TrkB-Targeted Therapy for Mucoepidermoid Carcinoma

 ,
,  , and
, and 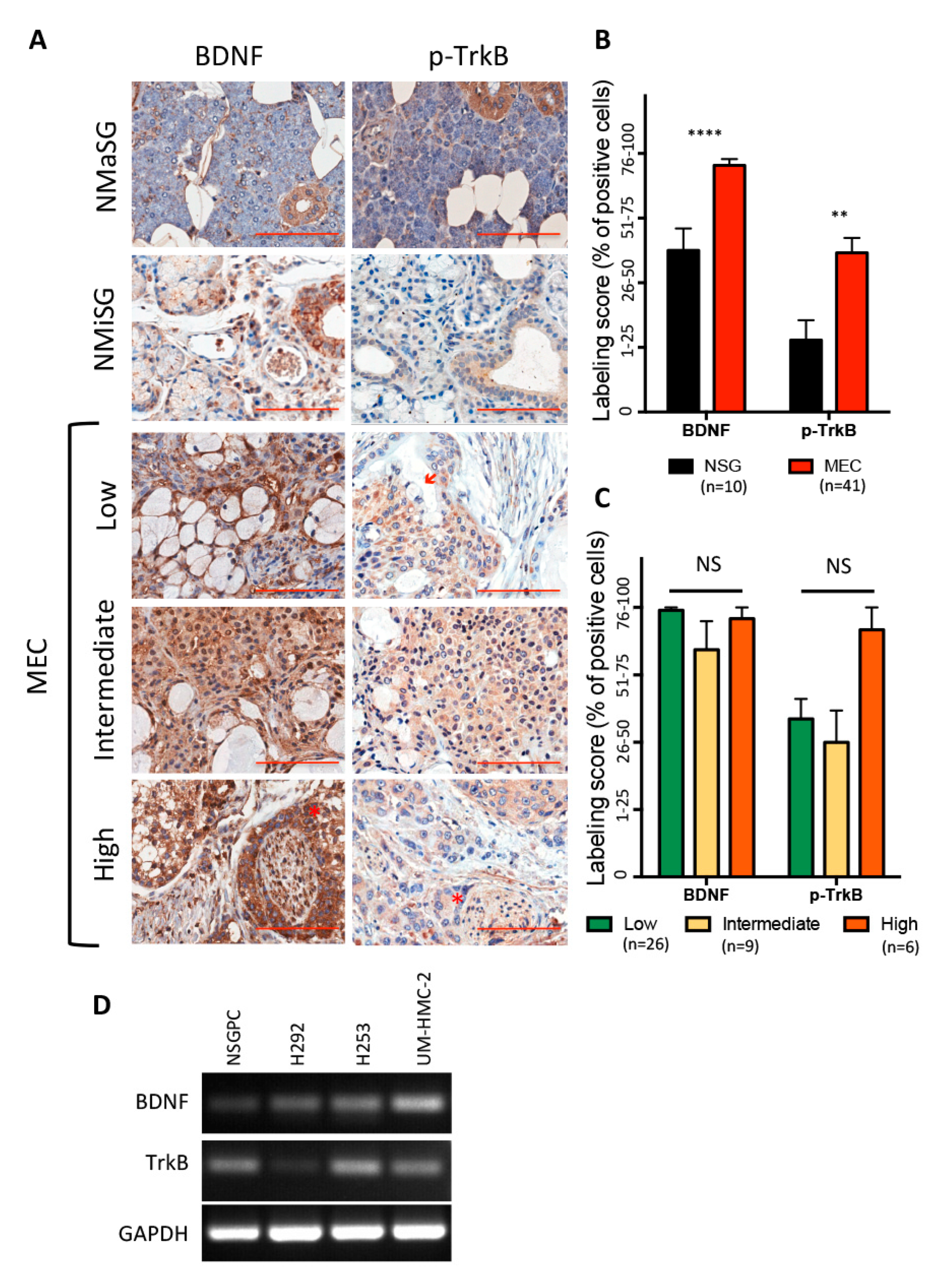{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Human Tissue Specimens
2.2. Immunohistochemistry
2.3. Cell Lines and Treatments
2.4. PCR
2.5. Transmission Electron Microscopy (TEM)
2.6. Scratch Assay
2.7. Transwell Invasion Assay
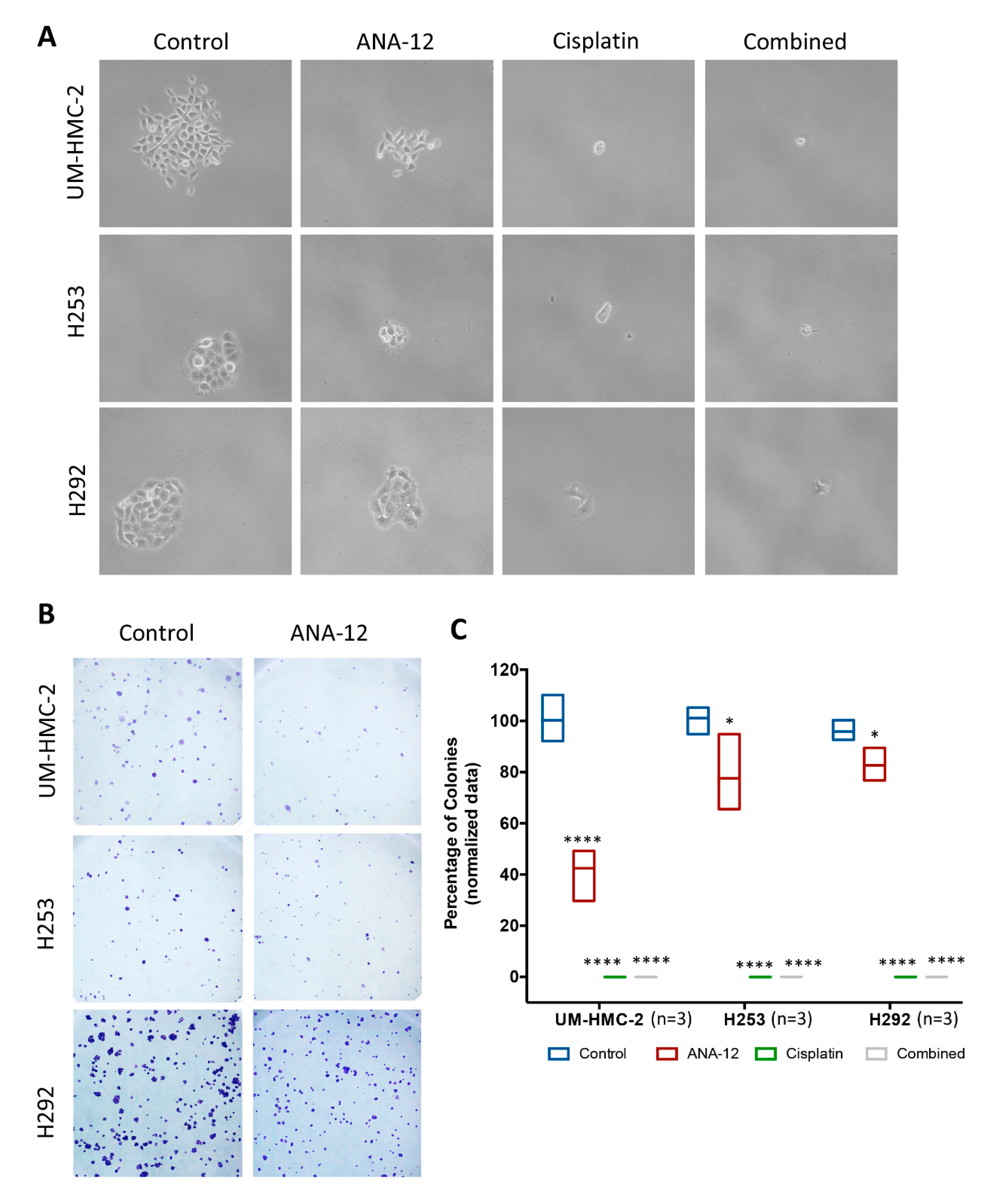
2.8. Clonogenic Assay
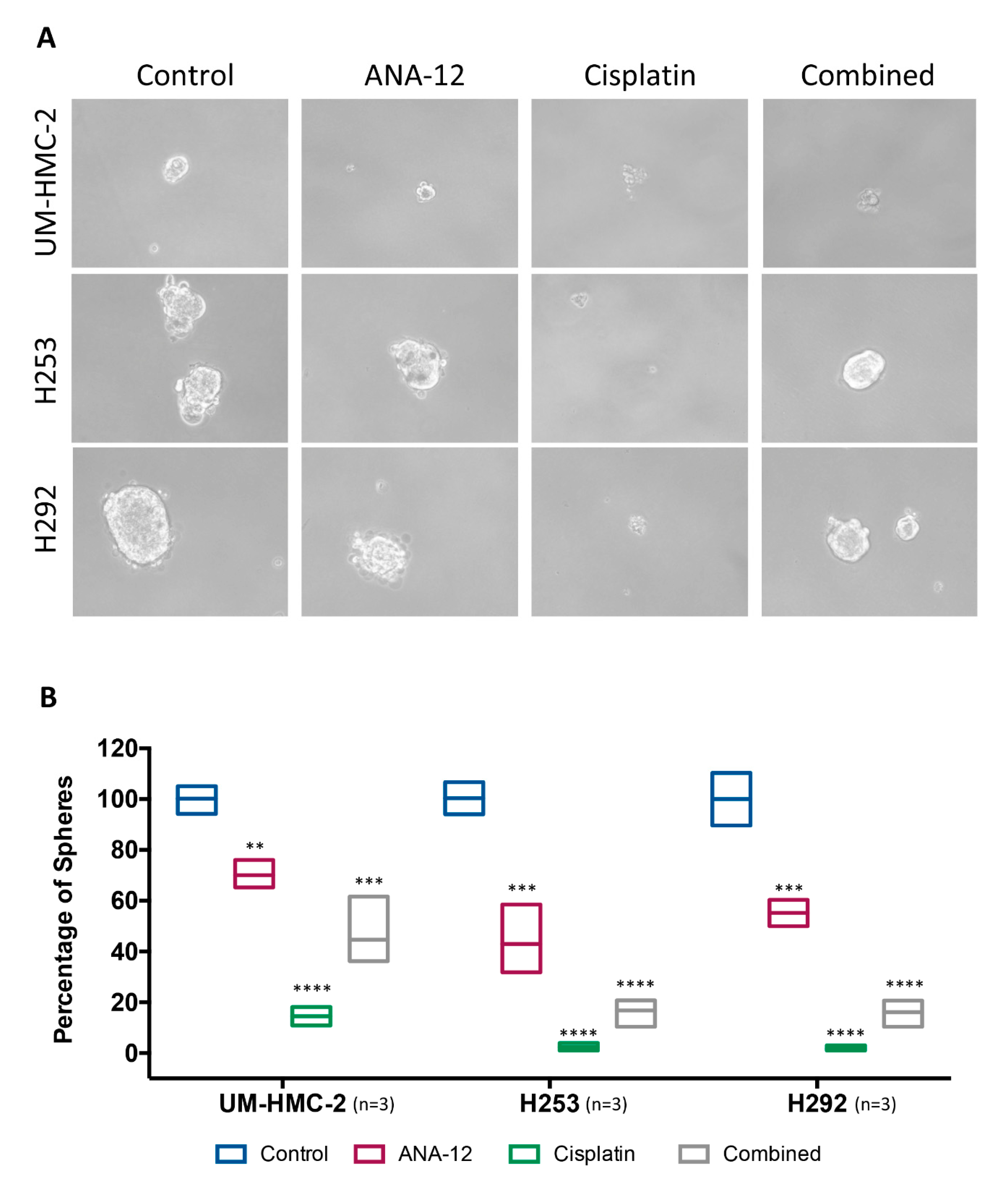
2.9. Spheroid Assay
2.10. Statistical Analysis
3. Results
3.1. BDNF/TrkB Pathway in Human Salivary Glands and Mucoepidermoid Carcinoma
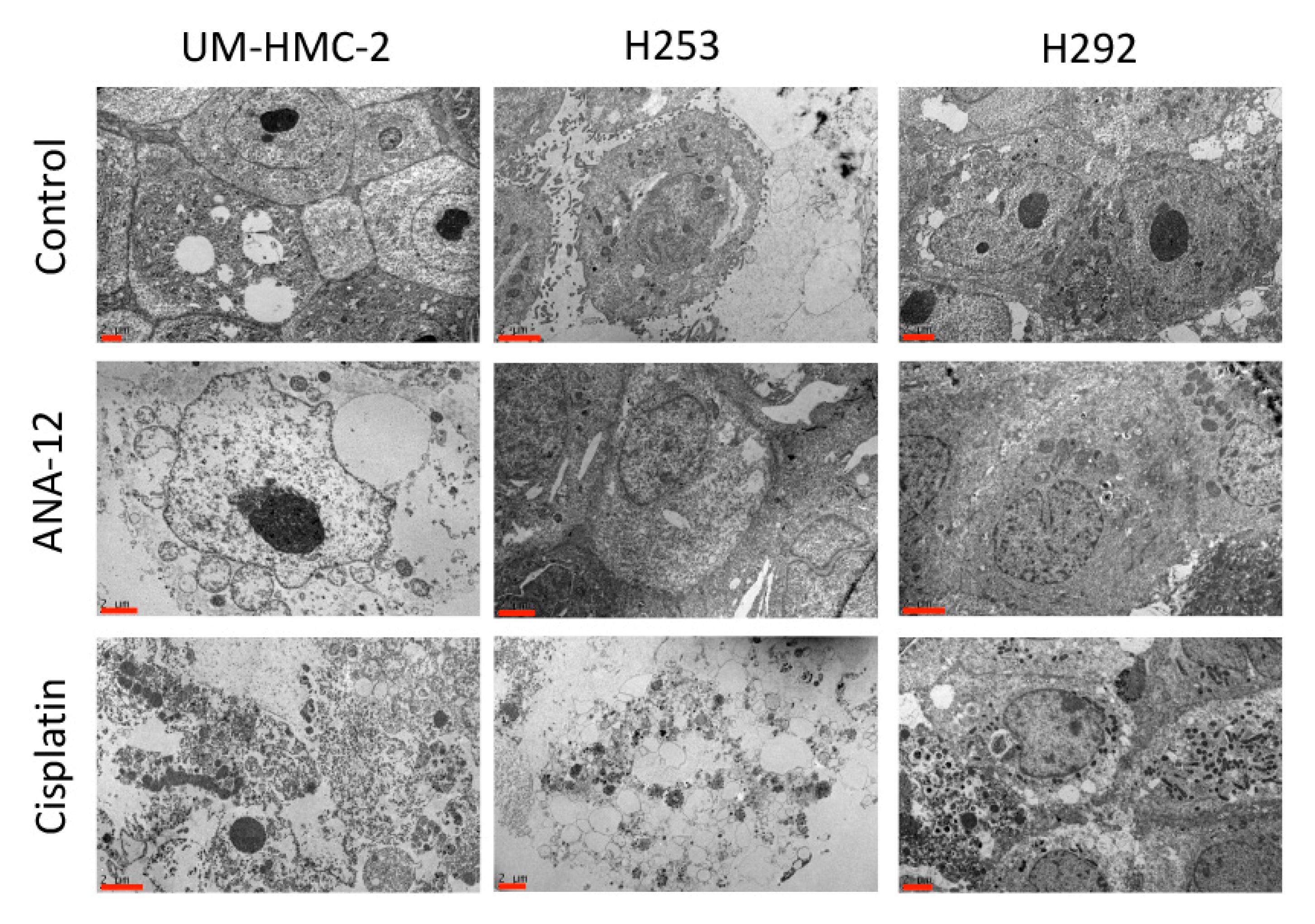
3.2. Effects of Cisplatin and TrkB Inhibition on Cell Ultrastructural Morphology
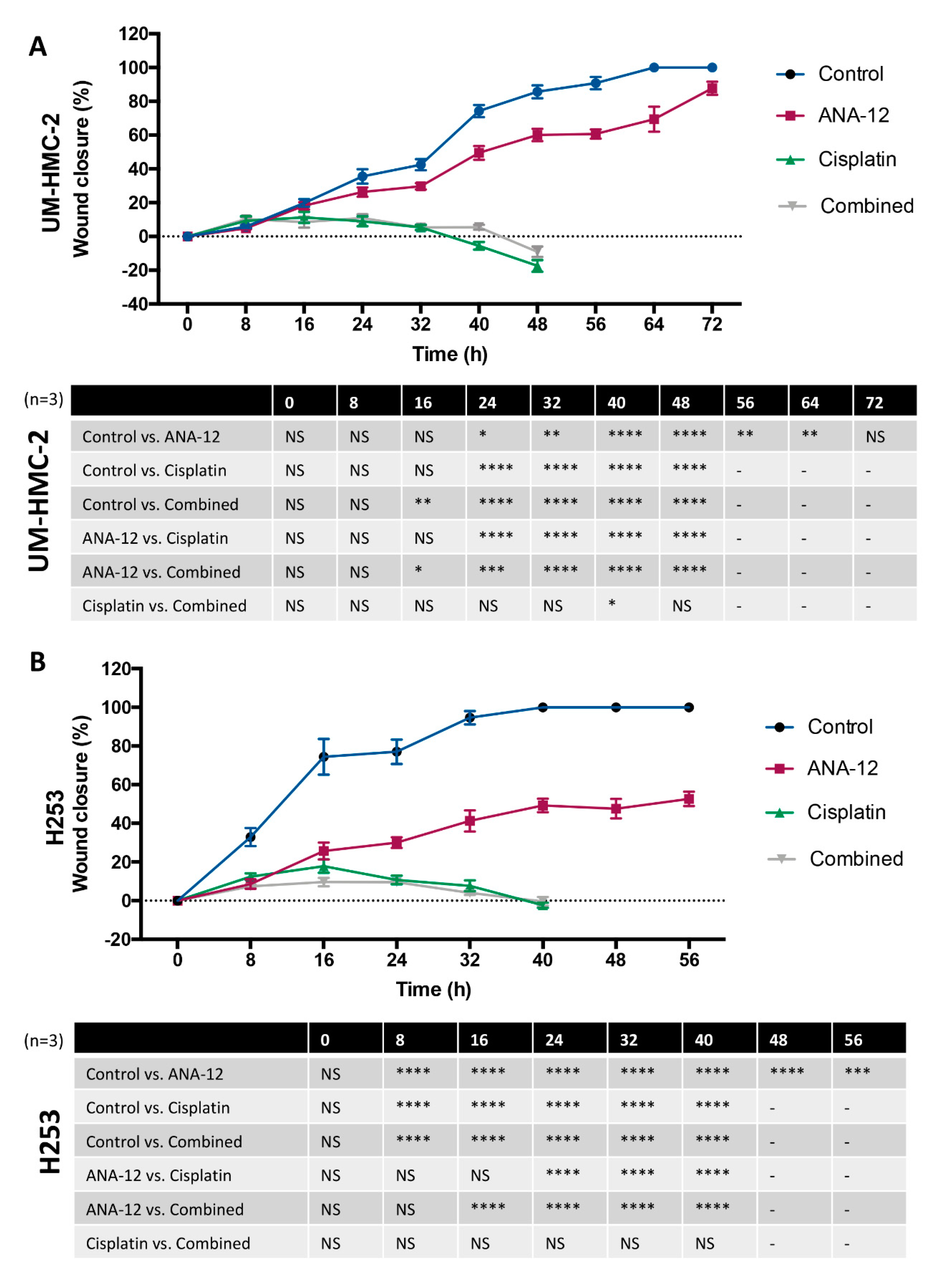
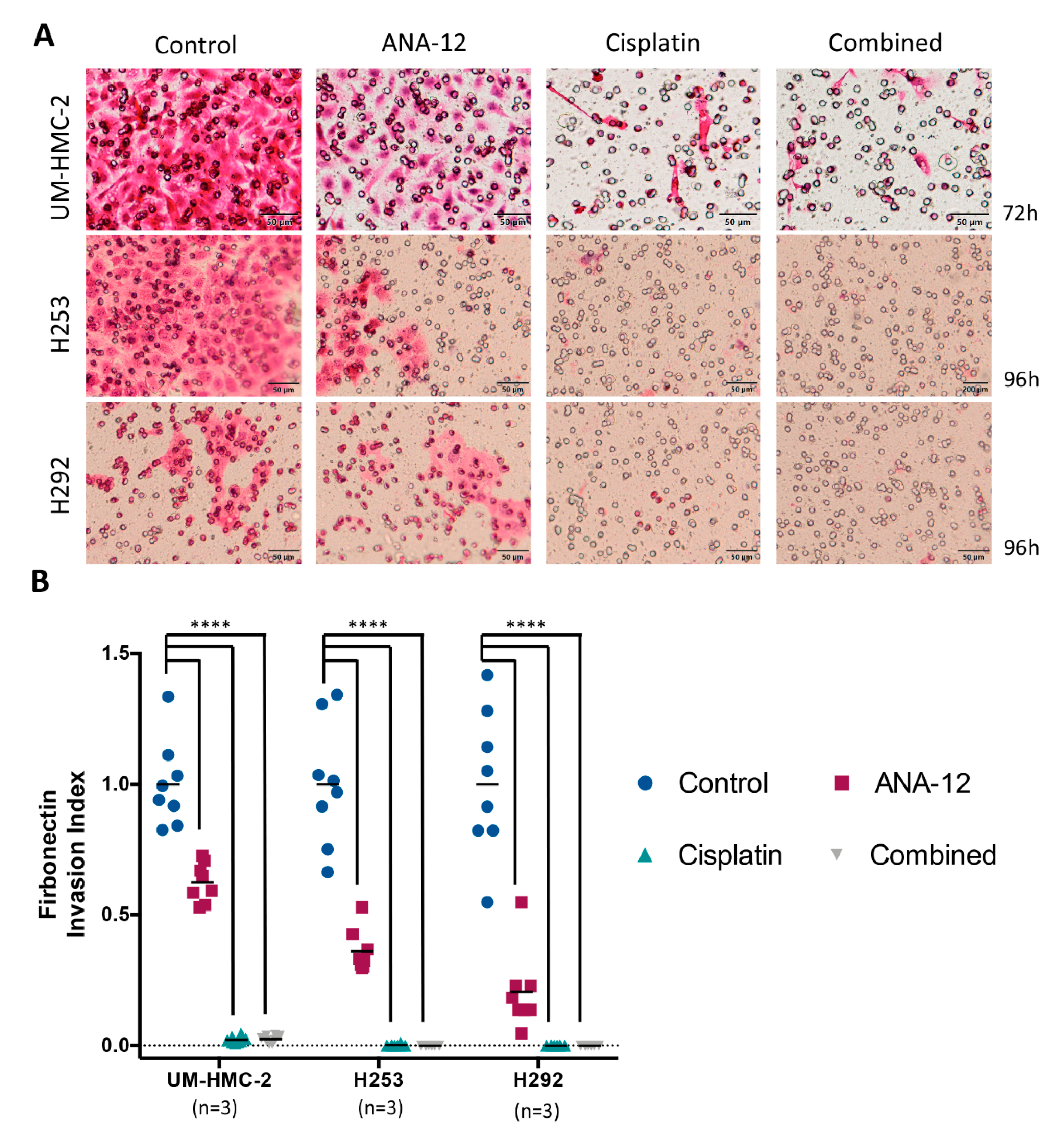
3.3. Effect of TrkB Inhibition and Cisplatin on MEC Cell Migration and Invasion
3.4. Effect of TrkB Inhibition and Cisplatin on MEC Cell Survival and CSC Number
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Tomorrow; International Agency for Research on Cancer: Lyon, France, 2018; Available online: https://gco.iarc.fr/tomorrow (accessed on 5 February 2020).
- El-Naggar, A.K.; Chan, J.K.C.; Grandis, J.R.; Takata, T.; Slootweg, P.J. WHO Classification of Head and Neck Tumours, 4th ed.; IARC: Lyon, France, 2017; Volume 9. [Google Scholar]
- Fonseca, F.P.; Carvalho Mde, V.; de Almeida, O.P.; Rangel, A.L.; Takizawa, M.C.; Bueno, A.G.; Vargas, P.A. Clinicopathologic analysis of 493 cases of salivary gland tumours in a Southern Brazilian population. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012, 114, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.; Craig, G.; Speight, P.M.; Franklin, C. The range and demographics of salivary gland tumours diagnosed in a UK population. Oral Oncol. 2008, 44, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Huo, Z.; Wu, H.; Li, J.; Li, S.; Wu, S.; Liu, Y.; Luo, Y.; Cao, J.; Zeng, X.; Liang, Z. Primary Pulmonary Mucoepidermoid Carcinoma: Histopathological and Moleculargenetic Studies of 26 Cases. PLoS ONE 2015, 10, e0143169. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.H.; Roberts, D.B.; El-Naggar, A.K.; Hanna, E.Y.; Garden, A.S.; Kies, M.S.; Weber, R.S.; Kupferman, M.E. Prognostic factors in mucoepidermoid carcinoma of the salivary glands. Cancer 2012, 118, 3928–3936. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, M.; Balm, A.J.; Smeele, L.E.; Wouters, M.W.; Van Dijk, B.A. An epidemiological evaluation of salivary gland cancer in the Netherlands (1989–2010). Cancer Epidemiol. 2015, 39, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.M.; Garcia, J.; Granchi, P.J.; Johnson, J.; Eisele, D.W. Late recurrence from salivary gland cancer. Cancer 2008, 112, 340–344. [Google Scholar] [CrossRef]
- Adams, A.; Warner, K.; Pearson, A.T.; Zhang, Z.; Kim, H.S.; Mochizuki, D.; Basura, G.; Helman, J.; Mantesso, A.; Castilho, R.M.; et al. ALDH/CD44 identifies uniquely tumourigenic cancer stem cells in salivary gland mucoepidermoid carcinomas. Oncotarget 2015, 6, 26633–26650. [Google Scholar] [CrossRef] [Green Version]
- Guimarães, D.M.; Almeida, L.O.; Martins, M.D.; Warner, K.A.; Silva, A.R.S.; Vargas, P.A.; Nunes, F.D.; Squarize, C.H.; Nör, J.E.; Castilho, R.M. Sensitizing mucoepidermoid carcinomas to chemotherapy by targeted disruption of cancer stem cells. Oncotarget 2016, 7, 42447–42460. [Google Scholar] [CrossRef]
- Wagner, V.P.; Martins, M.A.; Martins, M.D.; Warner, K.A.; Webber, L.P.; Squarize, C.H.; Nör, J.E.; Castilho, R.M. Overcoming adaptive resistance in mucoepidermoid carcinoma through inhibition of the IKK-β/IκBα/NFκB axis. Oncotarget 2016, 7, 73032. [Google Scholar] [CrossRef] [Green Version]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for Growth Factors in Cancer Progression. Physiology 2010, 25, 85–101. [Google Scholar] [CrossRef] [Green Version]
- De Moraes, J.K.; Wagner, V.P.; Fonseca, F.P.; Vargas, P.A.; De Farias, C.B.; Roesler, R.; Santos-Silva, A.R. Uncovering the role of brain-derived neurotrophic factor/tyrosine kinase receptor B signaling in head and neck malignancies. J. Oral Pathol. Med. 2017, 47, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; De Vitis, C.; Noto, A.; Fattore, L.; Mariotta, S.; Cherubini, E.; Roscilli, G.; Liguori, G.; Scognamiglio, G.; Rocco, G.; et al. TrkB is responsible for EMT transition in malignant pleural effusions derived cultures from adenocarcinoma of the lung. Cell Cycle 2013, 12, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhecke, E.; Adriaenssens, E.; Verbeke, S.; Meignan, S.; Germain, E.; Berteaux, N.; Nurcombe, V.; Le Bourhis, X.; Hondermarck, H. Brain-derived neurotrophic factor and neurotrophin-4/5 are expressed in breast cancer and can be targeted to inhibit tumour cell survival. Clin. Cancer Res. 2011, 17, 1741–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okugawa, Y.; Tanaka, K.; Inoue, Y.; Kawamura, M.; Kawamoto, A.; Hiro, J.; Saigusa, S.; Toiyama, Y.; Ohi, M.; Uchida, K.; et al. Brain-derived neurotrophic factor/tropomyosin-related kinase B pathway in gastric cancer. Br. J. Cancer 2013, 108, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Farias, C.B.; Rosemberg, D.B.; Heinen, T.E.; Koehler-Santos, P.; Abujamra, A.L.; Kapczinski, F.; Brunetto, A.L.; Ashton-Prolla, P.; Meurer, L.; Bogo, M.R.; et al. BDNF/TrkB Content and Interaction with Gastrin-Releasing Peptide Receptor Blockade in Colorectal Cancer. Oncology 2010, 79, 430–439. [Google Scholar] [CrossRef]
- Jia, S.; Wang, W.; Hu, Z.; Shan, C.; Wang, L.; Wu, B.; Yang, Z.; Yang, X.; Lei, D.-L. BDNF mediated TrkB activation contributes to the EMT progression and the poor prognosis in human salivary adenoid cystic carcinoma. Oral Oncol. 2015, 51, 64–70. [Google Scholar] [CrossRef]
- Shan, C.; Wei, J.; Hou, R.; Wu, B.; Yang, Z.; Wang, L.; Lei, D.; Yang, X. Schwann cells promote EMT and the Schwann-like differentiation of salivary adenoid cystic carcinoma cells via the BDNF/TrkB axis. Oncol. Rep. 2015, 35, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Warner, K.A.; Adams, A.; Bernardi, L.; Nor, C.; Finkel, K.A.; Zhang, Z.; McLean, S.A.; Helman, J.; Wolf, G.T.; Divi, V.; et al. Characterization of tumourigenic cell lines from the recurrence and lymph node metastasis of a human salivary mucoepidermoid carcinoma. Oral Oncol. 2013, 49, 1059–1066. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Barde, Y.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Liu, B.; Ji, R.; Jiang, X.; Yan, X.; Xin, Y. Targeting the BDNF/TrkB pathway for the treatment of tumours. Oncol Lett. 2019, 17, 2031–2039. [Google Scholar] [PubMed] [Green Version]
- De Vicente, J.C.; García-Suárez, O.; Esteban, I.; Santamaria, J.; Vega, J. Immunohistochemical localization of neurotrophins and neurotrophin receptors in human and mouse salivary glands. Ann. Anat. Anat. Anz. 1998, 180, 157–163. [Google Scholar] [CrossRef]
- Saruta, J.; Fujino, K.; To, M.; Tsukinoki, K. Expression and Localization of Brain-Derived Neurotrophic Factor (BDNF) mRNA and Protein in Human Submandibular Gland. Acta Histochem. Cytochem. 2012, 45, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Chernichenko, N.; Linkov, G.; Li, P.; Bakst, R.L.; Chen, C.-H.; He, S.; Yu, Y.A.; Chen, N.G.; Szalay, A.A.; Fong, Y.; et al. Oncolytic vaccinia virus therapy of salivary gland carcinoma. JAMA Otolaryngol. Neck Surg. 2013, 139, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Lagha, A.; Chraiet, N.; Ayadi, M.; Krimi, S.; Allani, B.; Rifi, H.; Raies, H.; Mezlini, A. Systemic therapy in the management of metastatic or advanced salivary gland cancers. Head Neck Oncol. 2012, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Chintakuntlawar, A.V.; Okuno, S.H.; Price, K.A. Systemic therapy for recurrent or metastatic salivary gland malignancies. Cancers Head Neck 2016, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Cazorla, M.; Prémont, J.; Mann, A.; Girard, N.; Kellendonk, C.; Rognan, D. Identification of a low–molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Investig. 2011, 121, 1846–1857. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Al-Bahlani, S.M.; Al-Dhahli, B.; Al-Adawi, K.; Al-Nabhani, A.; Al-Kindi, M. Platinum-Based Drugs Differentially Affect the Ultrastructure of Breast Cancer Cell Types. BioMed Res. Int. 2017, 2017, 3178794. [Google Scholar] [CrossRef]
- Huang, W.-C.; Hung, M.-C. Induction of Akt Activity by Chemotherapy Confers Acquired Resistance. J. Formos. Med Assoc. 2009, 108, 180–194. [Google Scholar] [CrossRef] [Green Version]
- Ramos, G.D.O.; Bernardi, L.; Lauxen, I.; Filho, M.S.; Horwitz, A.R.; Lamers, M.L. Fibronectin Modulates Cell Adhesion and Signaling to Promote Single Cell Migration of Highly Invasive Oral Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0151338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudenska, M.; Kratochvilova, M.; Vicar, T.; Gumulec, J.; Balvan, J.; Polanska, H.; Přibyl, J.; Masarik, M. Cisplatin enhances cell stiffness and decreases invasiveness rate in prostate cancer cells by actin accumulation. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, V.; Jain, M.V. In Vitro Tumourigenic Assay: Colony Forming Assay for Cancer Stem Cells. Methods Mol Biol. 2018, 1692, 89–95. [Google Scholar]
- Almeida, L.O.; Guimarães, D.M.; Squarize, C.H.; Castilho, R.M. Profiling the Behaviour of Distinct Populations of Head and Neck Cancer Stem Cells. Cancers 2016, 8, 7. [Google Scholar] [CrossRef]
- Almeida, L.O.; Abrahao, A.C.; Rosselli-Murai, L.K.; Giudice, F.S.; Zagni, C.; Leopoldino, A.M.; Squarize, C.H.; Castilho, R.M. NFκB mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (HNSCC). FEBS Open Bio 2014, 4, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, R.L.B.; Sousa, L.O.; Castilho, R.M.; Almeida, L.O. Cancer Stem Cells: Powerful Targets to Improve Current Anticancer Therapeutics. Stem Cells Int. 2019, 2019, 9618065. [Google Scholar]
- Thomaz, A.; Pinheiro, K.V.; Souza, B.K.; Gregianin, L.; Brunetto, A.L.; Brunetto, A.T.; de Farias, C.B.; Jaeger, M.D.C.; Ramaswamy, V.; Nör, C.; et al. Antitumour Activities and Cellular Changes Induced by TrkB Inhibition in Medulloblastoma. Front Pharmacol. 2019, 10, 698. [Google Scholar] [CrossRef] [Green Version]
- Wagner, V.P.; Martins, M.D.; Martins, M.A.T.; Almeida, L.O.; Warner, K.A.; Nör, J.E.; Squarize, C.H.; Castilho, R.M. Targeting histone deacetylase and NFκB signaling as a novel therapy for Mucoepidermoid Carcinomas. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- González-Arriagada, W.A.; Santos-Silva, A.R.; Ito, F.A.; Vargas, P.A.; Speight, P.M.; Bingle, L.; Lopes, M.A. Expression pattern of PLUNC proteins as an auxiliary tool for the diagnosis of high-grade mucoepidermoid carcinoma of the salivary gland. J. Oral Pathol. Med. 2012, 41, 589–597. [Google Scholar] [CrossRef]
- Pérez-De-Oliveira, M.E.; Wagner, V.P.; Araújo, A.L.D.; Santos-Silva, A.R.; Santos-Silva, A.R.; Bingle, L.; Vargas, P.A. Prognostic value of CRTC1-MAML2 translocation in salivary mucoepidermoid carcinoma: Systematic review and meta-analysis. J. Oral Pathol. Med. 2019, 49, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Mandel, A.; Ozdener, H.; Utermohlen, V. Identification of pro- and mature brain-derived neurotrophic factor in human saliva. Arch. Oral Biol. 2009, 54, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batsakis, J.G. Salivary gland neoplasia: An outcome of modified morphogenesis and cytodifferentiation. Oral Surgery Oral Med. Oral Pathol. 1980, 49, 229–232. [Google Scholar] [CrossRef]
- De Moraes, J.K.; Wagner, V.P.; Fonseca, F.P.; Amaral-Silva, G.K.D.; De Farias, C.B.; Pilar, E.F.S.; Gregianin, L.; Roesler, R.; Vargas, P.A.; Santos-Silva, A.R.; et al. Activation of BDNF/TrkB/Akt pathway is associated with aggressiveness and unfavorable survival in oral squamous cell carcinoma. Oral Dis. 2019, 25, 1925–1936. [Google Scholar] [CrossRef]
- Antunes, L.C.M.; Cartell, A.; De Farias, C.B.; Bakos, R.M.; Roesler, R.; Schwartsmann, G. Tropomyosin-Related Kinase Receptor and Neurotrophin Expression in Cutaneous Melanoma Is Associated with a Poor Prognosis and Decreased Survival. Oncology 2019, 97, 26–37. [Google Scholar] [CrossRef]
- Patani, N.; Jiang, W.; Mokbel, K. Brain-derived neurotrophic factor expression predicts adverse pathological & clinical outcomes in human breast cancer. Cancer Cell Int. 2011, 11, 23. [Google Scholar]
- Kimura, S.; Harada, T.; Ijichi, K.; Tanaka, K.; Liu, R.; Shibahara, D.; Kawano, Y.; Otsubo, K.; Yoneshima, Y.; Iwama, E.; et al. Expression of brain-derived neurotrophic factor and its receptor TrkB is associated with poor prognosis and a malignant phenotype in small cell lung cancer. Lung Cancer 2018, 120, 98–107. [Google Scholar] [CrossRef]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife 2013, 2, e00747. [Google Scholar] [CrossRef]
- Moriwaki, K.; Ayani, Y.; Kuwabara, H.; Terada, T.; Kawata, R.; Asahi, M. TRKB tyrosine kinase receptor is a potential therapeutic target for poorly differentiated oral squamous cell carcinoma. Oncotarget 2018, 9, 25225–25243. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Jiffar, T.; Kupferman, M.E. A Novel Role for BDNF-TrkB in the Regulation of Chemotherapy Resistance in Head and Neck Squamous Cell Carcinoma. PLoS ONE 2012, 7, e30246. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Ye, H.-Q.; Ren, Q.-C. Upregulation of the BDNF/TrKB pathway promotes epithelial-mesenchymal transition, as well as the migration and invasion of cervical cancer. Int. J. Oncol. 2017, 52, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.V.; Vanner, R.; Dirks, P.B.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Ma, Z.Y.; Zhou, Z.W.; Gao, W.C.; Du, Z.G.; Zhao, Z.H.; Li, Q.Q. The TrkB+ cancer stem cells contribute to post-chemotherapy recurrence of triple-negative breast cancers in an orthotopic mouse model. Oncogene 2014, 34, 761–770. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, V.P.; Martins, M.D.; Amoura, E.; Zanella, V.G.; Roesler, R.; de Farias, C.B.; Bingle, C.D.; Vargas, P.A.; Bingle, L. TrkB-Targeted Therapy for Mucoepidermoid Carcinoma. Biomedicines 2020, 8, 531. https://doi.org/10.3390/biomedicines8120531
Wagner VP, Martins MD, Amoura E, Zanella VG, Roesler R, de Farias CB, Bingle CD, Vargas PA, Bingle L. TrkB-Targeted Therapy for Mucoepidermoid Carcinoma. Biomedicines. 2020; 8(12):531. https://doi.org/10.3390/biomedicines8120531
Chicago/Turabian StyleWagner, Vivian P., Manoela D. Martins, Esra Amoura, Virgilio G. Zanella, Rafael Roesler, Caroline B. de Farias, Colin D. Bingle, Pablo A. Vargas, and Lynne Bingle. 2020. "TrkB-Targeted Therapy for Mucoepidermoid Carcinoma" Biomedicines 8, no. 12: 531. https://doi.org/10.3390/biomedicines8120531
APA StyleWagner, V. P., Martins, M. D., Amoura, E., Zanella, V. G., Roesler, R., de Farias, C. B., Bingle, C. D., Vargas, P. A., & Bingle, L. (2020). TrkB-Targeted Therapy for Mucoepidermoid Carcinoma. Biomedicines, 8(12), 531. https://doi.org/10.3390/biomedicines8120531