Targeting TGFβ Signaling to Address Fibrosis Using Antisense Oligonucleotides

 and
and
Abstract
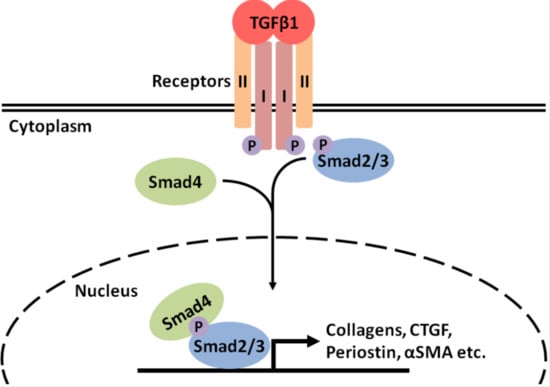
:
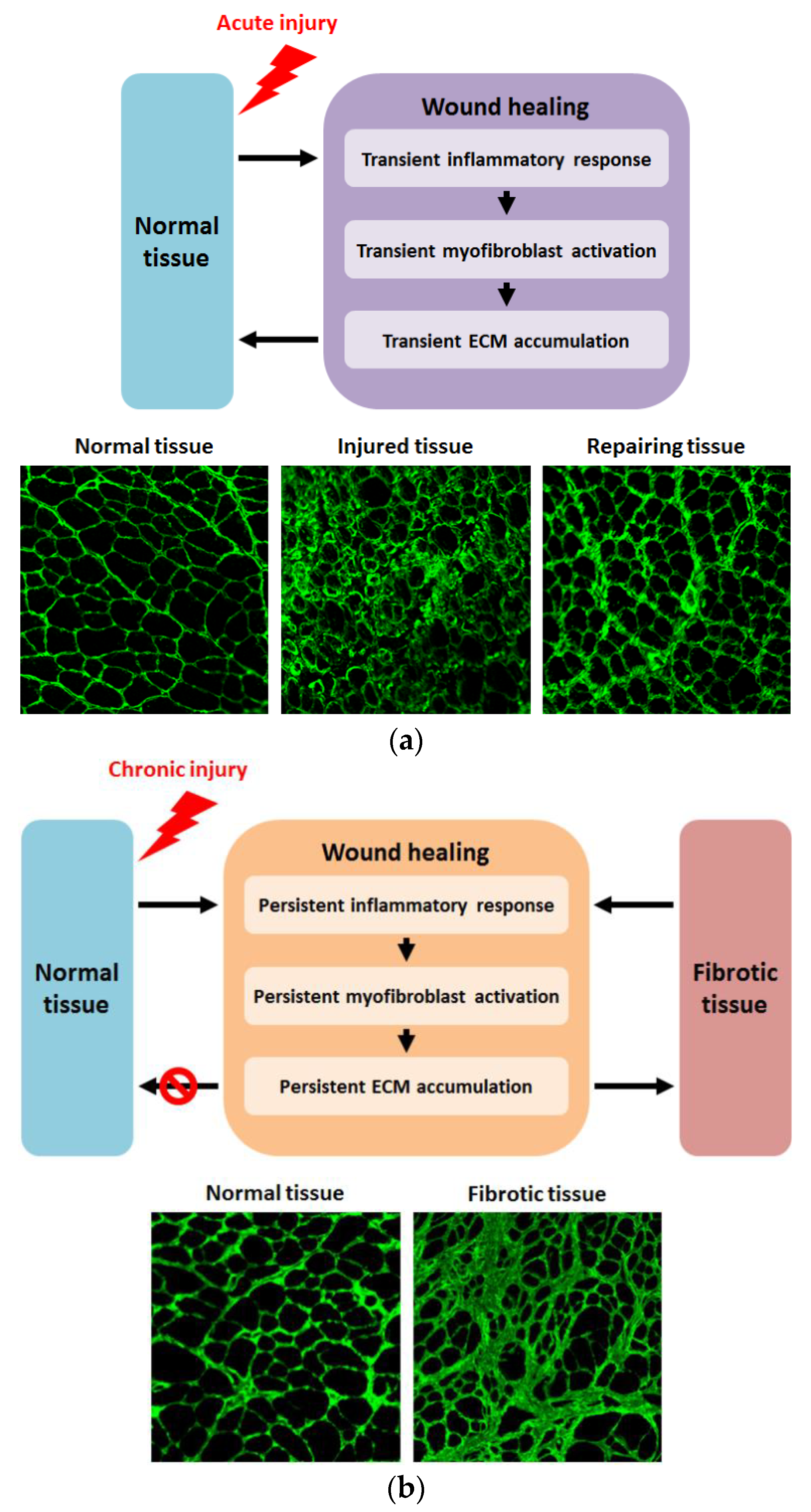
1. Introduction
2. Transforming Growth Factor β Signaling in Fibrosis
3. The Use of AOs to Target TGFβ Signaling to Inhibit Fibrosis
3.1. TGFβ1
3.2. Myostatin
3.3. ALK5
3.4. Smad3
3.5. Connexin43
3.6. miRs
4. The Use of AOs to Target Expression of Downstream Fibrotic Mediators of TGFβ1
4.1. CTGF
4.2. Periostin
4.3. TIMPs
5. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Kumar, S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. 2018, 93, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Walraven, M.; Hinz, B. Therapeutic approaches to control tissue repair and fibrosis: Extracellular matrix as a game changer. Matrix Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, H.; Kimura-Kuroda, J.; Komuta, Y.; Yoshioka, N.; Li, H.; Kawamura, K.; Raisman, G. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res. 2012, 349, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 373, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Murtha, L.A.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Knight, D.A.; Boyle, A.J. The processes and mechanisms of cardiac and pulmonary fibrosis. Front. Physiol. 2017, 8, 777. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, L.; Wang, B.; Yuan, M.; Zhu, R. Drugs and targets in fibrosis. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed]
- Horbelt, D.; Denkis, A.; Knaus, P. A portrait of transforming growth factor β superfamily signalling: Background matters. Int. J. Biochem. Cell Biol. 2012, 44, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 4, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Broekelmann, T.J.; Limper, A.H.; Colby, T.V.; McDonald, J.A. Transforming growth factor β1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [PubMed]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-β1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Weiner, F.R.; Flanders, K.C.; Giambrone, M.A.; Wind, R.; Biempica, L.; Zern, M.A. In vitro and in vivo association of transforming growth factor-beta 1 with hepatic fibrosis. J. Cell Biol. 1989, 108, 2477–2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayasaka, A.; Suzuki, N.; Fukuyama, E.; Kanda, Y. Plasma levels of transforming growth factor β1 in chronic liver disease. Clin. Chim. Acta 1996, 244, 117–119. [Google Scholar] [CrossRef]
- Mael-Ainin, M.; Abed, A.; Conway, S.J.; Dussaule, J.; Chatziantoniou, C. Inhibition of periostin expression protects against the development of renal inflammation and fibrosis. JASN 2014, 25, 1724–1736. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-β1. Mol. Genet. MeTab. 2000, 71, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Joyce, J.; Margulies, K.B.; Tsuda, T. Enhanced bioactive myocardial transforming growth factor-beta in advanced human heart failure. Circ. J. 2014, 78, 2711–2718. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Minota, S.; Sakurai, H.; Miyazono, K.; Yamada, A.; Kanazawa, I.; Kawai, M. Expression of transforming growth factor-beta 1 and its relation to endomysial fibrosis in progressive muscular dystrophy. Am. J. Pathol. 1994, 144, 221–226. [Google Scholar] [PubMed]
- Bernasconi, P.; Torchiana, E.; Confalonieri, P.; Brugnoni, R.; Barresi, R.; Mora, M.; Cornelio, F.; Morandi, L.; Mantegazza, R. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J. Clin. Investig. 1995, 96, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Ishitobi, M.; Haginoya, K.; Zhao, Y.; Ohnuma, A.; Minato, J.; Yanagisawa, T.; Tanabu, M.; Kikuchi, M.; Linuma, K. Elevated plasma levels of transforming growth factor beta1 in patients with muscular dystrophy. Neuroreport 2000, 11, 4033–4035. [Google Scholar] [CrossRef] [PubMed]
- Grobet, L.; Martin, L.J.; Poncelet, D.; Pirottin, D.; Brouwers, B.; Riquet, J.; Schoerberlein, A.; Dunner, S.; Menissier, F.; Massabanda, J.; et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat. Genet. 1997, 17, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Mcpherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hübner, C.; Riebel, T.; Kömen, W.; Braun, T.; Tobin, J.F.; Lee, S.J. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef] [PubMed]
- Mosher, D.S.; Quignon, P.; Bustamante, C.D.; Sutter, N.B.; Mellersh, C.S.; Parker, H.G.; Ostrander, E.A. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007, 3, e79. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, S.; Thomas, O.B.K.; Barton, E.R.; Morris, L.D.; Whittemore, L.; Ahima, R.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Yang, Z.; Liu, C.W.; Wang, R.; Tien, P.; Dale, R. Myostatin antisense RNA-mediated muscle growth in normal and cancer cachexia mice. Gene Ther. 2008, 15, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.C.; Lin, B.K. Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr. Opin. Support. Palliat. Care 2013, 7, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Butcher, J.T.; Ali, M.I.; Ma, M.W.; McCarthy, C.G.; Islam, B.N.; Fox, L.G.; Mintz, J.D.; Larion, S.; Fulton, D.J.; Stepp, D.W. Effect of myostatin deletion on cardiac and microvascular function. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Pirruccello-Straub, M.; Jackson, J.; Wawersik, S.; Webster, M.; Salta, L.; Long, K.; McConaughy, W.; Capili, A.; Boston, C.; Carven, G.; et al. Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Sci. Rep. 2018, 8, 2292. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Mcpherron, A.C.; Winik, N.; Lee, S. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Li, J.; Jiang, J.; Zhu, X.; Wang, B.; Li, J.; Xiao, X. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum. Gene Ther. 2008, 19, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.B.; Kollias, H.D.; Wagner, K.R. Myostatin directly regulates skeletal muscle fibrosis. J. Biol. Chem. 2008, 283, 19371–19378. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 386, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Al-Zaidy, S.A.; Sahenk, Z.; Rodino-Klapac, L.R.; Kaspar, B.; Mendell, J.R. Follistatin gene therapy improves ambulation in becker muscular dystrophy. J. Neuromuscul. Dis. 2015, 2, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-beta-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, V.Q.; Iacob, R.E.; Tian, Y.; Mcconaughy, W.; Jackson, J.; Su, Y.; Zhao, B.; Engen, J.R.; Pirruccello-Straub, M.; Springer, T.A. Tolloid cleavage activates latent GDF8 by priming the pro-complex for dissociation. EMBO J. 2018, 37, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Shinozaki, M.; Hara, T.; Furuya, T.; Miyazono, K. Two major Smad pathways in TGF-β superfamily signalling. Genes Cells 2002, 7, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4435. [Google Scholar] [PubMed]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrecchia, F.; Chu, M.; Mauviel, A. Identification of novel TGF-β/ Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 2001, 276, 17058–17062. [Google Scholar] [CrossRef] [PubMed]
- Presser, L.D.; McRae, S.; Waris, G. Activation of TGF-β1 promoter by hepatitis C virus-induced AP-1 and Sp1: Role of TGF-β1 in hepatic stellate cell activation and invasion. PLoS ONE 2013, 8, e56367. [Google Scholar] [CrossRef] [PubMed]
- Schonherr, E.; Jarvelainen, H.T.; Sandell, L.J.; Wight, T.N. Effects of platelet-derived growth factor and transforming growth factor-β1 on the synthesis of a large versican-like chondroitin sulfate proteoglycan by arterial smooth muscle cells. J. Biol. Chem. 1991, 266, 17640–17647. [Google Scholar] [PubMed]
- Dadlani, H.; Ballinger, M.L.; Osman, N.; Getachew, R.; Little, P.J. Smad and p38 MAP kinase-mediated signaling of proteoglycan synthesis in vascular smooth muscle. J. Biol. Chem. 2008, 283, 7844–7852. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Liu, X.; Ansari, D.O.; Lodish, H.F. Synergistic cooperation of TFE3 and Smad proteins in TGF-ß-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. 1998, 12, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin–TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotendorst, G.; Okochi, H.; Hayashi, N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ. 1996, 7, 469–480. [Google Scholar] [PubMed]
- Lasky, J.A.; Ortiz, L.A.; Tonthat, B.; Hoyle, G.W.; Corti, M.; Athas, G.; Lungarella, G.; Brody, A.; Friedman, M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am. J. Physiol. 1998, 275, 365–371. [Google Scholar] [CrossRef]
- Chen, Y.; Blom, I.E.; Sa, S.; Goldschmeding, R.; Abraham, D.J.; Leask, A. CTGF expression in mesangial cells: Involvement of SMADs, MAP kinase, and PKC. Kidney Int. 2002, 62, 1149–5119. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Dargere, D.; Bonvoust, F.; Vidaud, M.; Segarini, P.; Bedossa, P. Effects and regulation of connective tissue growth factor on hepatic stellate cells. Lab. Investig. 2002, 82, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Vial, C.; Zúñiga, LM.; Cabello-Verrugio, C.; Cañón, P.; Fadic, R.; Brandan, E. Skeletal muscle cells express the profibrotic cytokine connective tissue growth factor (CTGF/CCN2), which induces their dedifferentiation. J. Cell. Physiol. 2008, 215, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, J.; Elliott, C.; Wen, W.; Hamilton, D.; Conway, S. Spatiotemporal expression of periostin during skin development and incisional wound healing: Lessons for human fibrotic scar formation. J. Cell Commun. Signal. 2010, 4, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.K.; Bozyk, P.D.; Bentley, J.K.; Popova, A.P.; Birch, C.M.; Wilke, C.A.; Fry, C.D.; White, E.S.; Sisson, T.H.; Tayob, N.; et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. 2012, 303, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ng, Y.; Hill, P.A.; Nikolic-Paterson, D.J.; Mu, W.; Atkins, R.C.; Lan, H.Y. Transforming growth factor-β regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999, 56, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar] [PubMed]
- Bo Li, Z.; Zhang, J.; Wagner, K.R. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J. Cell Sci. 2012, 125, 3957–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asazuma-Nakamura, Y.; Dai, P.; Harada, Y.; Jiang, Y.; Hamaoka, K.; Takamatsu, T. Cx43 contributes to TGF-β signaling to regulate differentiation of cardiac fibroblasts into myofibroblasts. Exp. Cell Res. 2009, 315, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C. Connexin43: A protein from rat heart homologous to a gap junction protein from liver. J. Cell Biol. 1987, 105, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.H.; De Vuyst, E.; Leybaert, L. The gap junction cellular internet: Connexin hemichannels enter the signalling limelight. Biochem. J. 2006, 397, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.; Qiu, C.; Frank, S.; Wang, C.M.; Brown, T.; Green, C.R.; Becker, D.L. Limiting burn extension by transient inhibition of Connexin43 expression at the site of injury. Br. J. Plast. Surg. 2005, 58, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Muramatsu, T.; Uekusa, T.; Sasaki, H.; Shimono, M. Inhibition of connexin 43 expression and function in cultured rat dental pulp cells by antisense oligonucleotide. Cell Tissue Res. 2007, 329, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deva, N.; Zhang, J.; Green, C.; Danesh-Meyer, H. Connexin43 modulation inhibits scarring in a rabbit eye glaucoma trabeculectomy model. Inflammation 2012, 35, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Nakagami, T.; Tanaka, H.; Hitomi, T.; Takamatsu, T. Cx43 mediates TGF-β signaling through competitive Smads binding to microtubules. Mol. Biol. Cell 2007, 18, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, B.; Stanczyk, J.; Jungel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010, 62, 1733–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lu, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Urban, G.W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Putta, S.; Lanting, L.; Sun, G.; Lawson, G.; Kato, M.; Natarajan, R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, K.V.; Milosevic, J.; Kaminski, N. MicroRNAs in idiopathic pulmonary fibrosis. Transl. Res. 2011, 157, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Merkel, P.A.; Furst, D.E.; Khanna, D.; Emery, P.; Hsu, V.M.; Silliman, N.; Streisand, J.; Powell, J.; Akesson, A.; et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: A multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007, 56, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-β1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Llopiz, D.; Dotor, J.; Casares, N.; Bezunartea, J.; Díaz-Valdés, N.; Ruiz, M.; Aranda, F.; Berraondo, P.; Prieto, J.; Lasarte, J.J.; et al. Peptide inhibitors of transforming growth factor-β enhance the efficacy of antitumor immunotherapy. Int. J. Cancer 2009, 125, 2614–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Kuwano, K.; Maeyama, T.; Yoshimi, M.; Hamada, N.; Fukumoto, J.; Egashira, K.; Hiasa, K.; Takayama, K.; Nakanishi, Y. Gene transfer of soluble transforming growth factor type II receptor by in vivo electroporation attenuates lung injury and fibrosis. J. Clin. Pathol. 2007, 60, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Latres, E.; Pangilinan, J.; Miloscio, L.; Bauerlein, R.; Na, E.; Potocky, T.B.; Huang, Y.; Eckersdorff, M.; Rafique, A.; Mastaitis, J.; et al. Myostatin blockade with a fully human monoclonal antibody induces muscle hypertrophy and reverses muscle atrophy in young and aged mice. Skeletal Muscle 2015, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanovich, S.; Perkins, K.J.; Krag, T.O.; Whittemore, L.A.; Khurana, T.S. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005, 19, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, M.; Takehara, Y.; Sugino, H.; Matsumoto, M.; Hashimoto, O.; Hasegawa, Y.; Murakami, T.; Uezumi, A.; Takeda, S.; Noji, S.; et al. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 2008, 22, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Rodino-Klapac, L.R.; Haidet, A.M.; Kota, J.; Handy, C.; Kaspar, B.K.; Mendell, J.R. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve 2009, 39, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.; Walton, K.L.; Hagg, A.; Colgan, T.D.; Johnson, K.; Qian, H.; Gregorevic, P.; Harrison, C.A. Specific targeting of TGF-β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5266–E5275. [Google Scholar] [CrossRef] [PubMed]
- Pistilli, E.E.; Bogdanovich, S.; Goncalves, M.D.; Ahima, R.S.; Lachey, J.; Seehra, J.; Khurana, T. Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am. J. Pathol. 2011, 178, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Grygielko, E.T.; Martin, W.M.; Tweed, C.; Thornton, P.; Harling, J.; Brooks, D.P.; Laping, N.J. Inhibition of gene markers of fibrosis with a novel inhibitor of transforming growth factor-β type I receptor kinase in puromycin-induced nephritis. J. Pharmacol. Exp. Ther. 2005, 313, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; ten Dijke, P.; Laping, N.J. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Karkampouna, S.; Kruithof, B.P.; Kloen, P.; Obdeijn, M.C.; van Der Laan, A.M.A.; Tanke, H.J.; Kemaladewi, D.U.; Hoogaars, W.M.; Aartsma-Rus, A.; Clark, I.M.; et al. Novel Ex Vivo Culture Method for the Study of Dupuytren’s Disease: Effects of TGFβ Type 1 Receptor Modulation by Antisense Oligonucleotides. Mol. Ther. Nucleic Acids 2014, 3, e142. [Google Scholar] [CrossRef] [PubMed]
- Akagi, Y.; Isaka, Y.; Arai, M.; Kaneko, T.; Takenaka, M.; Moriyama, T.; Kaneda, Y.; Ando, A.; Orita, Y.; Kamada, T.; et al. Inhibition of TGF-beta 1 expression by antisense oligonucleotides suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1996, 50, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y.; Tsujie, M.; Ando, Y.; Nakamura, H.; Kaneda, Y.; Imai, E.; Hori, M. Transforming growth factor-beta 1 antisense oligodeoxynucleotides block interstitial fibrosis in unilateral ureteral obstruction. Kidney Int. 2000, 58, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Han, D.C.; Hoffman, B.B.; Hong, SW.; Guo, J.; Ziyadeh, F.N. Therapy with antisense TGF-β1 oligodeoxynucleotides reduces kidney weight and matrix mRNAs in diabetic mice. Am. J. Physiol. Renal. Physiol. 2000, 278, F628–F634. [Google Scholar] [CrossRef] [PubMed]
- Loiselle, A.E.; Yukata, K.; Geary, M.B.; Kondabolu, S.; Shi, S.; Jonason, J.H.; Awad, H.A.; O’Keefe, R.J. Development of antisense oligonucleotide (ASO) technology against Tgf-β signaling to prevent scarring during flexor tendon repair. J. Orthop. Res. 2015, 33, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, M.; Lahme, B.; Van de Leur, E.; Gressner, AM.; Weiskirchen, R. Adenoviral delivery of an antisense RNA complementary to the 3′ coding sequence of transforming growth factor-beta1 inhibits fibrogenic activities of hepatic stellate cells. Cell Growth Differ. 2002, 13, 265–273. [Google Scholar] [PubMed]
- Malerba, A.; Kang, J.K.; Mcclorey, G.; Saleh, A.F.; Popplewell, L.; Gait, M.J.; Wood, M.J.; Dickson, G. Dual myostatin and dystrophin exon skipping by morpholino nucleic acid oligomers conjugated to a cell-penetrating peptide is a promising therapeutic strategy for the treatment of Duchenne muscular dystrophy. Mol. Ther. Nucleic Acids 2012, 1, e62. [Google Scholar] [CrossRef] [PubMed]
- Lu-Nguyen, N.B.; Jarmin, S.A.; Saleh, A.F.; Popplewell, L.; Gait, M.J.; Dickson, G. Combination antisense treatment for destructive exon skipping of myostatin and open reading frame rescue of dystrophin in neonatal mdx mice. Mol. Ther. 2015, 23, 1341–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu-Nguyen, N.; Malerba, A.; Popplewell, L.; Schnell, F.; Hanson, G.; Dickson, G. Systemic antisense therapeutics for dystrophin and myostatin exon splice modulation improve muscle pathology of adult mdx mice. Mol. Ther. Nucleic Acids 2017, 6, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 2004, 20, 61–86. [Google Scholar] [CrossRef] [PubMed]
- Kemaladewi, D.U.; Pasteuning, S.; van Der Meulen, J.W.; van Heiningen, S.H.; van Ommen, G.; Ten Dijke, P.; Aartsma-Rus, A.; AC’t Hoen, P.; Hoogaars, W.M. Targeting TGF-β signaling by antisense oligonucleotide-mediated knockdown of TGF-β type I receptor. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Kemaladewi, D.U.; Hoogaars, W.M.H.; van Heiningen, S.H.; Terlouw, S.; de Gorter, D.J.J.; den Dunnen, J.T.; van Ommen, G.J.; Aartsma-Rus, A.; ten Dijke, P.; AC’t Hoen, P. Dual exon skipping in myostatin and dystrophin for Duchenne muscular dystrophy. BMC Med. Genom. 2011, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Kim, C.; Kim, Y. Cellular delivery of cationic lipid nanoparticle-based SMAD3 antisense oligonucleotides for the inhibition of collagen production in keloid fibroblasts. Eur. J. Pharm. Biopharm. 2012, 82, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; An, H.; Kim, W.; Gwon, M.; Gu, H.; Park, Y.; Park, K.K. Anti-fibrotic effects of synthetic oligodeoxynucleotide for TGF-β1 and Smad in an animal model of liver cirrhosis. Mol. Ther. Nucleic Acids 2017, 8, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.C.; Gross, J.; Fiedler, T.; Fischer, S.; Kissler, M.; Bussen, P.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Chau, B.N.; Xin, C.; Hartner, J.; Ren, S.; Castano, A.P.; Linn, G.; Li, J.; Tran, P.T.; Kaimal, V.; Huang, X.; et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 2012, 15, 121ra18. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 4, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res. 2012, 110, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Treguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, A.; Okochi, H.; Bradham, D.M.; Grotendorst, G.R. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol. Biol. Cell 1993, 4, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, A.; Nashiro, K.; Kikuchi, K.; Sato, S.; Ihn, H.; Gary, R.G.; Takehara, K. Significant correlation between connective tissue growth factor gene expression and skin sclerosis in tissue sections from patients with systemic sclerosis. J. Investig. Dermatol. 1995, 105, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, A.; Nashiro, K.; Kikuchi, K.; Sato, S.; Ihn, H.; Fujimoto, M.; Gary, R.G.; Takehara, K. Connective tissue growth factor gene expression in tissue sections from localized scleroderma, keloid, and other fibrotic skin disorders. J. Investig. Dermatol. 1996, 106, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Dargere, D.; Vidaud, M.; De Gouville, A.; Huet, S.; Martinez, V.; Gauthier, J.; Ba, N.; Sobesky, R.; Ratziu, V.; et al. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology 1999, 30, 968–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Aten, J.; Richard, J.B.; Barry, S.O.; Ton, J.R.; Jan, J.W.; Goldschmeding, R. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 1998, 53, 853–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Haginoya, K.; Wu, Y.; Chiba, Y.; Nakanishi, T.; Onuma, A.; Sato, Y.; Takigawa, M.; Iinuma, K.; Tsuchiya, S. Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J. Neurol. Sci. 2008, 267, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Adler, S.G.; Schwartz, S.; Williams, M.E.; Arauz-Pacheco, C.; Bolton, W.K.; Lee, T.; Li, D.; Neff, T.B.; Urquilla, P.R.; Sewell, K.L. Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin. J. Am. Soc. Nephrol. 2010, 5, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Usinger, W.; Nichols, B.; Gray, J.; Xu, L.; Seeley, T.W.; Brenner, M.; Guo, G.; Zhang, W.; Oliver, N.; et al. Cooperative interaction of CTGF and TGF-beta in animal models of fibrotic disease. Fibrogenes. Tissue Repair 2011, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.G.; Gutierrez, J.; Cabello-Verrugio, C.; Cabrera, D.; Lipson, K.; Goldschmeding, R.; Brandan, E. Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum. Mol. Genet. 2013, 22, 4938–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Scholand, M.B.; de Andrade, J.; Lancaster, L.; Mageto, Y.; Goldin, J.; Brown, K.K.; Flaherty, K.R.; Wencel, M.; Wanger, J.; et al. FG-3019 anti-connective tissue growth factor monoclonal antibody: Results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 47, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Liu, F.; Luo, J.; Yang, X.; Wen, H.; Su, Y.; Yan, K.; Li, Y.; Liang, Y. Effect of small interfering RNA on the expression of connective tissue growth factor and type I and III collagen in skin fibroblasts of patients with systemic sclerosis. Br. J. Dermatol. 2006, 155, 1145–1453. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xie, Q.; Shi, Y.; Li, D.; Zhang, M.; Jiang, S.; Zhou, H.; Lu, H.; Jin, Y. Inhibition of connective tissue growth factor by siRNA prevents liver fibrosis in rats. J. Gene Med. 2006, 8, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Xie, Y.; Peng, M.; Ma, L.; Zhou, Y.; Zhang, Y.; Kang, W.; Wang, J.; Bai, X.; Wang, P.; et al. Inhibition of connective tissue growth factor suppresses hepatic stellate cell activation in vitro and prevents liver fibrosis in vivo. Clin. Exp. Med. 2014, 14, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Sugawara, A.; Mukoyama, M.; Mori, K.; Makino, H.; Suganami, T.; Nagae, T.; Yahata, K.; Fujinaga, Y.; Tanaka, I.; et al. Role of connective tissue growth factor in profibrotic action of transforming growth factor-β: A potential target for preventing renal fibrosis. Am. J. Kidney Dis. 2001, 38, S134–S138. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Mukoyama, M.; Sugawara, A.; Mori, K.; Nagae, T.; Makino, H.; Suganami, T.; Yahata, K.; Fujinaga, Y.; Tanaka, I.; et al. Role of connective tissue growth factor in fibronectin expression and tubulointerstitial fibrosis. Am. J. Physiol. Renal. Physiol. 2002, 282, F933–F942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, H.; Mukoyama, M.; Nagae, T.; Mori, K.; Suganami, T.; Sawai, K.; Yoshioka, T.; Koshikawa, M.; Nishida, T.; Takigawa, M.; et al. Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2004, 15, 14301440. [Google Scholar] [CrossRef]
- Guha, M.; Xu, Z.; Tung, D.; Lanting, L.; Natarajan, R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007, 21, 3355–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisco, M.; Kryger, Z.B.; O’Shaughnessy, K.D.; Kim, P.S.; Schultz, G.S.; Ding, X.; Roy, N.K.; Dean, N.M.; Mustoe, T.A. Antisense inhibition of connective tissue growth factor (CTGF/CCN2) mRNA limits hypertrophic scarring without affecting wound healing in vivo. Wound Repair Regen. 2008, 16, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, S.; Kikuno, R.; Tezuka, K.; Amann, E. Osteoblast-specific factor 2: Cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem. J. 1993, 294, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Amizuka, N.; Takeshita, S.; Takamatsu, H.; Katsuura, M.; Ozawa, H.; Toyama, Y.; Bonewald, L.F.; Kudo, A. Identification and Characterization of a Novel Protein, Periostin, with Restricted Expression to Periosteum and Periodontal Ligament and Increased Expression by Transforming Growth Factor β. J. Bone Miner. Res. 1999, 14, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616. [Google Scholar] [CrossRef]
- Hamilton, D. Functional role of periostin in development and wound repair: Implications for connective tissue disease. J. Cell Commun. Signal. 2008, 2, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Hoersch, S.; Andrade-Navarro, M.A. Periostin shows increased evolutionary plasticity in its alternatively spliced region. BMC Evol. Biol. 2010, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetsch, S.C.; Hawke, T.J.; Gallardo, T.D.; Richardson, J.A.; Garry, D.J. Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol. Genom. 2003, 14, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson-Boeters, L.; Wen, W.; Hamilton, D. Periostin localizes to cells in normal skin, but is associated with the extracellular matrix during wound repair. J. Cell Commun. Signal. 2009, 3, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdemir, C.; Akpulat, U.; Sharafi, P.; Yıldız, Y.; Onbaşılar, İ.; Kocaefe, Ç. Periostin is temporally expressed as an extracellular matrix component in skeletal muscle regeneration and differentiation. Gene 2014, 553, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Stanton, L.W.; Garrard, L.J.; Damm, D.; Garrick, B.L.; Lam, A.; Kapoun, A.M.; Zheng, Q.; Protter, A.A.; Schreiner, G.F.; White, R.T. Altered patterns of gene expression in response to myocardial infarction. Circ. Res. 2000, 86, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Xu, J.; Kaiser, R.A.; Melendez, J.; Hambleton, M.; Sargent, M.A.; Lorts, A.; Brunskill, E.W.; Dorn, G.W.; Conway, S.J.; et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007, 101, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, M.; Nakamura, K.; Kii, I.; Kashima, T.; Amizuka, N.; Li, M.; Saito, M.; Fukuda, K.; Nishiyama, T.; Kitajima, S.; et al. Periostin is essential for cardiac healing after acute myocardial infarction. J. Exp. Med. 2008, 205, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wu, H.; Xia, W.; Chen, X.; Zhu, S.; Zhang, S.; Shao, Y.; Ma, W.; Yang, D.; Zhang, J. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J. Cardiol. 2014, 63, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Takayama, G.; Arima, K.; Kanaji, T.; Toda, S.; Tanaka, H.; Shoji, S.; Mckenzie, A.N.J.; Nagai, H.; Hotokebuchi, T.; Izuhara, K. Periostin: A novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J. Allergy Clin. Immunol. 2006, 118, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, S.S.; Yuan, S.; Innes, A.L.; Kerr, S.; Woodruff, P.G.; Hou, L.; Muller, S.J.; Fahy, J.V. Roles of epithelial cell- derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc. Natl. Acad. Sci. USA 2010, 107, 14170–14175. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.A.; Damon, B.; Mironov, V.; Kasyanov, V.; Ramamurthi, A.; Moreno-Rodriguez, R.; Trusk, T.; Potts, J.D.; Goodwin, R.L.; Davis, J.; et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J. Cell. Biochem. 2007, 101, 695–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J. Biol. Chem. 2010, 285, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Kii, I.; Saito, M.; Kudo, A. Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J. Biol. Chem. 2010, 285, 13294–13303. [Google Scholar] [CrossRef] [PubMed]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar] [PubMed]
- Bao, S.; Ouyang, G.; Bai, X.; Huang, Z.; Ma, C.; Liu, M.; Shao, R.; Anderson, R.M.; Rich, J.N.; Wang, X. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 2004, 5, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Baril, P.; Gangeswaran, R.; Mahon, P.C.; Caulee, K.; Kocher, H.M.; Harada, T.; Zhu, M.; Kalthoff, H.; Crnogorac-Jurcevic, T.; Lemoine, N.R. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: Role of the β4 integrin and the PI3k pathway. Oncogene 2007, 26, 2082–2094. [Google Scholar] [CrossRef] [PubMed]
- Butcher, J.T.; Norris, R.A.; Hoffman, S.; Mjaatvedt, C.H.; Markwald, R.R. Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI3-kinase. Dev. Biol. 2007, 302, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Um, J.; Park, J.; Nam, B.; Lee, J.; Jung, J.; Kim, Y.; Kim, S.; Park, J.; Wu, M.; Han, S.; et al. Periostin-binding DNA aptamer treatment attenuates renal fibrosis under diabetic conditions. Sci. Rep. 2017, 7, 8490. [Google Scholar] [CrossRef] [PubMed]
- Takai, S.; Yoshino, M.; Takao, K.; Yoshikawa, K.; Jin, D. Periostin antisense oligonucleotide prevents adhesion formation after surgery in mice. J. Pharmacol. Sci. 2017, 133, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Semba, T.; Sugihara, E.; Kamoshita, N.; Ueno, S.; Fukuda, K.; Yoshino, M.; Takao, K.; Yoshikawa, K.; Izuhara, K.; Arima, Y.; et al. Periostin antisense oligonucleotide suppresses bleomycin-induced formation of a lung premetastatic niche for melanoma. Cancer Sci. 2018, 109. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak-Wielgomas, K.; Dziegiel, P. The role of periostin in neoplastic processes. Folia Histochem. Cytobiol. 2015, 53, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Lister, N.C.; Clemson, M.; Morris, K.V. RNA-directed epigenetic silencing of periostin inhibits cell motility. R. Soc. Open Sci. 2015, 2, 140545. [Google Scholar] [CrossRef] [PubMed]
- Lagente, V.; Manoury, B.; Nenan, S.; Le Quement, C.; Martin-Chouly, C.; Boichot, E. Role of matrix metalloproteinases in the development of airway inflammation and remodeling. Braz. J. Med. Biol. Res. 2005, 38, 1521–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alameddine, H.S.; Morgan, J.E. Matrix metalloproteinases and tissue inhibitor of metalloproteinases in inflammation and fibrosis of skeletal muscles. J. Neuromuscul. Dis. 2016, 29, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.M.; Fridman, R. TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochem. J. 2003, 374, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Remacle, A.G.; Chernov, A.V.; Liu, H.; Shubayev, I.; Lai, C.; Dolkas, J.; Shiryaev, S.A.; Golubkov, V.S.; Mizisin, A.P.; et al. The MMP-9/TIMP-1 axis controls the status of differentiation and function of myelin-forming Schwann cells in nerve regeneration. PLoS ONE 2012, 7, e33664. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Mao, J.; Gao, L.; Liu, J.; Wu, T. Effect of antisense TIMP-1 cDNA on the expression of TIMP-1 and MMP-2 in lung tissue with pulmonary fibrosis induced by bleomycin. Mol. Med. Rep. 2013, 7, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.H.; Cheng, Y.Q.; Xie, Y.M.; Zhou, Y.X.; Cao, Y.Z. Inhibiting effect of antisense oligonucleotides phosphorthioate on gene expression of TIMP-1 in rat liver fibrosis. World J. Gastroenterol. 2001, 7, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.H.; Zhu, C.L.; Zhang, Y.F.; Yang, J.; Zhang, J.C.; Gao, R.T. Inhibitory effect of antisense oligonucleotide targeting TIMP-2 on immune-induced liver fibrosis. Dig. Dis. Sci. 2010, 55, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tan, W.; Zhang, L.; Tan, Q.; Yang, J. Beneficial impact of bFGF antisense therapy in a rat model of pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2015, 32, 22–31. [Google Scholar] [PubMed]
- Wang, J.H.; Newbury, L.J.; Knisely, A.S.; Monia, B.; Hendry, B.M.; Sharpe, C.C. Antisense knockdown of Kras inhibits fibrosis in a rat model of unilateral ureteric obstruction. Am. J. Pathol. 2012, 180, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Rossert, J.; Mauviel, A. Blocking sp1 transcription factor broadly inhibits extracellular matrix gene expression in vitro and in vivo: Implications for the treatment of tissue fibrosis. J. Investig. Dermatol. 2001, 116, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.; Zhang, X.; Tan, M.; Zheng, R.; Zhao, L. Intervention of transforming pulmonary fibrosis with NF-κB p65 antisense oligonucleotide. Int. J. Clin. Exp. Med. 2014, 7, 5252–5259. [Google Scholar] [PubMed]
- Lawrance, I.C.; Wu, F.; Leite, A.Z.; Willis, J.; West, G.A.; Fiocchi, C.; Chakravarti, S. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-κB. Gastroenterology 2003, 125, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.M.; Wang, Z.L.; Li, Z.H. STAT1 activation and STAT1-dependent immune-response gene ICAM-1 expression in alveolar macrophages of rats suffered from interstitial pulmonary fibrosis. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2003, 19, 3–6. [Google Scholar] [PubMed]
- Wettstein, G.; Bellaye, P.S.; Kolb, M.; Hammann, A.; Crestani, B.; Soler, P.; Marchal-Somme, J.; Hazoume, A.; Gauldie, J.; Gunther, A.; et al. Inhibition of HSP27 blocks fibrosis development and EMT features by promoting Snail degradation. FASEB J. 2013, 27, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McMillan, H.J.; Mah, J.K.; Tarnopolsky, M.; Selby, K.; McClure, T.; Wilson, D.M.; Sherman, M.L.; Escolar, D.; Attie, K.M. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2017, 55, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.M. Transforming growth factor beta: The good, the bad, and the ugly. J. Exp. Med. 1994, 180, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, D.P.; Dangott, L.J.; Lightfoot, J.T. Lessons learned from vivo-morpholinos: How to avoid vivo-morpholino toxicity. BioTechniques 2014, 56, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Desviat, L.R.; Smedsrød, B.; Piétri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J.A. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 7, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Castillo Alvarez, A.; Gauck, S.; Gait, M.J.; Coursindel, T.; Wood, M.J.A.; Lebleu, B.; Boisguerin, P. Cellular trafficking determines the exon skipping activity of Pip6a-PMO in mdx skeletal and cardiac muscle cells. Nucleic Acids Res. 2014, 42, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Aoki, Y.; Koo, T.; Mcclorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. Self-assembly into nanoparticles is essential for receptor mediated uptake of therapeutic antisense oligonucleotides. Nano Lett. 2015, 15, 4364–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Pro-Fibrotic Factor | Function | Model | References |
|---|---|---|---|
| TGFβ1 | Pro-fibrotic master—regulator | In vitro and in vivo rodent models of renal fibrosis | [92,93,94] |
| In vivo mouse model of tendon scarring | [95] | ||
| In vitro models of hepatic fibrosis | [96] | ||
| ALK5 | Component of canonical TGFβ1 receptor—TGFβ1 signaling | Ex vivo in cultures of Dupuytren’s patient tissue | [91] |
| In vivo mouse model of DMD | [102] | ||
| SMAD3 | Component of canonical TGFβ1 signaling pathway | In vitro culture of human keloid fibroblasts | [104] |
| In vivo mouse model of tendon scarring | [95] | ||
| In vitro and in vivo mouse models of renal fibrosis | [105] | ||
| Connexin43 | Component of gap junctions—pro-fibrotic factor | In vitro culture of rat cardiac fibroblasts | [63] |
| In vivo model of rabbit eye glaucoma trabeculectomy | [68] | ||
| In vivo neonatal mouse model of burn injury | [66] | ||
| miR-21 | Pro-fibrotic miR | In vivo mouse model of cardiac fibrosis | [106] |
| In vivo mouse models of renal fibrosis | [107] | ||
| In vivo mouse models of pulmonary fibrosis | [76] | ||
| miR-192 | Pro-fibrotic miR | In vivo mouse models of renal fibrosis | [75] |
| miR-208a | Pro-fibrotic miR | In vivo rodent models of vascular and cardiac fibrosis | [108,109] |
| miR-34 | Pro-fibrotic miR | In vivo mouse and rat models of cardiac fibrosis | [110,111] |
| CTGF | Downstream pro-fibrotic effector of TGFβ1 | In vivo neonatal mouse model of burn injury | [66] |
| In vivo rabbit model of hypertrophic scarring | [129] | ||
| In vivo mouse model of tendon scarring | [95] | ||
| Periostin | Downstream pro-fibrotic effector of TGFβ1 | In vivo rat model of renal injury | [17] |
| In vivo mouse model of renal fibrosis | [151] | ||
| In vivo mouse model of surgically induced adhesions | [152] | ||
| In vivo mouse model of pulmonary fibrosis | [153] | ||
| Cultures of multiple human cell lines | [155] | ||
| TIMPs | MMP inhibitors and pro-fibrotic factors | In vivo rat model of pulmonary fibrosis | [160] |
| In vivo rat model of hepatic fibrosis | [161,162] | ||
| bFGF * | Cytokine—pro-fibrotic factor | In vivo rat model of pulmonary fibrosis | [163] |
| Kras * | Monomeric GTPase—component of signal transduction pathways | In vivo rat model of renal fibrosis | [164] |
| Sp1 * | Transcription factor | In vitro culture of human dermal fibroblasts and in vivo in murine skin | [165] |
| NF-κB * | Transcription factor | In vivo mouse model of pulmonary fibrosis | [166] |
| In vivo mouse model of interstitial fibrosis | [167] | ||
| STAT1 * | Transcription factor | In vivo rat model of pulmonary fibrosis | [168] |
| HSP27 * | Chaperone | In vitro and in vivo rat model of pulmonary fibrosis | [169] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
March, J.T.; Golshirazi, G.; Cernisova, V.; Carr, H.; Leong, Y.; Lu-Nguyen, N.; Popplewell, L.J. Targeting TGFβ Signaling to Address Fibrosis Using Antisense Oligonucleotides. Biomedicines 2018, 6, 74. https://doi.org/10.3390/biomedicines6030074
March JT, Golshirazi G, Cernisova V, Carr H, Leong Y, Lu-Nguyen N, Popplewell LJ. Targeting TGFβ Signaling to Address Fibrosis Using Antisense Oligonucleotides. Biomedicines. 2018; 6(3):74. https://doi.org/10.3390/biomedicines6030074
Chicago/Turabian StyleMarch, James T., Golnoush Golshirazi, Viktorija Cernisova, Heidi Carr, Yee Leong, Ngoc Lu-Nguyen, and Linda J. Popplewell. 2018. "Targeting TGFβ Signaling to Address Fibrosis Using Antisense Oligonucleotides" Biomedicines 6, no. 3: 74. https://doi.org/10.3390/biomedicines6030074
APA StyleMarch, J. T., Golshirazi, G., Cernisova, V., Carr, H., Leong, Y., Lu-Nguyen, N., & Popplewell, L. J. (2018). Targeting TGFβ Signaling to Address Fibrosis Using Antisense Oligonucleotides. Biomedicines, 6(3), 74. https://doi.org/10.3390/biomedicines6030074