
Is Chronic Kidney Disease Due to Cadmium Exposure Inevitable and Can It Be Reversed?

Abstract
:1. Introduction
2. Environmental Cadmium Exposure and Risk of Chronic Kidney Disease
2.1. Findings from Systematic Reviews and Meta-Analyses
2.2. Exposure Levels of Concern
2.3. Impact of Normalisation of Urinary Excretion Rates of Cadmium and Albumin
2.4. Demonstrable Dose–Response Relationships
3. Cadmium and Albuminuria
3.1. Tubular Protein Reabsorption: Overview
3.2. Albuminuria in Cadmium-Exposed Subjects
3.3. Fractional Reductions in the Reabsorption of Albumin and β2M
3.4. Overall Effects of Cadmium Burden on Tubular Function
3.5. Implication of Albumin Reabsorption for the Delivery of Cadmium to Proximal Tubules
3.6. Summary on the Impact of Cadmium on Protein Reabsorptive Function
4. eGFR Decline and the Health Risk Assessment of Environmental Cadmium
4.1. Measurement of Kidney Burden of Cadmium
4.2. Cadmium Excretion and Glomerular Filtration Rate
4.3. The NOAEL Equivalent Value of Cadmium Burden
4.4. Evidence for the Threshold Level of Cadmium
5. Past and Present Health Threat of Environmental Cadmium
5.1. The WHO Exposure Guidelines and the Nephrotoxicity Threshold Level
5.2. β2-Microglobulinuria Is the Manifestation of Severe Kidney Pathologies
5.3. Cadmium and Renal Tubular Cell Death: Molecular Basis
5.3.1. Mitochondrial Dysfunction and Calcium Homeostasis Disruption
5.3.2. The Deprivation of Cellular Antioxidant Defence by Cadmium
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef]
- Murton, M.; Goff-Leggett, D.; Bobrowska, A.; Garcia Sanchez, J.J.; James, G.; Wittbrodt, E.; Nolan, S.; Sörstadius, E.; Pecoits-Filho, R.; Tuttle, K. Burden of chronic kidney disease by KDIGO categories of glomerular filtration rate and albuminuria: A Systematic review. Adv. Ther. 2021, 38, 180–200. [Google Scholar] [CrossRef]
- Rex, N.; Melk, A.; Schmitt, R. Cellular senescence and kidney aging. Clin. Sci. 2023, 137, 1805–1821. [Google Scholar] [CrossRef]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.W.; et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Gobe, G.C.; Phelps, K.R. Estimation of health risks associated with dietary cadmium exposure. Arch. Toxicol. 2023, 97, 329–358. [Google Scholar] [CrossRef]
- Fechner, C.; Hackethal, C.; Höpfner, T.; Dietrich, J.; Bloch, D.; Lindtner, O.; Sarvan, I. Results of the BfR MEAL Study: In Germany, mercury is mostly contained in fish and seafood while cadmium, lead, and nickel are present in a broad spectrum of foods. Food Chem. X 2022, 14, 100326. [Google Scholar] [CrossRef]
- Almerud, P.; Zamaratskaia, G.; Lindroos, A.K.; Bjermo, H.; Andersson, E.M.; Lundh, T.; Ankarberg, E.H.; Lignell, S. Cadmium, total mercury, and lead in blood and associations with diet, sociodemographic factors, and smoking in Swedish adolescents. Environ. Res. 2021, 197, 110991. [Google Scholar] [CrossRef]
- Pappas, R.S.; Fresquez, M.R.; Watson, C.H. Cigarette smoke cadmium breakthrough from traditional filters: Implications for exposure. J. Anal. Toxicol. 2015, 39, 45–51. [Google Scholar] [CrossRef]
- Kim, J.; Song, H.; Lee, J.; Kim, Y.J.; Chung, H.S.; Yu, J.M.; Jang, G.; Park, R.; Chung, W.; Oh, C.M.; et al. Smoking and passive smoking increases mortality through mediation effect of cadmium exposure in the United States. Sci. Rep. 2023, 13, 3878. [Google Scholar] [CrossRef]
- Thévenod, F.; Herbrechter, R.; Schlabs, C.; Pethe, A.; Lee, W.K.; Wolff, N.A.; Roussa, E. Role of the SLC22A17/lipocalin-2 receptor in renal endocytosis of proteins/metalloproteins: A focus on iron- and cadmium-binding proteins. Am. J. Physiol. Ren. Physiol. 2023, 325, F564–F577. [Google Scholar] [CrossRef]
- Rentschler, G.; Kippler, M.; Axmon, A.; Raqib, R.; Skerfving, S.; Vahter, M.; Broberg, K. Cadmium concentrations in human blood and urine are associated with polymorphisms in zinc transporter genes. Metallomics 2014, 6, 885–891. [Google Scholar] [CrossRef]
- Rentschler, G.; Kippler, M.; Axmon, A.; Raqib, R.; Ekström, E.C.; Skerfving, S.; Vahter, M.; Broberg, K. Polymorphisms in iron homeostasis genes and urinary cadmium concentrations among nonsmoking women in Argentina and Bangladesh. Environ. Health Perspect. 2013, 121, 467–472. [Google Scholar] [CrossRef]
- Ng, E.; Lind, P.M.; Lindgren, C.; Ingelsson, E.; Mahajan, A.; Morris, A.; Lind, L. Genome-wide association study of toxic metals and trace elements reveals novel associations. Hum. Mol. Genet. 2015, 24, 4739–4745. [Google Scholar] [CrossRef]
- Peng, X.; Li, C.; Zhao, D.; Huang, L. Associations of micronutrients exposure with cadmium body burden among population: A systematic review. Ecotoxicol. Environ. Saf. 2023, 256, 114878. [Google Scholar] [CrossRef]
- Satarug, S.; Baker, J.R.; Reilly, P.E.B.; Moore, M.R.; Williams, D.J. Cadmium levels in the lung, liver, kidney cortex, and urine samples from Australians without occupational exposure to metals. Arch. Environ. Health 2002, 57, 69–77. [Google Scholar] [CrossRef]
- Elinder, C.G.; Lind, B.; Kjellström, T.; Linnman, L.; Friberg, L. Cadmium in kidney cortex, liver, and pancreas from Swedish autopsies. Estimation of biological half time in kidney cortex, considering calorie intake and smoking habits. Arch. Environ. Health 1976, 31, 292–302. [Google Scholar] [CrossRef]
- Barregard, L.; Sallsten, G.; Lundh, T.; Mölne, J. Low-level exposure to lead, cadmium and mercury, and histopathological findings in kidney biopsies. Environ. Res. 2022, 211, 113119. [Google Scholar] [CrossRef]
- Argyropoulos, C.P.; Chen, S.S.; Ng, Y.-H.; Roumelioti, M.-E.; Shaffi, K.; Singh, P.P.; Tzamaloukas, A.H. Rediscovering Beta-2 microglobulin as a biomarker across the spectrum of kidney diseases. Front. Med. 2017, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Konta, T.; Mashima, Y.; Ichikawa, K.; Takasaki, S.; Ikeda, A.; Hoshikawa, M.; Suzuki, K.; Shibata, Y.; Watanabe, T.; et al. The association between renal tubular damage and rapid renal deterioration in the Japanese population: The Takahata study. Clin. Exp. Nephrol. 2011, 15, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Doccioli, C.; Sera, F.; Francavilla, A.; Cupisti, A.; Biggeri, A. Association of cadmium environmental exposure with chronic kidney disease: A systematic review and meta-analysis. Sci. Total Environ. 2024, 906, 167165. [Google Scholar] [CrossRef] [PubMed]
- Jalili, C.; Kazemi, M.; Cheng, H.; Mohammadi, H.; Babaei, A.; Taheri, E.; Moradi, S. Associations between exposure to heavy metals and the risk of chronic kidney disease: A systematic review and meta-analysis. Crit. Rev. Toxicol. 2021, 51, 165–182. [Google Scholar] [CrossRef]
- Byber, K.; Lison, D.; Verougstraete, V.; Dressel, H.; Hotz, P. Cadmium or cadmium compounds and chronic kidney disease in workers and the general population: A systematic review. Crit. Rev. Toxicol. 2016, 46, 191–240. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Gobe, G.C.; Yimthiang, S.; Buha Đorđević, A. Health risk in a geographic area of Thailand with endemic cadmium contamination: Focus on albuminuria. Toxics 2023, 11, 68. [Google Scholar] [CrossRef]
- Satarug, S.; Đorđević, A.B.; Yimthiang, S.; Vesey, D.A.; Gobe, G.C. The NOAEL equivalent of environmental cadmium exposure associated with GFR reduction and chronic kidney disease. Toxics 2022, 10, 614. [Google Scholar] [CrossRef]
- Myong, J.-P.; Kim, H.-R.; Baker, D.; Choi, B. Blood cadmium and moderate-to-severe glomerular dysfunction in Korean adults: Analysis of KNHANES 2005–2008 data. Int. Arch. Occup. Environ. Health 2012, 85, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Chung, J.H.; Kim, S.J.; Koh, E.S.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Shin, S.J. Blood lead and cadmium levels and renal function in Korean adults. Clin. Exp. Nephrol. 2014, 18, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.J.; Hung, C.H.; Wang, C.W.; Tu, H.P.; Li, C.H.; Tsai, C.C.; Lin, W.Y.; Chen, S.C.; Kuo, C.H. Associations among heavy metals and proteinuria and chronic kidney disease. Diagnostics 2021, 11, 282. [Google Scholar] [CrossRef]
- Feng, X.; Zhou, R.; Jiang, Q.; Wang, Y.; Chen, C. Analysis of cadmium accumulation in community adults and its correlation with low-grade albuminuria. Sci. Total Environ. 2022, 834, 155210. [Google Scholar] [CrossRef] [PubMed]
- Grau-Perez, M.; Pichler, G.; Galan-Chilet, I.; Briongos-Figuero, L.S.; Rentero-Garrido, P.; Lopez-Izquierdo, R.; Navas-Acien, A.; Weaver, V.; García-Barrera, T.; Gomez-Ariza, J.L.; et al. Urine cadmium levels and albuminuria in a general population from Spain: A gene-environment interaction analysis. Environ. Int. 2017, 106, 27–36. [Google Scholar] [CrossRef]
- Navas-Acien, A.; Tellez-Plaza, M.; Guallar, E.; Muntner, P.; Silbergeld, E.; Jaar, B.; Weaver, V. Blood cadmium and lead and chronic kidney disease in US adults: A joint analysis. Am. J. Epidemiol. 2009, 170, 1156–1164. [Google Scholar] [CrossRef]
- Ferraro, P.M.; Costanzi, S.; Naticchia, A.; Sturniolo, A.; Gambaro, G. Low level exposure to cadmium increases the risk of chronic kidney disease: Analysis of the NHANES 1999–2006. BMC Publ. Health 2010, 10, 304. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, J.M.; Ricardo, A.C.; Persky, V.; Turyk, M. Associations between blood cadmium concentration and kidney function in the U.S. population: Impact of sex, diabetes and hypertension. Environ. Res. 2018, 169, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Ma, Z.; Dang, Y.; Yang, Y.; Cao, S.; Ouyang, C.; Shi, X.; Pan, J.; Hu, X. Associations of urinary and blood cadmium concentrations with all-cause mortality in US adults with chronic kidney disease: A prospective cohort study. Environ. Sci. Pollut. Res. Int. 2023, 30, 61659–61671. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Caffrey, J.L.; Lin, J.W.; Bayliss, D.; Faramawi, M.F.; Bateson, T.F.; Sonawane, B. Increased risk of cancer mortality associated with cadmium exposures in older Americans with low zinc intake. J. Toxicol. Environ Health A 2013, 76, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Wang, C.W.; Chen, H.C.; Tu, H.P.; Chen, S.C.; Hung, C.H.; Kuo, C.H. Gender difference in the associations among heavy metals with red blood cell hemogram. Int. J. Environ. Res. Public Health 2021, 19, 189. [Google Scholar] [CrossRef] [PubMed]
- Olsén, L.; Lind, P.M.; Lind, L. Gender differences for associations between circulating levels of metals and coronary risk in the elderly. Int. J. Hyg. Environ. Health 2012, 215, 411–417. [Google Scholar] [CrossRef]
- Sun, H.; Wang, D.; Zhou, Z.; Ding, Z.; Chen, X.; Xu, Y.; Huang, L.; Tang, D. Association of cadmium in urine and blood with age in a general population with low environmental exposure. Chemosphere 2016, 156, 392–397. [Google Scholar] [CrossRef]
- Butler-Dawson, J.; James, K.A.; Krisher, L.; Jaramillo, D.; Dally, M.; Neumann, N.; Pilloni, D.; Cruz, A.; Asensio, C.; Johnson, R.J.; et al. Environmental metal exposures and kidney function of Guatemalan sugarcane workers. J. Expo. Sci. Environ. Epidemiol. 2022, 32, 461–471. [Google Scholar] [CrossRef]
- Win-Thu, M.; Myint-Thein, O.; Win-Shwe, T.-T.; Mar, O. Environmental cadmium exposure induces kidney tubular and glomerular dysfunction in the Myanmar adults. J. Toxicol. Sci. 2021, 46, 319–328. [Google Scholar] [CrossRef]
- Skröder, H.; Hawkesworth, S.; Kippler, M.; El Arifeen, S.; Wagatsuma, Y.; Moore, S.E.; Vahter, M. Kidney function and blood pressure in preschool-aged children exposed to cadmium and arsenic-potential alleviation by selenium. Environ. Res. 2015, 140, 205–213. [Google Scholar] [CrossRef]
- Rodríguez-López, E.; Tamayo-Ortiz, M.; Ariza, A.C.; Ortiz-Panozo, E.; Deierlein, A.L.; Pantic, I.; Tolentino, M.C.; Es-trada-Gutiérrez, G.; Parra-Hernández, S.; Espejel-Núñez, A.; et al. Early-life dietary cadmium exposure and kidney function in 9-year-old children from the PROGRESS cohort. Toxics 2020, 8, 83. [Google Scholar] [CrossRef]
- Phelps, K.R.; Gosmanova, E.O. A generic method for analysis of plasma concentrations. Clin. Nephrol. 2020, 94, 43–49. [Google Scholar] [CrossRef]
- Denic, A.; Elsherbiny, H.; Rule, A.D. In-vivo techniques for determining nephron number. Curr. Opin. Nephrol. Hypertens. 2019, 28, 545–551. [Google Scholar] [CrossRef]
- Soveri, I.; Berg, U.B.; Björk, J.; Elinder, C.G.; Grubb, A.; Mejare, I.; Sterner, G.; Bäck, S.E.; SBU GFR Review Group. Measuring GFR: A systematic review. Am. J. Kidney Dis. 2014, 64, 411–424. [Google Scholar] [CrossRef]
- White, C.A.; Allen, C.M.; Akbari, A.; Collier, C.P.; Holland, D.C.; Day, A.G.; Knoll, G.A. Comparison of the new and traditional CKD-EPI GFR estimation equations with urinary inulin clearance: A study of equation performance. Clin. Chim. Acta 2019, 488, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Vesey, D.A.; Ruangyuttikarn, W.; Nishijo, M.; Gobe, G.C.; Phelps, K.R. The source and pathophysiologic significance of excreted cadmium. Toxics 2019, 7, 55. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Gobe, G.C. Cadmium-induced proteinuria: Mechanistic insights from dose–effect analyses. Int. J. Mol. Sci. 2023, 24, 1893. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Stevens, L.A.; Scmid, C.H.; Zhang, Y.; Castro, A.F., III; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Vesey, D.A.; Gobe, G.C.; Phelps, K.R. The pathogenesis of albuminuria in cadmium nephropathy. Curr. Res. Toxicol. 2024, 6, 100140. [Google Scholar] [CrossRef] [PubMed]
- Navar, L.G.; Maddox, D.A.; Munger, K.A. Chapter 3: The renal circulations and glomerular filtration. In Brenner and Rector’s the Kidney, 11th ed.; Elsevier: Philadelphia, PA, USA, 2020; pp. 80–114. [Google Scholar]
- Gauthier, C.; Nguyen-Simonnet, H.; Vincent, C.; Revillard, J.-P.; Pellet, M.V. Renal tubular absorption of β2-microglobulin. Kidney Int. 1984, 26, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.G.W.; Lapsley, M.; Lee, P.J.; Pusey, C.D.; Scheinman, S.J.; Tam, F.W.K.; Thakker, R.V.; Unwin, R.J.; Wrong, O. Glomerular protein sieving and implications for renal failure in Fanconi syndrome. Kidney Int. 2001, 60, 1885–1892. [Google Scholar] [CrossRef]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef]
- Molitoris, B.A.; Sandoval, R.M.; Yadav, S.P.S.; Wagner, M.C. Albumin uptake and processing by the proximal tubule: Physiological, pathological, and therapeutic implications. Physiol. Rev. 2022, 102, 1625–1667. [Google Scholar] [CrossRef]
- Eshbach, M.L.; Weisz, O.A. Receptor-mediated endocytosis in the proximal tubule. Annu. Rev. Physiol. 2017, 79, 425–448. [Google Scholar] [CrossRef]
- Comper, W.D.; Vuchkova, J.; McCarthy, K.J. New insights into proteinuria/albuminuria. Front. Physiol. 2022, 13, 991756. [Google Scholar] [CrossRef]
- Benzing, T.; Salant, D. Insights into glomerular filtration and albuminuria. N. Engl. J. Med. 2021, 384, 1437–1446. [Google Scholar] [CrossRef]
- Clapp, W.L.; Park, C.H.; Madsen, K.M.; Tisher, C.C. Axial heterogeneity in the handling of albumin by the rabbit proximal tubule. Lab. Investig. 1988, 58, 549–558. [Google Scholar] [PubMed]
- Schuh, C.D.; Polesel, M.; Platonova, E.; Haenni, D.; Gassama, A.; Tokonami, N.; Ghazi, S.; Bugarski, M.; Devuyst, O.; Ziegler, U.; et al. Combined structural and functional imaging of the kidney reveals major axial differences in proximal tubule endocytosis. J. Am. Soc. Nephrol. 2018, 29, 2696–2712. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.C.; Sandoval, R.M.; Yadav, S.P.S.; Campos, S.B.; Rhodes, G.J.; Phillips, C.L.; Molitoris, B.A. Lrpap1 (RAP) inhibits proximal tubule clathrin mediated and clathrin independent endocytosis, ameliorating renal aminoglycoside nephrotoxicity. Kidney360 2023, 4, 591–605. [Google Scholar] [CrossRef]
- Satarug, S.; Phelps, K.R. Cadmium Exposure and Toxicity. In Metal Toxicology Handbook; Bagchi, D., Bagchi, M., Eds.; CRC Press: Boca Raton, FL, USA, 2021; pp. 219–274. [Google Scholar]
- Polesel, M.; Kaminska, M.; Haenni, D.; Bugarski, M.; Schuh, C.; Jankovic, N.; Kaech, A.; Mateos, J.M.; Berquez, M.; Hall, A.M. Spatiotemporal organisation of protein processing in the kidney. Nat. Commun. 2022, 13, 5732. [Google Scholar] [CrossRef] [PubMed]
- Sarav, M.; Wang, Y.; Hack, B.K.; Chang, A.; Jensen, M.; Bao, L.; Quigg, R.J. Renal FcRn reclaims albumin but facilitates elimination of IgG. J. Am. Soc. Nephrol. 2009, 20, 1941–1952. [Google Scholar] [CrossRef]
- Tenten, V.; Menzel, S.; Kunter, U.; Sicking, E.-M.; van Roeyen, C.R.C.; Sanden, S.K.; Kaldenbach, M.; Boor, P.; Fuss, A.; Uhlig, S.; et al. Albumin is recycled from the primary urine by tubular transcytosis. J. Am. Soc. Nephrol. 2013, 24, 1966–1980. [Google Scholar] [CrossRef] [PubMed]
- Tojo, A.; Endou, H. Intrarenal handling of proteins in rats using fractional micropuncture technique. Am. J. Physiol. 1992, 263, F601–F606. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, G.F.; Piscator, M.; Nordberg, M. On the distribution of cadmium in blood. Acta Pharmacol. Toxicol. 1971, 30, 289–295. [Google Scholar] [CrossRef]
- Gibson, M.A.; Sarpong-Kumankomah, S.; Nehzati, S.; George, G.N.; Gailer, J. Remarkable differences in the biochemical fate of Cd2+, Hg2+, CH3Hg+ and thimerosal in red blood cell lysate. Metallomics 2017, 9, 1060–1072. [Google Scholar] [CrossRef]
- Turell, L.; Radi, R.; Alvarez, B. The thiol pool in human plasma: The central contribution of albumin to redox processes. Free Radic. Biol. Med. 2013, 65, 244–253. [Google Scholar] [CrossRef]
- Fels, J.; Scharner, B.; Zarbock, R.; Zavala Guevara, I.P.; Lee, W.K.; Barbier, O.C.; Thévenod, F. Cadmium complexed with β2-microglobulin, albumin and lipocalin-2 rather than metallothionein cause megalin:cubilin dependent toxicity of the renal proximal tubule. Int. J. Mol. Sci. 2019, 20, 2379. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, M.; Jiang, L.; Song, L. New insight into molecular interaction of heavy metal pollutant–cadmium (II) with human serum albumin. Environ. Sci. Pollut. Res. 2014, 21, 6994–7005. [Google Scholar] [CrossRef] [PubMed]
- Gburek, J.; Konopska, B.; Gołąb, K. Renal handling of albumin-from early findings to current concepts. Int. J. Mol. Sci. 2021, 22, 5809. [Google Scholar] [CrossRef]
- Edwards, A.; Long, K.R.; Baty, C.J.; Shipman, K.E.; Weisz, O.A. Modelling normal and nephrotic axial uptake of albumin and other filtered proteins along the proximal tubule. J. Physiol. 2022, 600, 1933–1952. [Google Scholar] [CrossRef]
- Castrop, H.; Schießl, I.M. Novel routes of albumin passage across the glomerular filtration barrier. Acta Physiol. 2017, 219, 544–553. [Google Scholar] [CrossRef]
- Santoyo-Sánchez, M.P.; Pedraza-Chaverri, J.; Molina-Jijón, E.; Arreola-Mendoza, L.; Rodríguez-Muñoz, R.; Barbier, O.C. Impaired endocytosis in proximal tubule from subchronic exposure to cadmium involves angiotensin II type 1 and cubilin receptors. BMC Nephrol. 2013, 14, 211. [Google Scholar] [CrossRef]
- Li, L.; Dong, F.; Xu, D.; Du, L.; Yan, S.; Hu, H.; Lobe, C.G.; Yi, F.; Kapron, C.M.; Liu, J. Short-term, low-dose cadmium exposure induces hyperpermeability in human renal glomerular endothelial cells. J. Appl. Toxicol. 2016, 36, 257–265. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, L.; Tao, T.; Su, W.; Guo, Y.; Yu, H.; Qin, J. Assessment of cadmium-induced nephrotoxicity using a kidney-on-a-chip device. Toxicol. Res. 2017, 6, 372–380. [Google Scholar] [CrossRef]
- Gena, P.; Calamita, G.; Guggino, W.B. Cadmium impairs albumin reabsorption by down-regulating megalin and ClC5 channels in renal proximal tubule cells. Environ. Health Perspect. 2010, 118, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.W.; Pongsakul, P.; Saiyasitpanich, D.; Klinpholap, S. Elevated levels of cadmium and zinc in paddy soils and elevated levels of cadmium in rice grain downstream of a zinc mineralized area in Thailand: Implications for public health. Environ. Geochem. Health 2005, 27, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Suwatvitayakorn, P.; Ko, M.S.; Kim, K.W.; Chanpiwat, P. Human health risk assessment of cadmium exposure through rice consumption in cadmium-contaminated areas of the Mae Tao sub-district, Tak, Thailand. Environ. Geochem. Health 2020, 42, 2331–2344. [Google Scholar] [CrossRef]
- Swaddiwudhipong, W.; Nguntra, P.; Kaewnate, Y.; Mahasakpan, P.; Limpatanachote, P.; Aunjai, T.; Jeekeeree, W.; Punta, B.; Funkhiew, T.; Phopueng, I. Human health effects from cadmium exposure: Comparison between persons living in cadmium-contaminated and non-contaminated areas in northwestern Thailand. Southeast Asian J. Trop. Med. Publ. Health 2015, 46, 133–142. [Google Scholar]
- Nishijo, M.; Suwazono, Y.; Ruangyuttikarn, W.; Nambunmee, K.; Swaddiwudhipong, W.; Nogawa, K.; Nakagawa, H. Risk assessment for Thai population: Benchmark dose of urinary and blood cadmium levels for renal effects by hybrid approach of inhabitants living in polluted and non-polluted areas in Thailand. BMC Publ. Health 2014, 14, 702. [Google Scholar] [CrossRef]
- Guo, D.; Liu, R. Spectroscopic investigation of the effects of aqueous-phase prepared CdTe quantum dots on protein hemoglobin at the molecular level. J. Biochem. Mol. Toxicol. 2017, 31, 21953. [Google Scholar] [CrossRef]
- Sopjani, M.; Föller, M.; Dreischer, P.; Lang, F. Stimulation of eryptosis by cadmium ions. Cell Physiol. Biochem. 2008, 22, 245–252. [Google Scholar] [CrossRef]
- Lang, F.; Bissinger, R.; Abed, M.; Artunc, F. Eryptosis—The neglected cause of anemia in end stage renal disease. Kidney Blood Press. Res. 2017, 42, 749–760. [Google Scholar] [CrossRef]
- Horiguchi, H.; Oguma, E.; Kayama, F. Cadmium induces anemia through interdependent progress of hemolysis, body iron accumulation, and insufficient erythropoietin production in rats. Toxicol. Sci. 2011, 122, 198–210. [Google Scholar] [CrossRef]
- Gray, R.D.; Stroupe, S.D. Kinetics and mechanism of bilirubin binding to human serum albumin. J. Biol. Chem. 1978, 253, 4370–4377. [Google Scholar] [CrossRef]
- Petersen, C.E.; Ha, C.-E.; Harohalli, K.; Feix, J.B.; Bhagavan, N.V. A dynamic model for bilirubin binding to human serum albumin. J. Biol. Chem. 2000, 275, 20985–20995. [Google Scholar] [CrossRef]
- Torra, M.; To-Figueras, J.; Brunet, M.; Rodamilans, M.; Corbella, J. Total and methionein-bound cadmium in the liver and the kidney of a population in Barcelona (Spain). Bull. Environ. Contam. Toxicol. 1994, 53, 509–515. [Google Scholar] [CrossRef]
- Yoshida, M.; Ohta, H.; Yamauchi, Y.; Seki, Y.; Sagi, M.; Yamazaki, K.; Sumi, Y. Age-dependent changes in metallothionein levels in liver and kidney of the Japanese. Biol. Trace Elem. Res. 1998, 63, 167–175. [Google Scholar] [CrossRef]
- DelRaso, N.J.; Foy, B.D.; Gearhart, J.M.; Frazier, J.M. Cadmium uptake kinetics in rat hepatocytes: Correction for albumin binding. Toxicol. Sci. 2003, 72, 19–30. [Google Scholar] [CrossRef]
- Chen, M.; Guo, H.; Liu, Y.; Zhang, Q. Structural changes of human serum albumin induced by calcium acetate. J. Biochem. Mol. Toxicol. 2014, 28, 281–287. [Google Scholar] [CrossRef]
- EFSA. European Food Safety Agency, Statement on tolerable weekly intake for cadmium. EFSA J. 2011, 9, 1975. [Google Scholar]
- EFSA Scientific Committee. Update: Use of the benchmark dose approach in risk assessment. EFSA J. 2017, 15, 4658. [Google Scholar]
- Wong, C.; Roberts, S.M.; Saab, I.N. Review of regulatory reference values and background levels for heavy metals in the human diet. Regul. Toxicol. Pharmacol. 2022, 130, 105122. [Google Scholar] [CrossRef]
- Lyon, T.D.; Aughey, E.; Scott, R.; Fell, G.S. Cadmium concentrations in human kidney in the UK: 1978–1993. J. Environ. Monit. 1999, 1, 227–231. [Google Scholar] [CrossRef]
- Benedetti, J.L.; Samuel, O.; Dewailly, E.; Gingras, S.; Lefebvre, M.A. Levels of cadmium in kidney and liver tissues among a Canadian population (province of Quebec). J. Toxicol. Environ. Health 1999, 56, 145–163. [Google Scholar] [CrossRef]
- Johansen, P.; Mulvad, G.; Pedersen, H.S.; Hansen, J.C.; Riget, F. Accumulation of cadmium in livers and kidneys in Greenlanders. Sci. Total Environ. 2006, 372, 58–63. [Google Scholar] [CrossRef]
- Barregard, L.; Fabricius-Lagging, E.; Lundh, T.; Mölne, J.; Wallin, M.; Olausson, M.; Modigh, C.; Sallsten, G. Cadmium, mercury, and lead in kidney cortex of living kidney donors: Impact of different exposure sources. Environ. Res. 2010, 110, 47–54. [Google Scholar] [CrossRef]
- Schaefer, H.R.; Flannery, B.M.; Crosby, L.M.; Pouillot, R.; Farakos, S.M.S.; Van Doren, J.M.; Dennis, S.; Fitzpatrick, S.; Middleton, K. Reassessment of the cadmium toxicological reference value for use in human health assessments of foods. Regul. Toxicol. Pharmacol. 2023, 144, 105487. [Google Scholar] [CrossRef]
- Fleming, R.E.; Bacon, B.R. Orchestration of iron homeostasis. N. Engl. J. Med. 2005, 352, 1741–1744. [Google Scholar] [CrossRef]
- Camaschella, C. Iron-deficiency anemia. N. Engl. J. Med. 2015, 373, 485–486. [Google Scholar] [CrossRef]
- Weaver, V.M.; Kim, N.S.; Jaar, B.G.; Schwartz, B.S.; Parsons, P.J.; Steuerwald, A.J.; Todd, A.C.; Simon, D.; Lee, B.K. Associations of low-level urine cadmium with kidney function in lead workers. Occup. Environ. Med. 2011, 68, 250–256. [Google Scholar] [CrossRef]
- Jin, R.; Zhu, X.; Shrubsole, M.J.; Yu, C.; Xia, Z.; Dai, Q. Associations of renal function with urinary excretion of metals: Evidence from NHANES 2003–2012. Environ. Int. 2018, 121, 1355–1362. [Google Scholar] [CrossRef]
- Buser, M.C.; Ingber, S.Z.; Raines, N.; Fowler, D.A.; Scinicariello, F. Urinary and blood cadmium and lead and kidney function: NHANES 2007–2012. Int. J. Hyg. Environ. Health 2016, 219, 261–267. [Google Scholar] [CrossRef]
- Akesson, A.; Lundh, T.; Vahter, M.; Bjellerup, P.; Lidfeldt, J.; Nerbrand, C.; Samsioe, G.; Strömberg, U.; Skerfving, S. Tubular and glomerular kidney effects in Swedish women with low environmental cadmium exposure. Environ. Health Perspect. 2005, 113, 1627–1631. [Google Scholar] [CrossRef]
- Satarug, S.; Gobe, G.C.; Ujjin, P.; Vesey, D.A. A comparison of the nephrotoxicity of low doses of cadmium and lead. Toxics 2020, 8, 18. [Google Scholar] [CrossRef]
- Saito, H.; Shioji, R.; Hurukawa, Y.; Nagai, K.; Arikawa, T. Cadmium-induced proximal tubular dysfunction in a cadmium-polluted area. Contrib. Nephrol. 1977, 6, 1–12. [Google Scholar]
- Roels, H.A.; Lauwerys, R.R.; Buchet, J.P.; Bernard, A.M.; Vos, A.; Oversteyns, M. Health significance of cadmium induced renal dysfunction: A five year follow up. Br. J. Ind. Med. 1989, 46, 755–764. [Google Scholar] [CrossRef]
- Järup, L.; Persson, B.; Elinder, C.G. Decreased glomerular filtration rate in solderers exposed to cadmium. Occup. Environ. Med. 1995, 52, 818–822. [Google Scholar] [CrossRef]
- Swaddiwudhipong, W.; Limpatanachote, P.; Mahasakpan, P.; Krintratun, S.; Punta, B.; Funkhiew, T. Progress in cadmium-related health effects in persons with high environmental exposure in northwestern Thailand: A five-year follow-up. Environ. Res. 2012, 112, 194–198. [Google Scholar] [CrossRef]
- Schnaper, H.W. The tubulointerstitial pathophysiology of progressive kidney disease. Adv. Chronic Kidney Dis. 2017, 24, 107–116. [Google Scholar] [CrossRef]
- Chevalier, R.L. The proximal tubule is the primary target of injury and progression of kidney disease: Role of the glomerulotubular junction. Am. J. Physiol. Ren. Physiol. 2016, 311, F145–F161. [Google Scholar] [CrossRef]
- Moffett, D.B.; Mumtaz, M.M.; Sullivan, D.W., Jr.; Whittaker, M.H. Chapter 13, General Considerations of Dose-Effect and Dose-Response Relationships. In Handbook on the Toxicology of Metals, 5th ed.; Nordberg, G., Costa, M., Eds.; Academic Press: Cambridge, MA, USA, 2022; Volume I: General Considerations, pp. 299–317. [Google Scholar]
- Satarug, S.; Vesey, D.A.; Khamphaya, T.; Pouyfung, P.; Gobe, G.C.; Yimthiang, S. Estimation of the cadmium nephrotoxicity threshold from loss of glomerular filtration rate and albuminuria. Toxics 2023, 11, 755. [Google Scholar] [CrossRef]
- Aoshima, K. Epidemiology and tubular dysfunction in the inhabitants of a cadmium-polluted area in the Jinzu River basin in Toyama Prefecture. Tohoku J. Exp. Med. 1987, 152, 151–172. [Google Scholar] [CrossRef]
- JECFA. In Proceedings of the Joint FAO/WHO Expert Committee on Food Additives and Contaminants, Seventy-Third Meeting, Geneva, Switzerland, 8–17 June 2010; In Summary and Conclusions; JECFA/73/SC; Food and Agriculture Organization of the United Nations/World Health Organization: Geneva, Switzerland, 2011; Available online: https://apps.who.int/iris/handle/10665/44521 (accessed on 12 February 2024).
- Codex Alimentarius, CODEX STAN 193–1995, General Standard for Contaminants and Toxins in Food and Feed. Available online: http://www.fao.org/fileadmin/user_upload/livestockgov/documents/1_CXS_193e.pdf (accessed on 12 February 2024).
- Nogawa, K.; Sakurai, M.; Ishizaki, M.; Kido, T.; Nakagawa, H.; Suwazono, Y. Threshold limit values of the cadmium concentration in rice in the development of itai-itai disease using benchmark dose analysis. J. Appl. Toxicol. 2017, 37, 962–966. [Google Scholar] [CrossRef]
- Nishijo, M.; Nogawa, K.; Suwazono, Y.; Kido, T.; Sakurai, M.; Nakagawa, H. Lifetime cadmium exposure and mortality for renal diseases in residents of the cadmium-polluted Kakehashi River Basin in Japan. Toxics 2020, 8, 81. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, Y.; Su, J.; Bai, Z.; Li, T.; Wu, Y. Maximum cadmium limits establishment strategy based on the dietary exposure estimation: An example from Chinese populations and subgroups. Environ. Sci. Pollut. Res. Int. 2018, 25, 18762–18771. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Nishijo, M.; Ruangyuttikarn, W.; Gobe, G.C. The inverse association of glomerular function and urinary β2-MG excretion and its implications for cadmium health risk assessment. Environ. Res. 2019, 173, 40–47. [Google Scholar] [CrossRef]
- Schardijn, G.H.C.; Statius van Eps, L.W. β2-microglobulin: Its significance in the evaluation of renal function. Kidney Int. 1987, 32, 635–641. [Google Scholar] [CrossRef]
- Hall, P.W., III; Chung-Park, M.; Vacca, C.V.; London, M.; Crowley, A.Q. The renal handling of beta2-microglobulin in the dog. Kidney Int. 1982, 22, 156–161. [Google Scholar] [CrossRef]
- Portman, R.J.; Kissane, J.M.; Robson, A.M. Use of β2-microglobulin to diagnose tubulo-interstitial renal lesions in children. Kidney Int. 1986, 30, 91–98. [Google Scholar] [CrossRef]
- Bernard, A. Renal dysfunction induced by cadmium: Biomarkers of critical effects. Biometals 2004, 17, 519–523. [Google Scholar] [CrossRef]
- Nogawa, K.; Kobayashi, E.; Honda, R. A study of the relationship between cadmium concentrations in urine and renal effects of cadmium. Environ. Health Perspect. 1979, 28, 161–168. [Google Scholar] [CrossRef]
- Ikeda, M.; Ezaki, T.; Moriguchi, J.; Fukui, Y.; Ukai, H.; Okamoto, S.; Sakurai, H. The threshold cadmium level that causes a substantial increase in β2-microglobulin in urine of general populations. Tohoku J. Exp. Med. 2005, 205, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Peterson, P.A.; Evrin, P.-E.; Berggard, I. Differentiation of glomerular, tubular, and normal proteinuria: Determinations of urinary excretion of β2-microglobulin, albumin, and total protein. J. Clin. Investig. 1969, 48, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.K.; Thévenod, F. Cell organelles as targets of mammalian cadmium toxicity. Arch. Toxicol. 2020, 94, 1017–1049. [Google Scholar] [CrossRef]
- Thévenod, F.; Lee, W.K.; Garrick, M.D. Iron and cadmium entry into renal mitochondria: Physiological and toxicological implications. Front. Cell Dev. Biol. 2020, 8, 848. [Google Scholar] [CrossRef]
- Ning, B.; Guo, C.; Kong, A.; Li, K.; Xie, Y.; Shi, H.; Gu, J. Calcium signaling mediates cell death and crosstalk with autophagy in kidney disease. Cells 2021, 10, 3204. [Google Scholar] [CrossRef]
- Liu, F.; Li, Z.F.; Wang, Z.Y.; Wang, L. Role of subcellular calcium redistribution in regulating apoptosis and autophagy in cadmium-exposed primary rat proximal tubular cells. J. Inorg. Biochem. 2016, 164, 99–109. [Google Scholar] [CrossRef]
- Li, K.; Guo, C.; Ruan, J.; Ning, B.; Wong, C.K.-C.; Shi, H.; Gu, J. Cadmium disrupted ER Ca2+ homeostasis by inhibiting SERCA2 expression and activity to induce apoptosis in renal proximal tubular cells. Int. J. Mol. Sci. 2023, 24, 5979. [Google Scholar] [CrossRef]
- Takeda, T.A.; Mu, A.; Tai, T.T.; Kitajima, S.; Taketani, S. Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci. Rep. 2015, 5, 10488. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A bilirubin-inducible fluorescent protein from eel muscle. Cell 2013, 153, 1602–1611. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
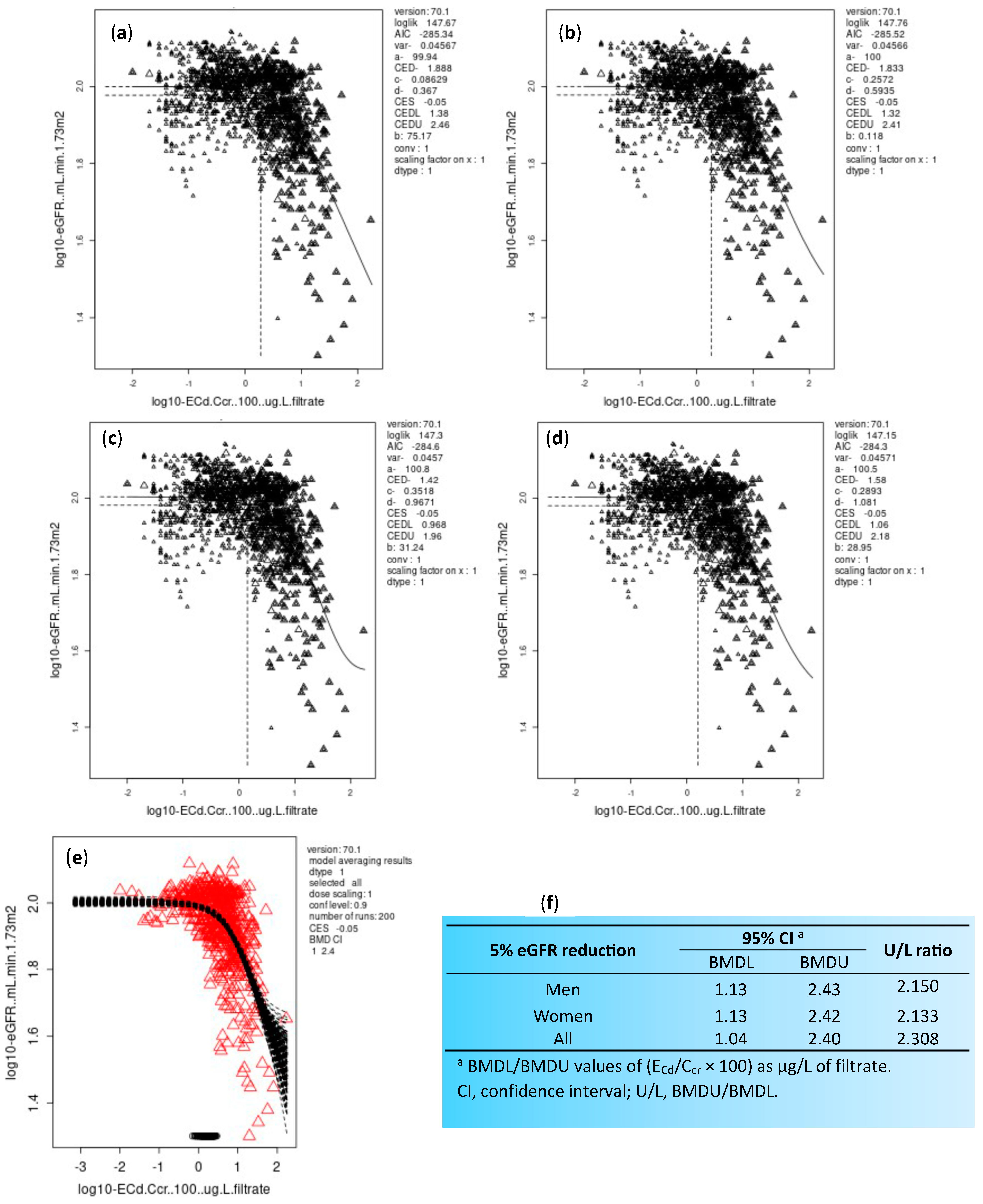{kind=link}
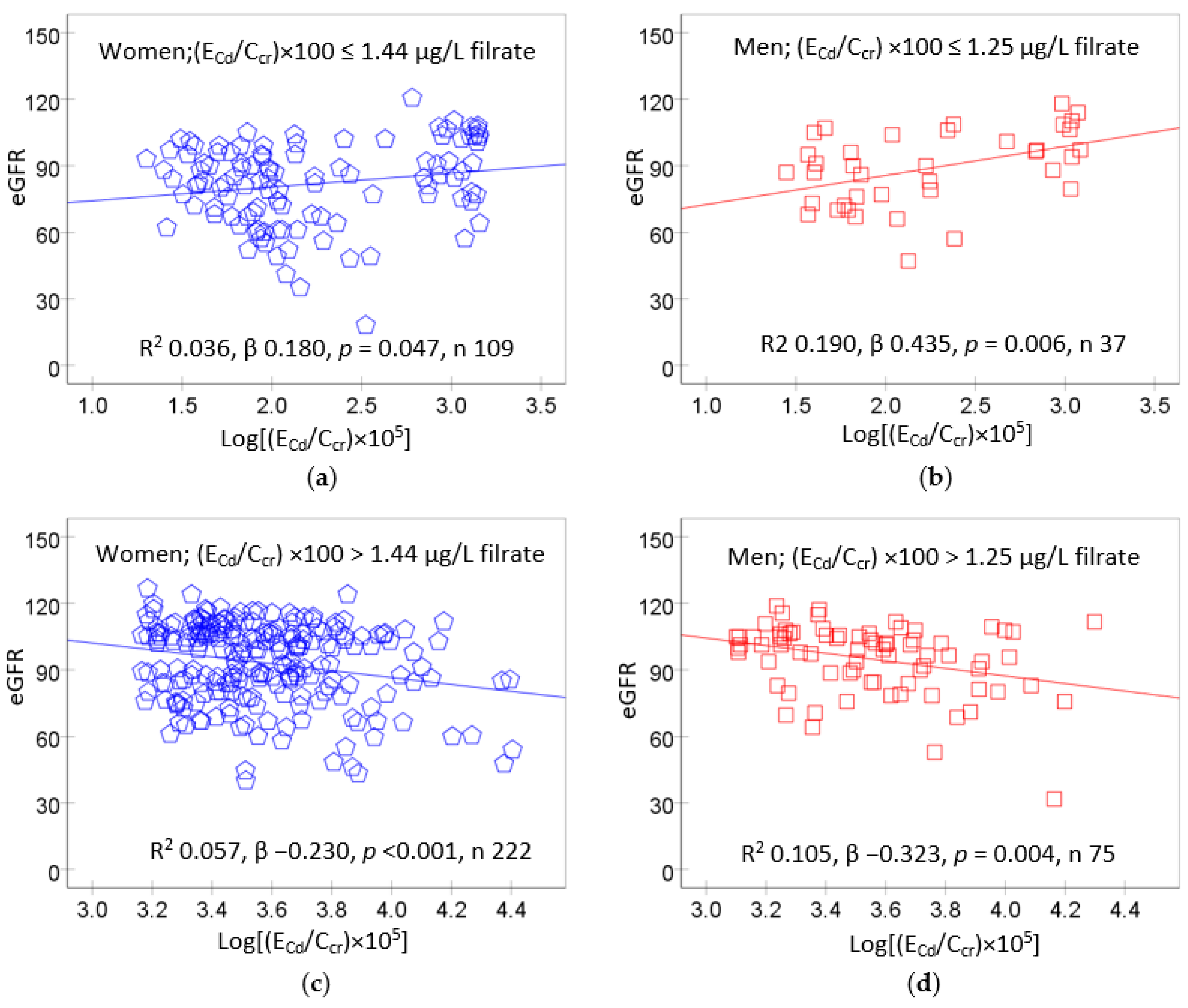{kind=link}
| Study Location | Cadmium Exposure Metrics and Effects Observed | Reference |
|---|---|---|
| Thailand, n 1189 16–87 years mean age 43.2 years | Risk of low eGFR increased 6.2-fold and 10.6-fold, comparing urinary Cd levels 0.38–2.49 and ≥2.5 µg/g creatinine with ≤0.37 µg/g creatinine, respectively. | Satarug et al., 2022 [24]. |
| Korea, n 2992 20–65 years | Increased risk of low eGFR (OR 1.97) in women was associated with blood Cd levels > 1.74 μg/L. | Myong et al., 2012 [25] |
| Korea, n 2005 ≥20 years | Increased risk of low eGFR (OR 1.93) was associated with blood Cd in the top quartile (mean, 2.08 μg/L). | Chung et al., 2014 [26] |
| Taiwan, n 2447 mean age 55.1 years | Increased risk of proteinuria was associated with urinary Cd (OR 2.67) and copper (OR 1.94). Mean urinary Cd in subjects with proteinuria (1.1 μg/L) was 27.3% higher than those without proteinuria. | Tsai et al., 2021 [27] |
| China n 683 (64.7% women) mean age 57.4 years | Risk of elevated albumin excretion increased 2.98-fold comparing urinary Cd levels ≤ 0.32 with >1.72 µg/g creatinine. | Feng et al., 2022 [28] |
| Spain, n 1397 age 18–85 years | Increased risks of albuminuria 1.58-fold and 4.54-fold were associated with urinary Cd levels > 0.27 and >0.54 µg/g creatinine, respectively. | Grau-Perez et al., 2017 [29] |
| United States NHANES 1999–2006 n 14,778, ≥20 years | Blood Cd levels ≥ 0.6 μg/L were associated with a low eGFR (OR 1.32), albuminuria (OR 1.92) and a low eGFR plus albuminuria (OR 2.91). | Navas-Acien et al., 2009 [30] |
| United States NHANES 1999–2006 n 5426, ≥20 years | Blood Cd levels > 1 µg/L plus urinary Cd levels > 1 µg/g creatinine was associated with albuminuria (OR 1.63). Blood Cd levels > 1 µg/L were associated with a low eGFR (OR 1.48) and albuminuria (OR 1.41). | Ferraro et al., 2010 [31] |
| United States NHANES 2007–2012 n 12,577, ≥20 years | Blood Cd levels > 0.61 μg/L were associated with a low eGFR (OR 1.80) and albuminuria (OR 1.60). eGFR reduction due to Cd was more pronounced in diabetic and hypertensive people, or both. | Madrigal et al., 2019 [32] |
| United States NHANES, 1999–2014 A cohort, n 1825 with CKD Follow-up period, 6.8 years | Urinary Cd levels ≥ 0.60 μg/g creatinine were associated with all-cause mortality (HR 1.75; 95%CI: 1.28, 2.39). Blood Cd levels ≥ 0.70 μg/L were associated all-cause mortality (HR 1.59; 95%CI: 1.17, 2.15). Linear dose–response relationships were observed between urinary and blood Cd levels and all-cause mortality. | Zhang et al., 2023 [33] |
| Parameter | All Subjects n = 215 | eGFR, mL/min/1.73 m2 | p | ||
|---|---|---|---|---|---|
| >90, n = 33 | 61–90, n = 131 | ≤60, n = 51 | |||
| Age, years | 57.0 ± 11.1 | 49.4 ± 9.4 | 55.6 ± 9.6 | 65.6 ± 10.6 | <0.001 |
| BMI, kg/m2 | 21.4 ± 3.6 | 21.2 ± 3.2 | 21.3 ± 3.5 | 21.7 ± 4.3 | 0.822 |
| eGFR, mL/min/1.73 m2 | 71.6 ± 19.4 | 100.4 ± 8.3 | 74.6 ± 8.2 | 45.4 ± 11.3 | <0.001 |
| Plasma creatinine, mg/dL | 1.07 ± 0.35 | 0.79 ± 0.13 | 0.98 ± 0.14 | 1.50 ± 0.44 | <0.001 |
| Urine creatinine, mg/dL | 118.4 ± 62.2 | 99.1 ± 53.1 | 116.8 ± 60.2 | 135.2 ± 69.4 | 0.054 |
| Urine Cd, µg/L | 11.85 ± 12.28 | 11.18 ± 18.70 | 10.56 ± 8.05 | 15.61 ± 15.31 | 0.079 |
| Urine β2M, mg/L | 4.92 ± 17.43 | 0.20 ± 0.36 | 1.18 ± 4.02 | 17.57 ± 32.31 | <0.001 |
| Urine α1M, mg/L | 13.09 ± 18.68 | 5.66 ± 6.17 | 8.37 ± 7.91 | 30.04 ± 30.31 | <0.001 |
| Urine albumin, mg/L | 25.57 ± 70.59 | 7.62 ± 7.29 | 22.64 ± 76.57 | 44.72 ± 73.74 | <0.001 |
| Urine protein, mg/L | 85.4 ± 199.1 | 14.9 ± 22.6 | 56.2 ± 144.6 | 206.2 ± 307.7 | <0.001 |
| Normalised to Ecr as Ex/Ecr | |||||
| ECd/Ecr, µg/g creatinine | 10.43 ± 8.02 | 10.26 ± 10.35 | 9.98 ± 6.79 | 11.69 ± 9.20 | 0.641 |
| Eβ2M/Ecr, mg/g creatinine | 4.87 ± 16.55 | 0.23 ± 0.37 | 1.66 ± 9.72 | 16.13 ± 27.49 | <0.001 |
| Eα1M/Ecr, mg/g creatinine | 11.34 ± 15.00 | 5.78 ± 4.95 | 7.53 ± 6.30 | 24.72 ± 24.57 | <0.001 |
| EAlb/Ecr, mg/g creatinine | 23.21 ± 55.07 | 10.47 ± 15.68 | 20.71 ± 59.50 | 37.88 ± 57.23 | <0.001 |
| EProt/Ecr, mg/g creatinine | 78.25 ± 174.96 | 16.73 ± 24.54 | 57.98 ± 149.26 | 170.13 ± 246.01 | <0.001 |
| Normalized to Ccr as Ex/Ccr | |||||
| (ECd/Ccr) × 100, µg/L filtrate | 11.27 ± 9.89 | 8.10 ± 9.06 | 9.67 ± 6.60 | 17.44 ± 14.17 | <0.001 |
| (Eβ2M/Ccr) × 100, mg/L filtrate | 7.74 ± 29.06 | 0.18 ± 0.28 | 1.82 ± 11.58 | 27.82 ± 52.20 | <0.001 |
| (Eα1M/Ccr) × 100, mg/L filtrate | 15.00 ± 28.25 | 4.46 ± 3.59 | 7.45 ± 6.63 | 41.20 ± 48.68 | <0.001 |
| (EAlb/Ccr) × 100, mg/L filtrate | 29.06 ± 75.93 | 7.50 ± 9.83 | 20.23 ± 56.82 | 65.68 ± 119.75 | <0.001 |
| (EProt/Ccr) × 100, mg/L filtrate | 109.9 ± 316.8 | 13.0 ± 19.1 | 56.3 ± 141.0 | 310.2 ± 568.2 | <0.001 |
| Independent Variables | Albuminuria | β2-microglobulinuria | Low eGFR |
|---|---|---|---|
| POR (95% CI) | POR (95% CI) | POR (95% CI) | |
| Age, years | 1.053 (1.024, 1.082) *** | 1.008 (0.988, 1.030) | 1.143 (1.104, 1.184) *** |
| BMI, kg/m2 | 1.009 (0.935, 1.089) | 0.987 (0.937, 1.039) | 1.073 (0.982, 1.173) |
| Gender | 0.959 (0.534, 1.722) | 0.974 (0.640, 1.484) | 0.904 (0.438, 1.846) |
| Smoking | 1.903 (1.021, 3.547) | 1.087 (0.720, 1.1641) | 1.232 (0.583, 2.605) |
| Hypertension | 1.815 (1.051, 3.134) | 1.262 (0.867, 1.839) | 1.474 (0.750, 2.894) |
| ECd/Ecr, µg/g creatinine | |||
| <2 | Referent | Referent | Referent |
| 2−4.99 | 0.799 (0.402, 1.586) | 1.120 (0.687, 1.826) | 1.959 (0.928, 4.133) |
| 5−9.99 | 1.080 (0.526, 2.216) | 1.417 (0.875, 2.296) | 3.463 (1.466, 8.179) ** |
| ≥10 | 1.093 (0.380, 3.139) | 1.807 (0.910, 3.587) | 3.382 (0.862, 13.27) |
| Independent Variables/Factors | Albuminuria | β2-microglobulinuria | Low eGFR |
|---|---|---|---|
| POR (95% CI) | POR (95% CI) | POR (95% CI) | |
| Age, years | 1.050 (1.021, 1.079) ** | 1.008 (0.986, 1.030) | 1.135 (1.094, 1.178) *** |
| BMI, kg/m2 | 1.017 (0.946, 1.093) | 0.989 (0.939, 1.043) | 1.083 (0.984, 1.192) |
| Gender | 1.196 (0.676, 2.116) | 0.962 (0.627, 1.474) | 1.258 (0.576, 2.744) |
| Smoking | 2.009 (1.118, 3.619) * | 1.011 (0.667, 1.534) | 1.280 (0.589, 2.782) |
| Hypertension | 1.912 (1.129, 3.237) * | 1.328 (0.907, 1.945) | 2.063 (0.992, 4.294) |
| (ECd/Ccr) × 100, µg/L filtrate | |||
| <2 | Referent | Referent | Referent |
| 2−4.99 | 1.764 (0.886, 3.514) | 1.914 (1.100, 3.330) * | 5.704 (2.414, 13.48) *** |
| 5−9.99 | 1.950 (1.009, 3.766) * | 1.744 (1.030, 2.951) * | 10.35 (4.160, 25.76) *** |
| ≥10 | 2.849 (1.136, 7.146) * | 2.462 (1.320, 4.595) ** | 18.06 (3.702, 88.15) *** |
| PTC Status | Protein | Filtration Rate | Excretion Rate | Catabolic Rate | Transcytosis Rate |
|---|---|---|---|---|---|
| Normal | Albumin | 60 gm/d | 20 mg/d | 2.980 gm/d | 57 gm/d |
| β2M | 300 mg/d | 100 μg/d | 299.9 mg/d | 0 | |
| Cd intoxicated | Albumin | 60 gm/d | 50 mg/d | 2.950 gm/d | 57 gm/d |
| β2M | 300 mg/d | 1000 μg/d | 299 mg/d | 0 |
| Country of Origin | Cadmium Content, µg/g Wet Tissue Weight | Reference |
|---|---|---|
| Australia, Autopsy, n 61, 2–89 years | The percentage of kidney Cd content ≥ 50 µg/g was 3.3%. a Mean lung, liver and kidney Cd were 0.13, 0.95 and 15.45 µg/g, respectively. Mean kidney Cd was 16 times higher than that in the liver. Peak hepatic and renal Cd levels were 1.5 and 25.9 µg/g. | Satarug et al. [15] |
| United Kingdom, Autopsy, n 2700, nationwide (1978–1993) | The percentage of kidney Cd content ≥ 50 µg/g was 3.9%. Mean kidney Cd content was 19 µg/g. Peak renal Cd level was 23 µg/g. | Lyon et al. [95] |
| Canada (Quebec) Autopsy, n 314 | Respective mean liver (kidney) Cd in smokers, ex-smokers and non-smokers were 2.5 (34.5), 1.4 (20.3) and 0.7 (7.0) µg/g. Mean liver Cd in female smokers was higher than male smokers (3.6 vs. 2.2 µg/g). Peak hepatic and renal Cd levels were 2.2 and 44.2 µg/g. | Benedetti et al. [96] |
| Greenland Autopsy, n 95, 19–89 years | Mean (range) liver Cd content was 5.3 (0.3–24.3) μg/g. Mean (range) kidney Cd content was 43.8 (6.7–126) μg/g. Peak hepatic and renal Cd levels were 1.97 and 22.3 μg/g. | Johansen et al. [97] |
| Sweden Kidney transplant donors, n 109, 24–70 years, median age 51. | Median kidney Cd was 12.9 μg/g. In non-smokers, the renal Cd accumulation rate was 3.9 μg/g for every 10-year increase in age. An additional 3.7 μg/g accumulation rate for every 10-year increase in smoking. In women who had serum ferritin levels ≤ 20 µg/L (depleted iron stores), the renal Cd accumulation rate was 4.5 μg/g for every 10-year increase in age. | Barregard et al. [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satarug, S. Is Chronic Kidney Disease Due to Cadmium Exposure Inevitable and Can It Be Reversed? Biomedicines 2024, 12, 718. https://doi.org/10.3390/biomedicines12040718
Satarug S. Is Chronic Kidney Disease Due to Cadmium Exposure Inevitable and Can It Be Reversed? Biomedicines. 2024; 12(4):718. https://doi.org/10.3390/biomedicines12040718
Chicago/Turabian StyleSatarug, Soisungwan. 2024. "Is Chronic Kidney Disease Due to Cadmium Exposure Inevitable and Can It Be Reversed?" Biomedicines 12, no. 4: 718. https://doi.org/10.3390/biomedicines12040718
APA StyleSatarug, S. (2024). Is Chronic Kidney Disease Due to Cadmium Exposure Inevitable and Can It Be Reversed? Biomedicines, 12(4), 718. https://doi.org/10.3390/biomedicines12040718