M. High glucose activates rat pancreatic stellate cells through protein kinase C and p38 mitogen-activated protein kinase pathway. Pancreas 2007, 34, 364–372. [Google Scholar] [CrossRef]
- Ryu, G.R.; Lee, E.; Chun, H.-J.; Yoon, K.-H.; Ko, S.-H.; Ahn, Y.-B.; Song, K.-H. Oxidative stress plays a role in high glucose-induced activation of pancreatic stellate cells. Biochem. Biophys. Res. Commun. 2013, 439, 258–263. [Google Scholar] [CrossRef]
- Kim, J.-W.; Park, S.-Y.; You, Y.-H.; Ham, D.-S.; Lee, S.-H.; Yang, H.K.; Jeong, I.-K.; Ko, S.-H.; Yoon, K.-H. Suppression of ROS Production by Exendin-4 in PSC Attenuates the High Glucose-Induced Islet Fibrosis. PLoS ONE 2016, 11, e0163187. [Google Scholar] [CrossRef] [PubMed]
- Kiss, K.; Baghy, K.; Spisák, S.; Szanyi, S.; Tulassay, Z.; Zalatnai, A.; Löhr, J.M.; Jesenofsky, R.; Kovalszky, I.; Firneisz, G. Chronic hyperglycemia induces trans-differentiation of human pancreatic stellate cells and enhances the malignant molecular communication with human pancreatic cancer cells. PLoS ONE 2015, 10, e0128059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bai, J.; Zhou, Q.; Hu, Y.; Wang, Q.; Yang, L.; Chen, H.; An, H.; Zhou, C.; Wang, Y.; et al. Glutathione prevents high glucose-induced pancreatic fibrosis by suppressing pancreatic stellate cell activation via the ROS/TGFβ/SMAD pathway. Cell Death Dis. 2022, 13, 440. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Nagashio, Y.; Asaumi, H.; Nomiyama, Y.; Taguchi, M.; Tashiro, M.; Kihara, Y.; Nakamura, H.; Otsuki, M. Pressure activates rat pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1175–G1181. [Google Scholar] [CrossRef] [PubMed]
- Asaumi, H.; Watanabe, S.; Taguchi, M.; Tashiro, M.; Otsuki, M. Externally applied pressure activates pancreatic stellate cells through the generation of intracellular reactive oxygen species. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G972–G978. [Google Scholar] [CrossRef] [PubMed]
- Radoslavova, S.; Fels, B.; Pethö, Z.; Gruner, M.; Ruck, T.; Meuth, S.G.; Folcher, A.; Prevarskaya, N.; Schwab, A.; Ouadid-Ahidouch, H. TRPC1 channels regulate the activation of pancreatic stellate cells through ERK1/2 and SMAD2 pathways and perpetuate their pressure-mediated activation. Cell Calcium 2022, 106, 102621. [Google Scholar] [CrossRef]
- Swain, S.M.; Romac, J.M.J.; Vigna, S.R.; Liddle, R.A. Piezo1-mediated stellate cell activation causes pressure-induced pancreatic fibrosis in mice. JCI Insight 2022, 7, e158288. [Google Scholar] [CrossRef]
- Loeck, T.; Rugi, M.; Todesca, L.M.; Soret, B.; Neumann, I.; Schimmelpfennig, S.; Najder, K.; Pethő, Z.; Farfariello, V.; Prevarskaya, N.; et al. The context-dependent role of the Na+/Ca2+-exchanger (NCX) in pancreatic stellate cell migration. Pflug. Arch. Eur. J. Physiol. 2023, 475, 1225–1240. [Google Scholar] [CrossRef]
- An, J.; Jiang, T.; Qi, L.; Xie, K. Acinar cells and the development of pancreatic fibrosis. Cytokine Growth Factor Rev. 2023, 71–72, 40–53. [Google Scholar] [CrossRef]
- Schlesinger, Y.; Yosefov-Levi, O.; Kolodkin-Gal, D.; Granit, R.Z.; Peters, L.; Kalifa, R.; Xia, L.; Nasereddin, A.; Shiff, I.; Amran, O.; et al. Single-cell transcriptomes of pancreatic preinvasive lesions and cancer reveal acinar metaplastic cells’ heterogeneity. Nat. Commun. 2020, 11, 4516. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Fine, D.R. Fibrogenesis in the pancreas after acinar cell injury. Scand. J. Surg. 2005, 94, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, M.; Nipper, M.; Deng, J.; Sharkey, F.E.; Johnson, R.L.; Crawford, H.C.; Chen, Y.; Wang, P. Activation of the intrinsic fibroinflammatory program in adult pancreatic acinar cells triggered by Hippo signaling disruption. PLoS Biol. 2019, 17, e3000418. [Google Scholar] [CrossRef] [PubMed]
- Bläuer, M.; Laaninen, M.; Sand, J.; Laukkarinen, J. Reciprocal stimulation of pancreatic acinar and stellate cells in a novel long-term in vitro co-culture model. Pancreatology 2016, 16, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Kim, H. Cerulein pancreatitis: Oxidative stress, inflammation, and apoptosis. Gut Liver 2008, 2, 74–80. [Google Scholar] [CrossRef]
- Szabó, V.; Csákány-Papp, N.; Görög, M.; Madácsy, T.; Varga, Á.; Kiss, A.; Tél, B.; Jójárt, B.; Crul, T.; Dudás, K.; et al. Orai1 calcium channel inhibition prevents progression of chronic pancreatitis. JCI Insight 2023, 8, e167645. [Google Scholar] [CrossRef]
- Xue, R.; Zhou, J.; Wu, J.; Meng, Q.; Gong, J.; Shen, L. P-element-Induced Wimpy-Testis-Like Protein 1 Regulates the Activation of Pancreatic Stellate Cells Through the PI3K/AKT/mTOR Signaling Pathway. Dig. Dis. Sci. 2023, 68, 1339–1350. [Google Scholar] [CrossRef]
- Charrier, A.L.; Brigstock, D.R. Connective tissue growth factor production by activated pancreatic stellate cells in mouse alcoholic chronic pancreatitis. Lab. Investig. 2010, 90, 1179–1188. [Google Scholar] [CrossRef]
- di Mola, F.F.; Friess, H.; Martignoni, M.E.; Di Sebastiano, P.; Zimmermann, A.; Innocenti, P.; Graber, H.; Gold, L.I.; Korc, M.; Büchler, M.W. Connective tissue growth factor is a regulator for fibrosis in human chronic pancreatitis. Ann. Surg. 1999, 230, 63–71. [Google Scholar] [CrossRef]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef]
- Karger, A.; Fitzner, B.; Brock, P.; Sparmann, G.; Emmrich, J.; Liebe, S.; Jaster, R. Molecular insights into connective tissue growth factor action in rat pancreatic stellate cells. Cell. Signal. 2008, 20, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.W.; Zhang, Y.P.; Qiao, M.M.; Fu, H.; Yuan, Y.Z. The study of regulation of connective tissue growth factor gene promoter by transforming growth factor beta1 in pancreatic stellate cells. Zhonghua Yi Xue Za Zhi 2004, 84, 1240–1242. [Google Scholar] [PubMed]
- Spanehl, L.; Revskij, D.; Bannert, K.; Ehlers, L.; Jaster, R. YAP activates pancreatic stellate cells and enhances pancreatic fibrosis. Hepatobiliary Pancreat. Dis. Int. 2022, 21, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Simeone, D.M.; Zhang, L.; Graziano, K.; Nicke, B.; Pham, T.; Schaefer, C.; Logsdon, C.D. Smad4 mediates activation of mitogen-activated protein kinases by TGF-beta in pancreatic acinar cells. Am. J. Physiol. Cell. Physiol. 2001, 281, C311–C319. [Google Scholar] [CrossRef]
- Bockman, D.E.; Merlino, G. Cytological changes in the pancreas of transgenic mice overexpressing transforming growth factor alpha. Gastroenterology 1992, 103, 1883–1892. [Google Scholar] [CrossRef]
- Komar, H.M.; Serpa, G.; Kerscher, C.; Schwoegl, E.; Mace, T.A.; Jin, M.; Yang, M.C.; Chen, C.S.; Bloomston, M.; Ostrowski, M.C.; et al. Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci. Rep. 2017, 7, 1787. [Google Scholar] [CrossRef]
- Bhatia, V.; Rastellini, C.; Han, S.; Aronson, J.F.; Greeley, G.H., Jr.; Falzon, M. Acinar cell-specific knockout of the PTHrP gene decreases the proinflammatory and profibrotic responses in pancreatitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 307, G533–G549. [Google Scholar] [CrossRef]
- Bläuer, M.; Laaninen, M.; Sand, J.; Laukkarinen, J. Wnt/β-catenin signalling plays diverse functions during the process of fibrotic remodelling in the exocrine pancreas. Pancreatology 2019, 19, 252–257. [Google Scholar] [CrossRef]
- Keefe, M.D.; Wang, H.; De La, O.J.; Khan, A.; Firpo, M.A.; Murtaugh, L.C. β-catenin is selectively required for the expansion and regeneration of mature pancreatic acinar cells in mice. Dis. Model. Mech. 2012, 5, 503–514. [Google Scholar] [CrossRef]
- Xiao, W.; Jiang, W.; Shen, J.; Yin, G.; Fan, Y.; Wu, D.; Qiu, L.; Yu, G.; Xing, M.; Hu, G.; et al. Retinoic Acid Ameliorates Pancreatic Fibrosis and Inhibits the Activation of Pancreatic Stellate Cells in Mice with Experimental Chronic Pancreatitis via Suppressing the Wnt/β-Catenin Signaling Pathway. PLoS ONE 2015, 10, e0141462. [Google Scholar] [CrossRef]
- Hoque, R.; Sohail, M.; Malik, A.; Sarwar, S.; Luo, Y.; Shah, A.; Barrat, F.; Flavell, R.; Gorelick, F.; Husain, S.; et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011, 141, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, H.; Jing, Q.; Yang, Y.; Xue, D.; Hao, C.; Zhang, W. Regulation of Pancreatic Fibrosis by Acinar Cell-Derived Exosomal miR-130a-3p via Targeting of Stellate Cell PPAR-γ. J. Inflamm. Res. 2021, 14, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Kandikattu, H.K.; Venkateshaiah, S.U.; Mishra, A. Chronic Pancreatitis and the Development of Pancreatic Cancer. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 1182–1210. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef]
- Shek, F.W.-T.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.F.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef]
- Zheng, M.; Li, H.; Sun, L.; Brigstock, D.R.; Gao, R. Interleukin-6 participates in human pancreatic stellate cell activation and collagen I production via TGF-β1/Smad pathway. Cytokine 2021, 143, 155536. [Google Scholar] [CrossRef]
- Weber, C. Pancreatitis: Alternatively activated macrophages mediate fibrosis. Nat. Reviews. Gastroenterol. Hepatol. 2015, 12, 372. [Google Scholar] [CrossRef]
- Gerasimenko, J.V.; Petersen, O.H.; Gerasimenko, O.V. SARS-CoV-2 S Protein Subunit 1 Elicits Ca2+ Influx—Dependent Ca2+ Signals in Pancreatic Stellate Cells and Macrophages In Situ. Function 2022, 3, zqac002. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Thompson, C.; Ganguly, K.; Singh, S.; Batra, S.K.; Kumar, S. Alcohol and Smoking Mediated Modulations in Adaptive Immunity in Pancreatitis. Cells 2020, 9, 1880. [Google Scholar] [CrossRef] [PubMed]
- Lighaam, L.C.; Aalberse, R.C.; Rispens, T. IgG4-Related Fibrotic Diseases from an Immunological Perspective: Regulators out of Control? Int. J. Rheumatol. 2012, 2012, 789164. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.S.; Keogh, G.W.; Apte, M.V.; Moran, C.S.; Stewart, N.L.; Crawford, D.H.; Pirola, R.C.; McCaughan, G.W.; Ramm, G.A.; Wilson, J.S. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am. J. Pathol. 1999, 155, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Luttenberger, T.; Schmid-Kotsas, A.; Menke, A.; Siech, M.; Beger, H.; Adler, G.; Grünert, A.; Bachem, M.G. Platelet-derived growth factors stimulate proliferation and extracellular matrix synthesis of pancreatic stellate cells: Implications in pathogenesis of pancreas fibrosis. Lab. Investig. 2000, 80, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Vogelmann, R.; Ruf, D.; Wagner, M.; Adler, G.; Menke, A. Effects of fibrogenic mediators on the development of pancreatic fibrosis in a TGF-beta 1 transgenic mouse model. Am. J. Physiol.-Gastrointest. Liver Physiol. 2001, 280, G164–G172. [Google Scholar] [CrossRef] [PubMed]
- Sarper, M.; Cortes, E.; Lieberthal, T.J.; Del Río Hernández, A. ATRA modulates mechanical activation of TGF-β by pancreatic stellate cells. Sci. Rep. 2016, 6, 27639. [Google Scholar] [CrossRef]
- Xu, X.F.; Liu, F.; Xin, J.Q.; Fan, J.W.; Wu, N.; Zhu, L.J.; Duan, L.F.; Li, Y.Y.; Zhang, H. Respective roles of the mitogen-activated protein kinase (MAPK) family members in pancreatic stellate cell activation induced by transforming growth factor-β1 (TGF-β1). Biochem. Biophys. Res. Commun. 2018, 501, 365–373. [Google Scholar] [CrossRef]
- Jaster, R.; Sparmann, G.; Emmrich, J.; Liebe, S. Extracellular signal regulated kinases are key mediators of mitogenic signals in rat pancreatic stellate cells. Gut 2002, 51, 579–584. [Google Scholar] [CrossRef]
- Tahara, H.; Sato, K.; Yamazaki, Y.; Ohyama, T.; Horiguchi, N.; Hashizume, H.; Kakizaki, S.; Takagi, H.; Ozaki, I.; Arai, H.; et al. Transforming growth factor-α activates pancreatic stellate cells and may be involved in matrix metalloproteinase-1 upregulation. Lab. Investig. 2013, 93, 720–732. [Google Scholar] [CrossRef]
- Lee, H.; Lim, C.; Lee, J.; Kim, N.; Bang, S.; Lee, H.; Min, B.; Park, G.; Noda, M.; Stetler-Stevenson, W.G.; et al. TGF-beta signaling preserves RECK expression in activated pancreatic stellate cells. J. Cell. Biochem. 2008, 104, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Xu, X.F.; Xin, J.Q.; Fan, J.W.; Wei, Y.Y.; Peng, Q.X.; Duan, L.F.; Wang, W.; Zhang, H. The effects of nuclear factor-kappa B in pancreatic stellate cells on inflammation and fibrosis of chronic pancreatitis. J. Cell. Mol. Med. 2021, 25, 2213–2227. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, N.; Miyata, T.; Ohnishi, H.; Yasuda, H.; Tamada, K.; Ueda, N.; Mashima, H.; Sugano, K. Activin A is an autocrine activator of rat pancreatic stellate cells: Potential therapeutic role of follistatin for pancreatic fibrosis. Gut 2003, 52, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cao, Y.; Yang, W.; Duan, C.; Aronson, J.F.; Rastellini, C.; Chao, C.; Hellmich, M.R.; Ko, T.C. BMP2 inhibits TGF-β-induced pancreatic stellate cell activation and extracellular matrix formation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G804–G813. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Lei, X.F.; Miyauchi, A.; Noguchi, M.; Omoto, T.; Haraguchi, S.; Miyazaki, T.; Miyazaki, A.; Kim-Kaneyama, J.R. Hic-5 is required for activation of pancreatic stellate cells and development of pancreatic fibrosis in chronic pancreatitis. Sci. Rep. 2020, 10, 19105. [Google Scholar] [CrossRef] [PubMed]
- Hama, K.; Ohnishi, H.; Aoki, H.; Kita, H.; Yamamoto, H.; Osawa, H.; Sato, K.; Tamada, K.; Mashima, H.; Yasuda, H.; et al. Angiotensin II promotes the proliferation of activated pancreatic stellate cells by Smad7 induction through a protein kinase C pathway. Biochem. Biophys. Res. Commun. 2006, 340, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Schmid-Kotsas, A.; Zhao, J.S.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Adler, G.; Waltenberger, J.; Grunert, A.; Bachem, M.G. Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am. J. Physiol.-Cell Physiol. 2001, 281, C532–C543. [Google Scholar] [CrossRef]
- Grotendorst, G.R. Connective tissue growth factor: A mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 1997, 8, 171–179. [Google Scholar] [CrossRef]
- Nelson, D.R.; Lauwers, G.Y.; Lau, J.Y.; Davis, G.L. Interleukin 10 treatment reduces fibrosis in patients with chronic hepatitis C: A pilot trial of interferon nonresponders. Gastroenterology 2000, 118, 655–660. [Google Scholar] [CrossRef]
- Demols, A.; Van Laethem, J.L.; Quertinmont, E.; Degraef, C.; Delhaye, M.; Geerts, A.; Deviere, J. Endogenous interleukin-10 modulates fibrosis and regeneration in experimental chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G1105–G1112. [Google Scholar] [CrossRef]
- Mews, P.; Phillips, P.; Fahmy, R.; Korsten, M.; Pirola, R.; Wilson, J.; Apte, M. Pancreatic stellate cells respond to inflammatory cytokines: Potential role in chronic pancreatitis. Gut 2002, 50, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Satoh, K.; Kanno, A.; Shimosegawa, T. Nuclear expression of interleukin-33 in pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G821–G832. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Mashima, H.; Ohnishi, H.; Sugano, K. IL-13 promotes the proliferation of rat pancreatic stellate cells through the suppression of NF-kappaB/TGF-beta1 pathway. Biochem. Biophys. Res. Commun. 2010, 393, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Satoh, M.; Kume, K.; Shimosegawa, T. Differential roles of signaling pathways for proliferation and migration of rat pancreatic stellate cells. Tohoku J. Exp. Med. 2003, 199, 69–84. [Google Scholar] [CrossRef]
- Storck, H.; Hild, B.; Schimmelpfennig, S.; Sargin, S.; Nielsen, N.; Zaccagnino, A.; Budde, T.; Novak, I.; Kalthoff, H.; Schwab, A. Ion channels in control of pancreatic stellate cell migration. Oncotarget 2017, 8, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Klonowski-Stumpe, H.; Reinehr, R.; Fischer, R.; Warskulat, U.; Lüthen, R.; Häussinger, D. Production and effects of endothelin-1 in rat pancreatic stellate cells. Pancreas 2003, 27, 67–74. [Google Scholar] [CrossRef]
- Masamune, A.; Satoh, M.; Kikuta, K.; Suzuki, N.; Shimosegawa, T. Endothelin-1 stimulates contraction and migration of rat pancreatic stellate cells. World J. Gastroenterol. 2005, 11, 6144–6151. [Google Scholar] [CrossRef]
- Charo, C.; Holla, V.; Arumugam, T.; Hwang, R.; Yang, P.; Dubois, R.N.; Menter, D.G.; Logsdon, C.D.; Ramachandran, V. Prostaglandin E2 regulates pancreatic stellate cell activity via the EP4 receptor. Pancreas 2013, 42, 467–474. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Hamada, S.; Shimosegawa, T. Fibrinogen induces cytokine and collagen production in pancreatic stellate cells. Gut 2009, 58, 550–559. [Google Scholar] [CrossRef]
- Hu, Y.; Wan, R.; Yu, G.; Shen, J.; Ni, J.; Yin, G.; Xing, M.; Chen, C.; Fan, Y.; Xiao, W.; et al. Imbalance of Wnt/Dkk negative feedback promotes persistent activation of pancreatic stellate cells in chronic pancreatitis. PLoS ONE 2014, 9, e95145. [Google Scholar] [CrossRef]
- Cui, L.; Li, C.; Gao, G.; Zhuo, Y.; Yang, L.; Cui, N.; Zhang, S. FTY720 inhibits the activation of pancreatic stellate cells by promoting apoptosis and suppressing autophagy via the AMPK/mTOR pathway. Life Sci. 2019, 217, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.A.; Lim, J.W.; Kim, H. Docosahexaenoic Acid Inhibits Cytokine Expression by Reducing Reactive Oxygen Species in Pancreatic Stellate Cells. J. Cancer Prev. 2021, 26, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Dong, B.; Qi, L.; Wei, Y.; Zhang, Y.; Cai, X.; Zhang, Q.; Li, J.; Li, L. Inhibitory Smads suppress pancreatic stellate cell activation through negative feedback in chronic pancreatitis. Ann. Transl. Med. 2021, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Cui, L.H.; Zhang, L.Q.; Yang, L.; Zhuo, Y.Z.; Cui, N.Q.; Zhang, S.K. Role of NLR family pyrin domain-containing 3 inflammasome in the activation of pancreatic stellate cells. Exp. Cell Res. 2021, 404, 112634. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.F.; Lin, H.; Liu, D.C.; Zhu, X.Y.; Huang, N.; Wei, Y.X.; Li, L. Heat shock protein 90 inhibitor ameliorates pancreatic fibrosis by degradation of transforming growth factor-β receptor. Cell. Signal. 2021, 84, 110001. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.P.; Zeng, J.H.; Lin, X.; Ni, Y.H.; Jiang, C.S.; Li, D.Z.; He, X.J.; Wang, R.; Wang, W. Puerarin Ameliorates Caerulein-Induced Chronic Pancreatitis via Inhibition of MAPK Signaling Pathway. Front. Pharmacol. 2021, 12, 686992. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Jin, L.; Ju, D.; Lu, Z.; Wang, C.; Guo, X.; Zhao, H.; Shen, S.; Cheng, Z.; Shen, J.; et al. The pancreatic clock is a key determinant of pancreatic fibrosis progression and exocrine dysfunction. Sci. Transl. Med. 2022, 14, eabn3586. [Google Scholar] [CrossRef]
- Ng, B.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Shekeran, S.G.; Goh, J.W.T.; Tan, J.; Kuthubudeen, F.; Lim, S.Y.; Xie, C.; et al. IL11 Activates Pancreatic Stellate Cells and Causes Pancreatic Inflammation, Fibrosis and Atrophy in a Mouse Model of Pancreatitis. Int. J. Mol. Sci. 2022, 23, 3549. [Google Scholar] [CrossRef]
- Bansod, S.; Saifi, M.A.; Godugu, C. Inhibition of discoidin domain receptors by imatinib prevented pancreatic fibrosis demonstrated in experimental chronic pancreatitis model. Sci. Rep. 2021, 11, 12894. [Google Scholar] [CrossRef]
- Cui, L.; Li, C.; Zhang, G.; Zhang, L.; Yao, G.; Zhuo, Y.; Cui, N.; Zhang, S. S1P/S1PR2 promote pancreatic stellate cell activation and pancreatic fibrosis in chronic pancreatitis by regulating autophagy and the NLRP3 inflammasome. Chem. Biol. Interact. 2023, 380, 110541. [Google Scholar] [CrossRef]
- Di Fazio, P.; Mielke, S.; Böhm, I.T.; Buchholz, M.; Matrood, S.; Schuppan, D.; Wissniowski, T. Toll-like receptor 5 tunes hepatic and pancreatic stellate cells activation. BMJ Open Gastroenterol. 2023, 10, e001148. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Wang, L.J.; Dong, Z.Q.; Wang, P.Y.; Lv, Y.W.; Wang, D.; Hu, L.H. Nintedanib Alleviates Chronic Pancreatitis by Inhibiting the Activation of Pancreatic Stellate Cells via the JAK/STAT3 and ERK1/2 Pathways. Dig. Dis. Sci. 2023, 68, 3644–3659. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yin, L.; Chen, Z.; Waldron, R.T.; Lugea, A.; Lin, Y.; Zhai, X.; Wen, L.; Han, Y.P.; Pandol, S.J.; et al. The unique pancreatic stellate cell gene expression signatures are associated with the progression from acute to chronic pancreatitis. Comput. Struct. Biotechnol. J. 2021, 19, 6375–6385. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Guo, J.; Qi, B.; Xiao, H. TGF-β1 induced proliferation, migration, and ECM accumulation through the SNHG11/miR-34b/LIF pathway in human pancreatic stellate cells. Endocr. J. 2021, 68, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, G.; Yang, W.; Chen, H.; Hu, J.; Zhao, Z.; Cheng, C.; Li, G.; Xie, Y.; Li, Y.; et al. Lnc-PFAR facilitates autophagy and exacerbates pancreatic fibrosis by reducing pre-miR-141 maturation in chronic pancreatitis. Cell Death Dis. 2021, 12, 996. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, M.; Li, X.; Long, Y.; Chen, K.; Wang, X.; Zhong, M.; Cheng, W.; Tian, X.; Wang, P.; et al. Inflammatory-miR-301a circuitry drives mTOR and Stat3-dependent PSC activation in chronic pancreatitis and PanIN. Mol. Ther. Nucleic Acids 2022, 27, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Almanzar, V.M.D.; Shah, K.; LaComb, J.F.; Mojumdar, A.; Patel, H.R.; Cheung, J.; Tang, M.; Ju, J.; Bialkowska, A.B. 5-FU-miR-15a Inhibits Activation of Pancreatic Stellate Cells by Reducing YAP1 and BCL-2 Levels In Vitro. Int. J. Mol. Sci. 2023, 24, 3954. [Google Scholar] [CrossRef]
- Chen, W.; Imasaka, M.; Lee, M.; Fukui, H.; Nishiura, H.; Ohmuraya, M. Reg family proteins contribute to inflammation and pancreatic stellate cells activation in chronic pancreatitis. Sci. Rep. 2023, 13, 12201. [Google Scholar] [CrossRef]
- Lin, H.; Ye, Z.; Xu, R.; Li, X.E.; Sun, B. The transcription factor JUN is a major regulator of quiescent pancreatic stellate cell maintenance. Gene 2023, 851, 147000. [Google Scholar] [CrossRef]
- Alhobayb, T.; Peravali, R.; Ashkar, M. The Relationship between Acute and Chronic Pancreatitis with Pancreatic Adenocarcinoma: Review. Diseases 2021, 9, 93. [Google Scholar] [CrossRef]
- Jin, G.; Hong, W.; Guo, Y.; Bai, Y.; Chen, B. Molecular Mechanism of Pancreatic Stellate Cells Activation in Chronic Pancreatitis and Pancreatic Cancer. J. Cancer 2020, 11, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Iovanna, J.; Santofimia-Castaño, P. Targeting Fibrosis: The Bridge That Connects Pancreatitis and Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 4970. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Regulatory Mediators | Primary Mechanisms of Agents on Pancreatic Stellate Cells |
|---|---|
| FTY720 | FTY720 activates the mTOR pathway to mediate apoptosis in PSCs. |
| DHA | DHA reduces ROS in PSCs to inhibit the expression of autocrine cytokines. |
| I-Smads | I-Smads inhibit intracellular signaling of TGF-β in PSCs, and the activation of Smad6 and Smad7 can downregulate activation markers in PSCs. |
| MCC950 | MCC950 inhibits the activation of NLRP3 in PSCs by suppressing the TGF-β1/Smad3 pathway. |
| 17AAG | 17AAG degrades TGFβRII through the ubiquitin-proteasome pathway, thereby inhibiting TGFβ1-induced activation of PSCs and extracellular matrix accumulation. |
| Puerarin | Puerarin inhibits the activation of MAPK family proteins (JNK1/2, ERK1/2, and p38 MAPK) in PSCs. |
| Mitoquinone (MitoQ) | MitoQ balances free radical levels and intracellular antioxidant system levels, thereby inhibiting PSC activation. |
| Imatinib (IMT) | IMT inhibits the TGF-β1/Smad signaling pathway in PSC activation. |
| S1P | Modulating the binding of S1P to S1PR2 can regulate autophagy and NLRP3 inflammasome-promoted activation of PSCs. |
| TLRs | Regulation of Toll-like receptors (TLRs) can mitigate the effects of TGF-β. |
| Nintedanib (Ninte) | Ninte inhibits the activation and proliferation of PSCs through the JAK/STAT3 and ERK1/2 pathways. |
| SPARC | In the recovery phase of recurrent acute pancreatitis and during the activation of PSCs in chronic pancreatitis, the SPARC gene is highly expressed. |
| miR-34b | The silencing of SNHG11 attenuates PSC proliferation, migration, and matrix accumulation through the miR-34b/LIF axis. |
| miR-141 | Regulation of long non-coding RNAs (LncRNAs) may play a role in controlling PSC autophagy and activation. |
| miR-301a | The silencing of miR-301a mediates effective inhibition of the Tsc1/mTOR and Gadd45g/Stat3 pathways, thereby maintaining PSC activation and fibrosis. |
| miR-15a | The miRNA modification of miR-15a to 5-FU-miR-15a significantly reduces the viability, proliferation, and migration of PSCs. |
| Reg1-3 | Deletion of the Reg gene in mice can lead to reduced pancreatic parenchyma loss, decreased collagen deposition, and reduced expression of inflammatory cytokines in chronic pancreatitis. |
| JUN | JUN is a key transcription factor in maintaining the quiescent state of PSCs. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, F.; Pan, Y.; Wu, D. Activation and Regulation of Pancreatic Stellate Cells in Chronic Pancreatic Fibrosis: A Potential Therapeutic Approach for Chronic Pancreatitis. Biomedicines 2024, 12, 108. https://doi.org/10.3390/biomedicines12010108
Kong F, Pan Y, Wu D. Activation and Regulation of Pancreatic Stellate Cells in Chronic Pancreatic Fibrosis: A Potential Therapeutic Approach for Chronic Pancreatitis. Biomedicines. 2024; 12(1):108. https://doi.org/10.3390/biomedicines12010108
Chicago/Turabian StyleKong, Fanyi, Yingyu Pan, and Dong Wu. 2024. "Activation and Regulation of Pancreatic Stellate Cells in Chronic Pancreatic Fibrosis: A Potential Therapeutic Approach for Chronic Pancreatitis" Biomedicines 12, no. 1: 108. https://doi.org/10.3390/biomedicines12010108
APA StyleKong, F., Pan, Y., & Wu, D. (2024). Activation and Regulation of Pancreatic Stellate Cells in Chronic Pancreatic Fibrosis: A Potential Therapeutic Approach for Chronic Pancreatitis. Biomedicines, 12(1), 108. https://doi.org/10.3390/biomedicines12010108