
Anti-Differentiation Effect of Oncogenic Met Receptor in Terminally-Differentiated Myotubes

 ,
,
Abstract
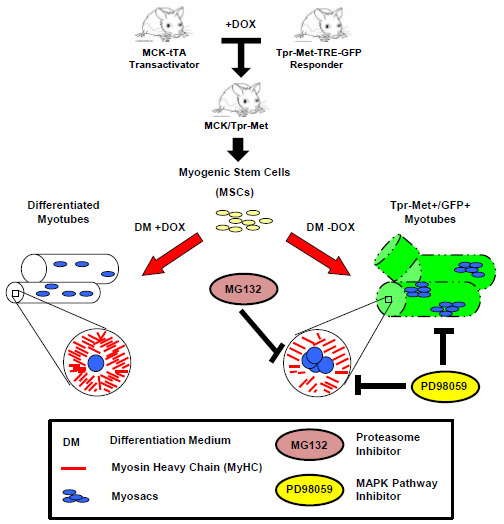
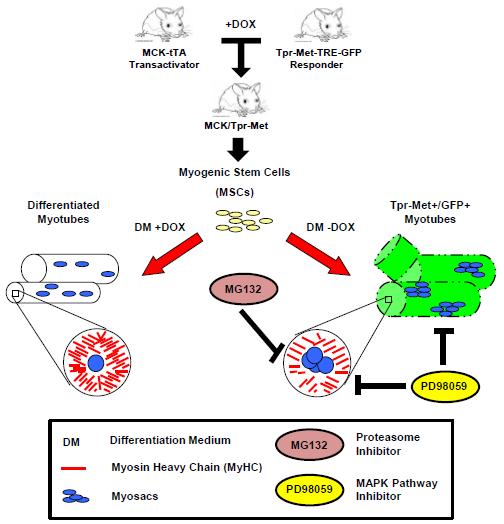
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
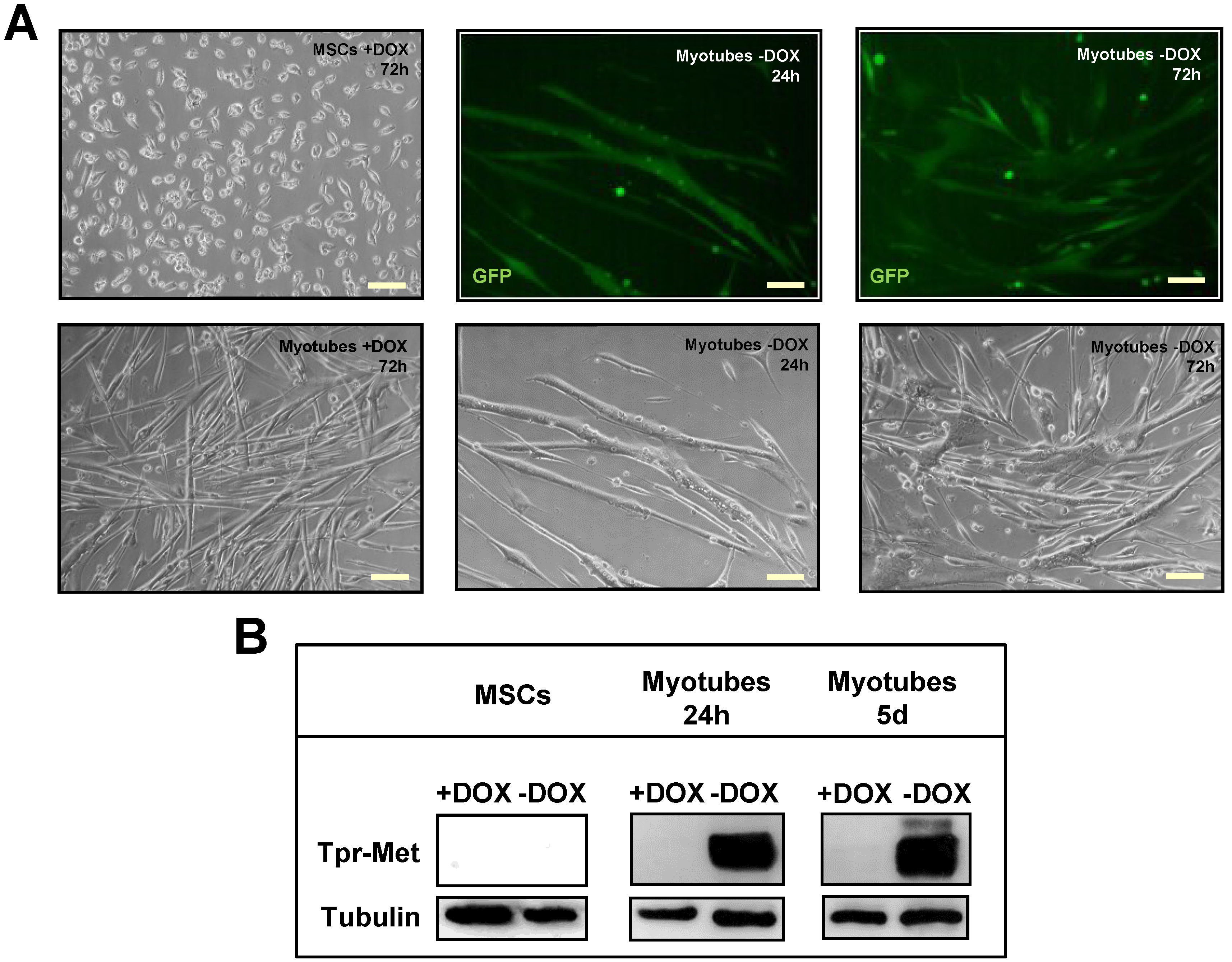
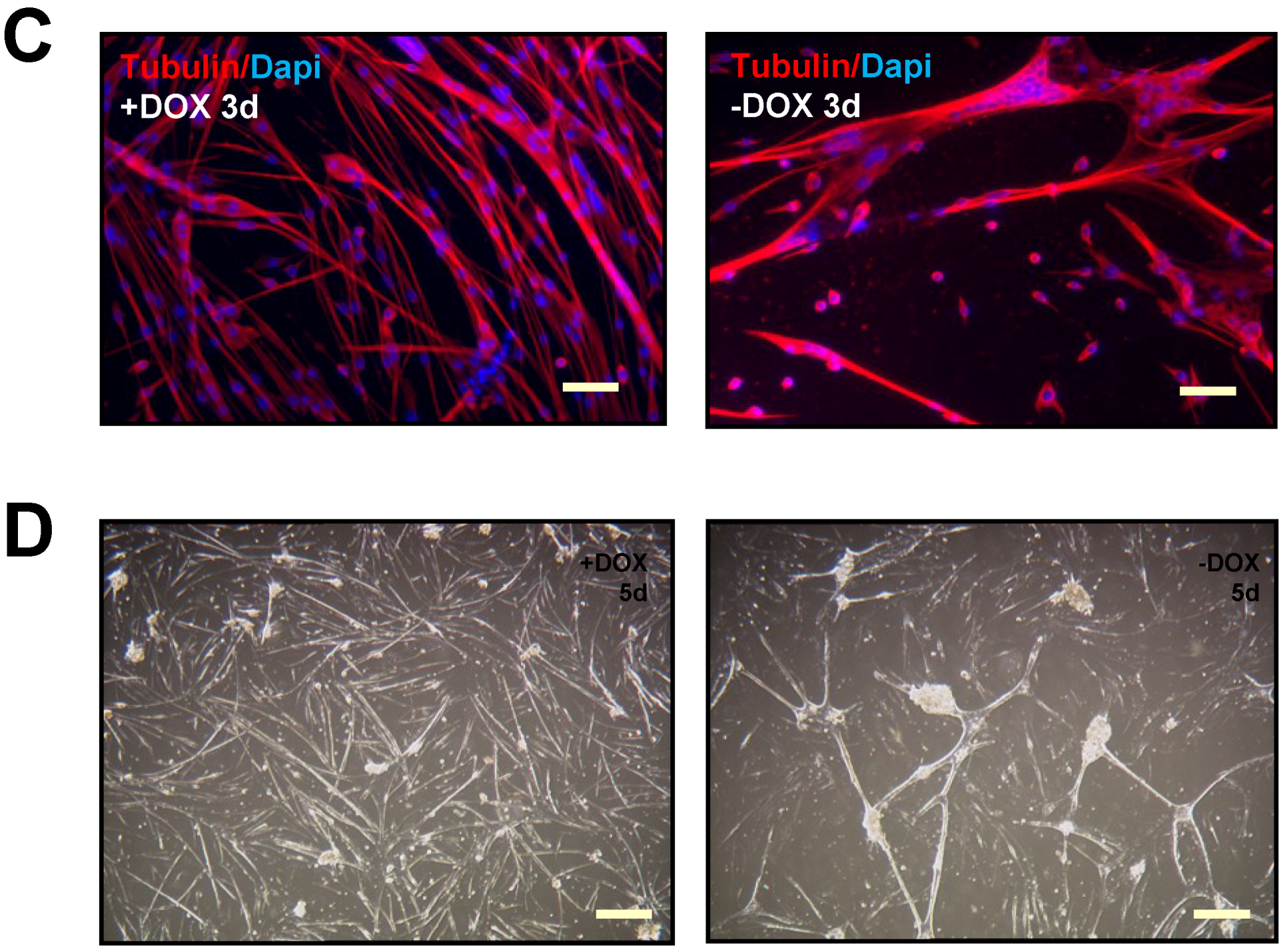
2.1. Terminally Differentiated Myotubes from Tpr–Met Mice form Gigantic Myosacs and Collapse



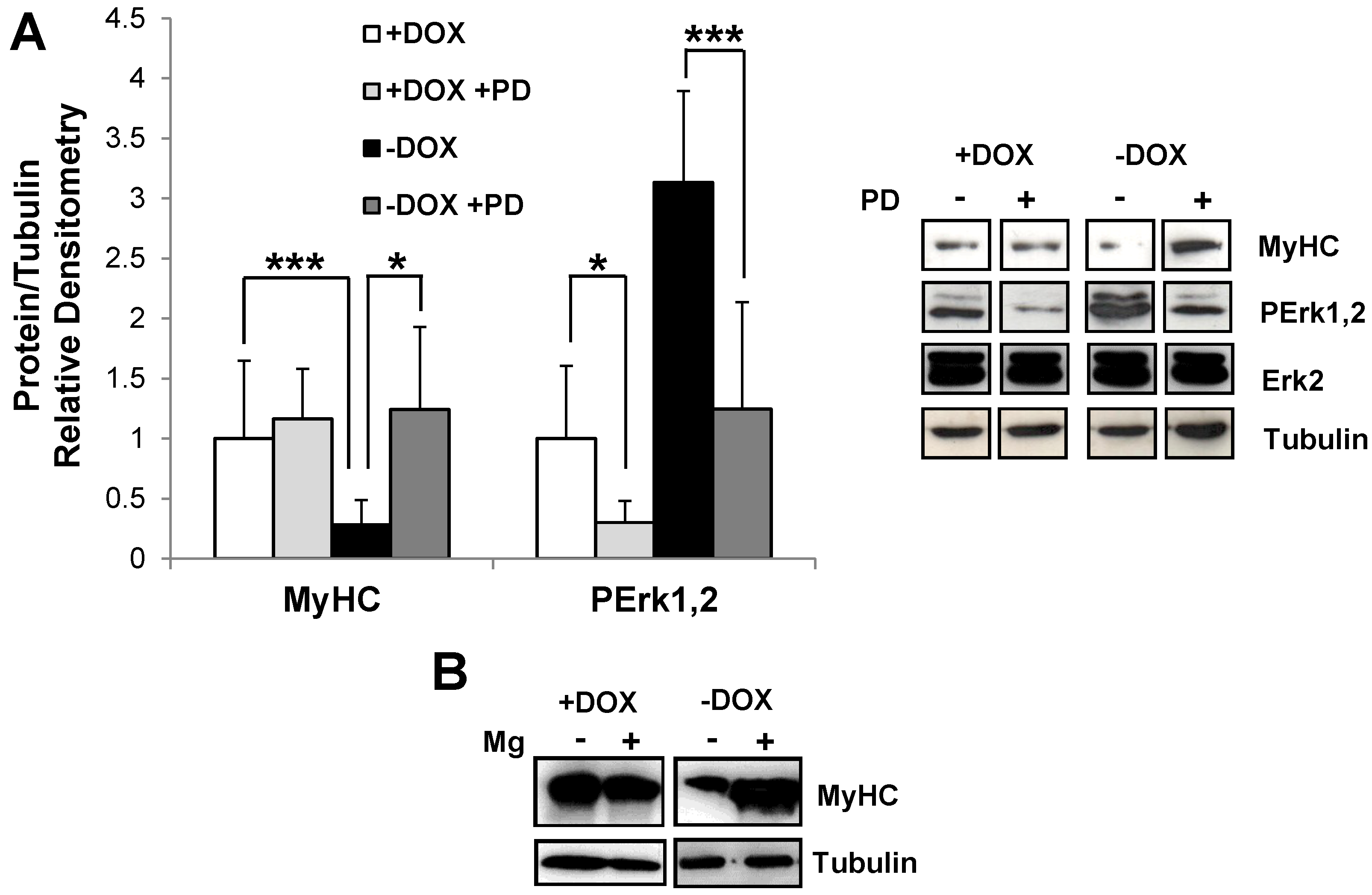
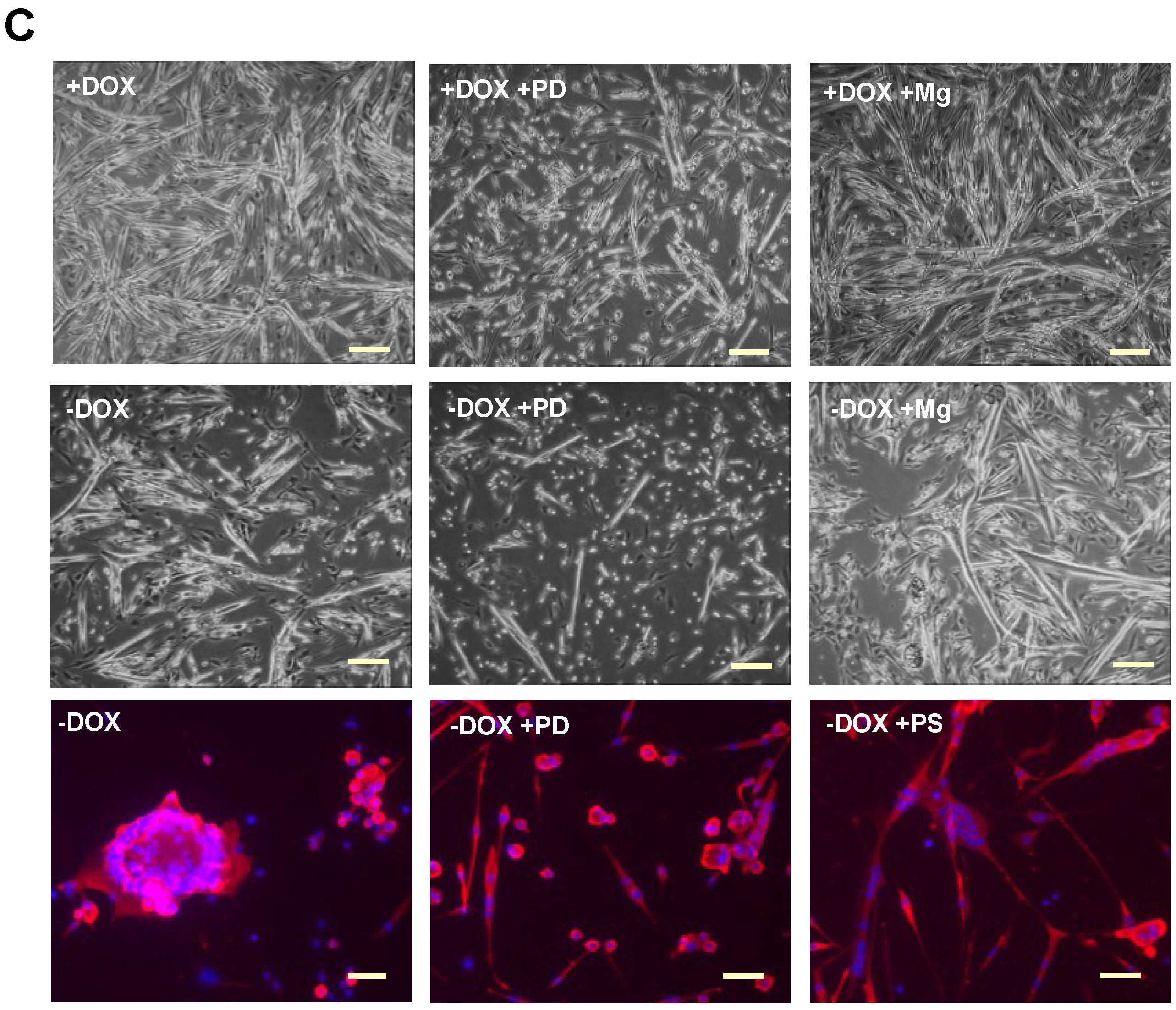
2.2. Induction of Tpr–Met in Myotubes Downregulates MyHC through Erk1,2 MAPK Activation and Proteasome-Dependent Degradation


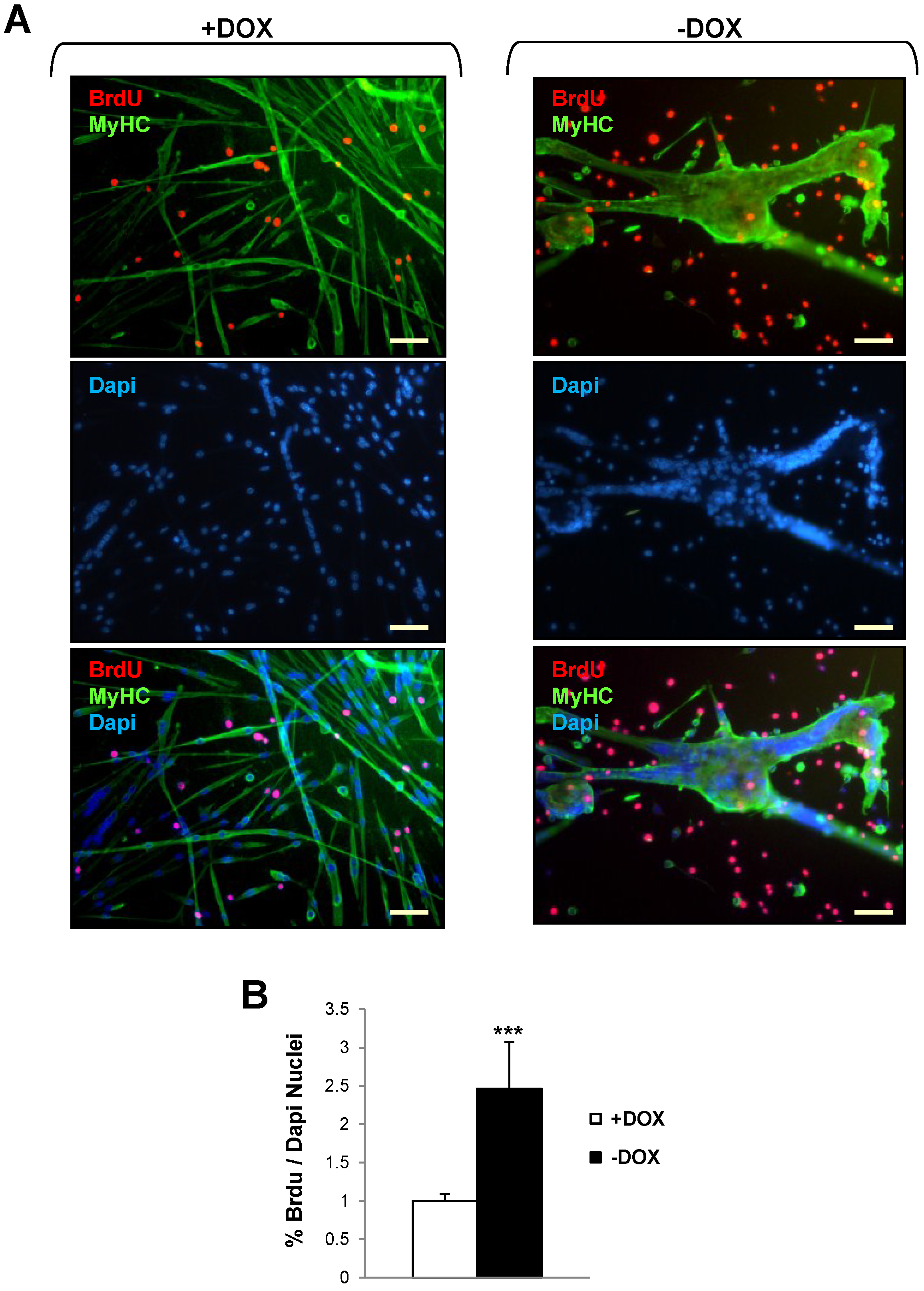
2.3. Induction of Tpr–Met Fails to Elicit DNA Endoreplication in Myotubes

3. Discussion
4. Materials and Methods
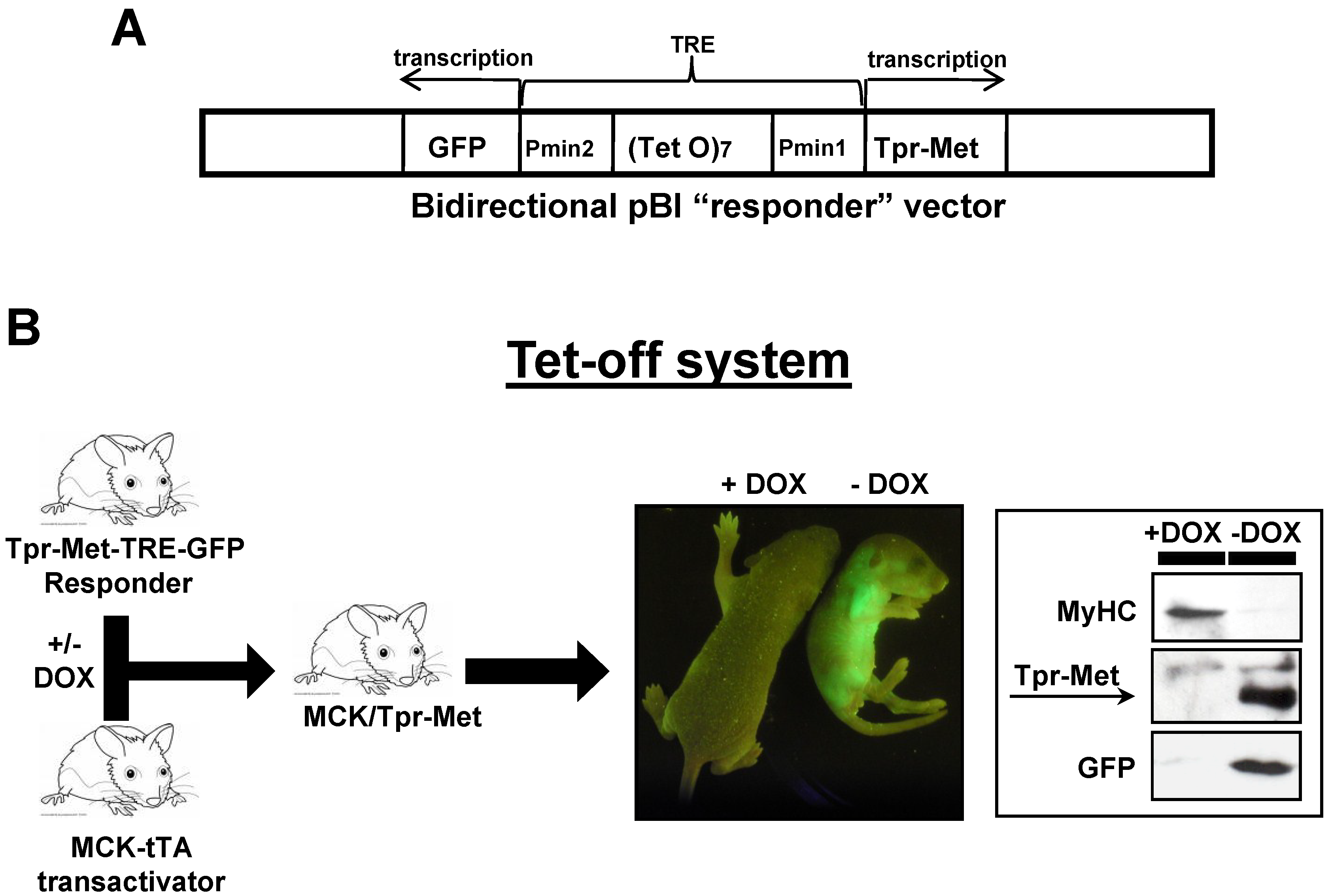
4.1. Bitransgenic Mice
4.2. Mouse Myogenic Culture Conditions and Differentiation Protocol
4.3. Inhibitors and Reagents
4.4. Western Blot (WB) Analysis
4.5. Immunofluorescence (IF)
4.6. BrdU Incorporation Assay
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Olson, E.N.; Klein, W.H. BHLH factors in muscle development: Dead lines and commitments, what to leave in and what to leave out. Genes Dev. 1994, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, R. Chemotaxis of skeletal muscle satellite cells. Dev. Dyn. 1997, 208, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, D.D.; Wold, B.J. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev. Biol. 1997, 191, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Gal-Levi, R.; Leshem, Y.; Aoki, S.; Nakamura, T.; Halevy, O. Hepatocyte growth factor plays a dual role in regulating skeletal muscle satellite cell proliferation and differentiation. Biochim. Biophys. Acta 1998, 1402, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, R.; Anderson, J.E.; Nevoret, C.J.; Halevy, O.; Allen, R.E. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev. Biol. 1998, 194, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; King, K.Y.; Brett, J.O.; Cromie, M.J.; Charville, G.W.; Maguire, K.K.; Brunson, C.; Mastey, N.; Liu, L.; Tsai, C.R.; et al. MTORC1 controls the adaptive transition of quiescent stem cells from G(0) to G(Alert). Nature 2014, 510, 393–396. [Google Scholar] [PubMed]
- Yamada, M.; Tatsumi, R.; Yamanouchi, K.; Hosoyama, T.; Shiratsuchi, S.; Sato, A.; Mizunoya, W.; Ikeuchi, Y.; Furuse, M.; Allen, R.E. High concentrations of HGF inhibit skeletal muscle satellite cell proliferation in vitro by inducing expression of myostatin: A possible mechanism for reestablishing satellite cell quiescence in vivo. Am. J. Physiol. Cell Physiol. 2010, 298, C465–C476. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, S.; Giordano, S.; Sthandier, O.; Gambarotta, G.; Maione, R.; Comoglio, P.; Amati, P. A natural hepatocyte growth factor/scatter factor autocrine loop in myoblast cells and the effect of the constitutive met kinase activation on myogenic differentiation. J. Cell Biol. 1997, 137, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Leshem, Y.; Spicer, D.B.; Gal-Levi, R.; Halevy, O. Hepatocyte growth factor (HGF) inhibits skeletal muscle cell differentiation: A role for the BHLH protein twist and the Cdk inhibitor P27. J. Cell Physiol. 2000, 184, 101–109. [Google Scholar] [PubMed]
- Miller, K.J.; Thaloor, D.; Matteson, S.; Pavlath, G.K. Hepatocyte growth factor affects satellite cell activation and differentiation in regenerating skeletal muscle. Am. J. Physiol. Cell Physiol. 2000, 278, C174–C181. [Google Scholar] [PubMed]
- Dean, M.; Park, M.; Vande Woude, G.F. Characterization of the rearranged Tpr–Met oncogene breakpoint. Mol. Cell Biol. 1987, 7, 921–924. [Google Scholar] [PubMed]
- Crepaldi, T.; Bersani, F.; Scuoppo, C.; Accornero, P.; Prunotto, C.; Taulli, R.; Forni, P.E.; Leo, C.; Chiarle, R.; Griffiths, J.; et al. Conditional activation of MET in differentiated skeletal muscle induces atrophy. J. Biol. Chem. 2007, 282, 6812–6822. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, K.; Raben, N.; Loeffler, L.; Parker, T.; Rochon, P.J.; Lee, E.; Danning, C.; Wada, R.; Thompson, C.; Bahtiyar, G.; et al. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc. Natl. Acad. Sci. USA 2000, 97, 9209–9214. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; Dalla, Z.P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, N.J.; Peterson, B.K.; Jacobson, L.A. Activation of Ras and the mitogen-activated protein kinase pathway promotes protein degradation in muscle cells of caenorhabditis elegans. Mol. Cell Biol. 2002, 22, 4181–4188. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, N.J.; Jacobson, L.A. Activated EGL-15 FGF receptor promotes protein degradation in muscles of caenorhabditis elegans. EMBO J. 2003, 22, 5058–5067. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Tisdale, M.J. Signal transduction pathways involved in proteolysis-inducing factor induced proteasome expression in murine myotubes. Br. J. Cancer 2003, 89, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Groves, N.; Schindeler, A.; Yeoh, T.; Biben, C.; Wang, C.C.; Sparrow, D.B.; Barnett, L.; Jenkins, N.A.; Copeland, N.G.; et al. The small muscle-specific protein csl modifies cell shape and promotes myocyte fusion in an insulin-like growth factor 1-dependent manner. J. Cell Biol. 2001, 153, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Yotov, W.V.; StArnaud, R. Differential splicing-in of a proline-rich exon converts alpha NAC into a muscle-specific transcription factor. Genes Dev. 1996, 10, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.M.; Tonks, N.K. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science 1997, 278, 1288–1291. [Google Scholar] [PubMed]
- Li, J.; Johnson, S.E. ERK2 is required for efficient terminal differentiation of skeletal myoblasts. Biochem. Biophs. Res. Commun. 2006, 345, 1425–1433. [Google Scholar]
- Allen, R.E.; Sheehan, S.M.; Taylor, R.G.; Kendall, T.L.; Rice, G.M. Hepatocyte growth-factor activates quiescent skeletal-muscle satellite cells in vitro. J. Cell Physiol. 1995, 165, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Helt, A.M.; Galloway, D.A. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 2003, 24, 159–169. [Google Scholar] [PubMed]
- Cardoso, M.C.; Leonhardt, H.; Nadal-Ginard, B. Reversal of terminal differentiation and control of dna replication: Cyclin A and Cdk2 specifically localize at subnuclear sites of dna replication. Cell 1993, 74, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, M.; Soddu, S.; Sacchi, A.; Tato, F. Adenovirus infection induces reentry into the cell cycle of terminally differentiated skeletal muscle cells. Ann. N. Y. Acad. Sci. 1995, 752, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Nadal-Ginard, B. Reversal of myogenic terminal differentiation by SV40 large T antigen results in mitosis and apoptosis. J. Cell Sci. 1998, 111, 1081–1093. [Google Scholar] [PubMed]
- Ghersa, P.; Gobert, R.P.; Sattonnet-Roche, P.; Richards, C.A.; Merlo, P.E.; Hooft, V.H. Highly controlled gene expression using combinations of a tissue-specific promoter, recombinant adenovirus and a tetracycline-regulatable transcription factor. Gene Ther. 1998, 5, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sala, V.; Gallo, S.; Gatti, S.; Vigna, E.; Ponzetto, A.; Crepaldi, T. Anti-Differentiation Effect of Oncogenic Met Receptor in Terminally-Differentiated Myotubes. Biomedicines 2015, 3, 124-137. https://doi.org/10.3390/biomedicines3010124
Sala V, Gallo S, Gatti S, Vigna E, Ponzetto A, Crepaldi T. Anti-Differentiation Effect of Oncogenic Met Receptor in Terminally-Differentiated Myotubes. Biomedicines. 2015; 3(1):124-137. https://doi.org/10.3390/biomedicines3010124
Chicago/Turabian StyleSala, Valentina, Simona Gallo, Stefano Gatti, Elisa Vigna, Antonio Ponzetto, and Tiziana Crepaldi. 2015. "Anti-Differentiation Effect of Oncogenic Met Receptor in Terminally-Differentiated Myotubes" Biomedicines 3, no. 1: 124-137. https://doi.org/10.3390/biomedicines3010124
APA StyleSala, V., Gallo, S., Gatti, S., Vigna, E., Ponzetto, A., & Crepaldi, T. (2015). Anti-Differentiation Effect of Oncogenic Met Receptor in Terminally-Differentiated Myotubes. Biomedicines, 3(1), 124-137. https://doi.org/10.3390/biomedicines3010124