BAY 41-2272 Attenuates CTGF Expression via sGC/cGMP-Independent Pathway in TGFβ1-Activated Hepatic Stellate Cells

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Reagents
2.2. Isolation of Mouse Primary HSCs
2.3. Cell Viability
2.4. Western Blotting
2.5. Immunofluorescence Staining
2.6. RNA Isolation and Quantitative Real-Time PCR
2.7. Determination of Intracellular cGMP Levels
2.8. sGC Activity Assay
2.9. Statistical Analysis
3. Results
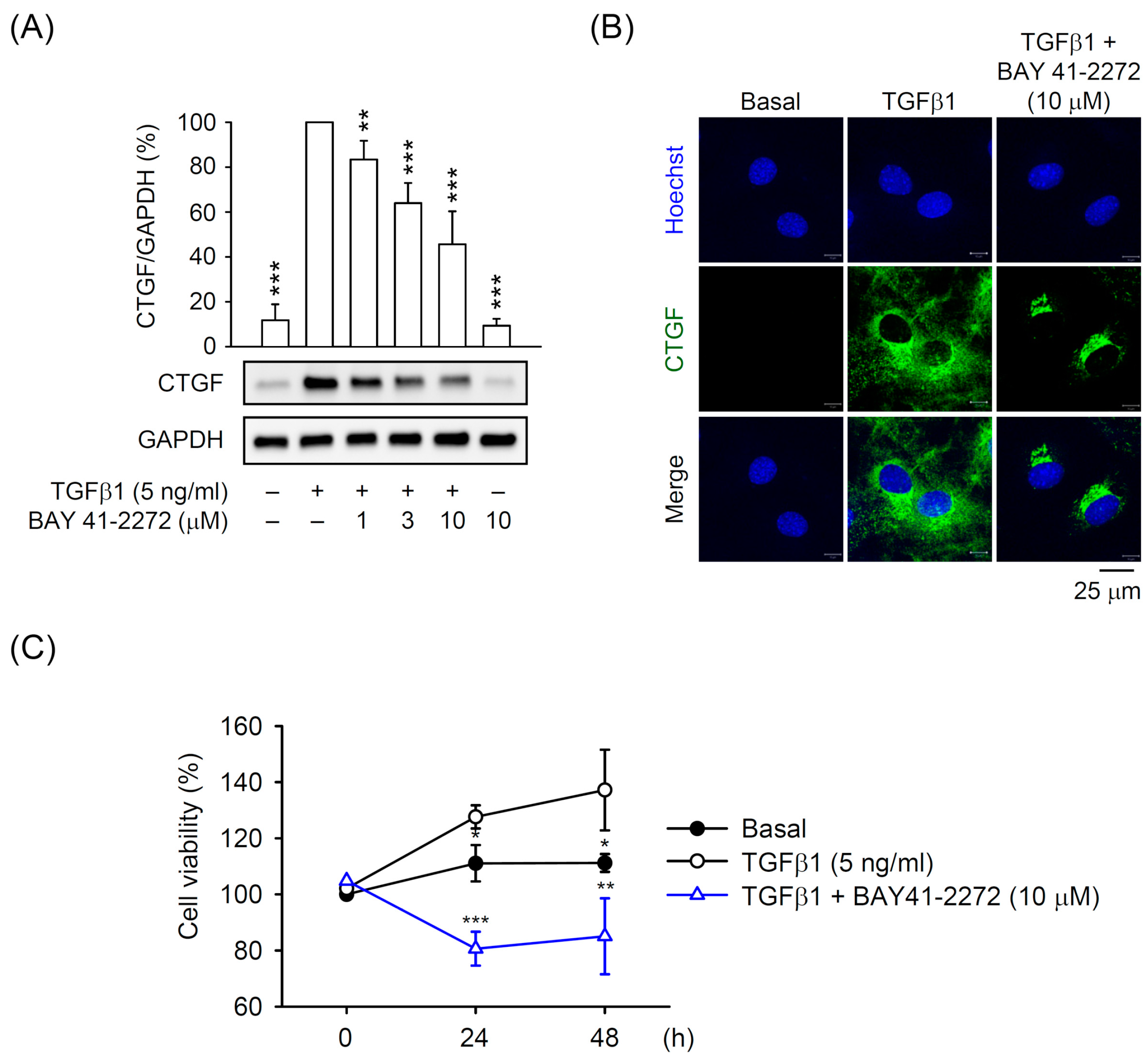
3.1. BAY 41-2272 sGC Stimulator Inhibited TGFβ1-Induced CTGF Expression and Cell Proliferation in Primary HSCs
3.2. The BAY 41-2272-Inhibited CTGF Expression and Cell Proliferation Was not via sGC/cGMP Pathway in TGFβ1-Activated Primary HSCs
3.3. PDE9 Modulated the BAY 41-2272-Mediated sGC/cGMP Signaling But not CTGF Inhibition in Primary HSCs
3.4. The TGFβ1-Induced CTGF Expression is Independent of cGMP Formation in Primary HSCs
3.5. BAY 41-2272 Selectively Inhibited the TGFβ1-Induced Akt Activation in Primary HSCs
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, J.; Wang, J.; Zhou, Q.; Yang, B.; He, Q.; Weng, Q. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharmacol. Res. 2020, 155, 104720. [Google Scholar] [CrossRef] [PubMed]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 9, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Deng, X.; Liang, J. Modulation of hepatic stellate cells and reversibility of hepatic fibrosis. Exp. Cell Res. 2017, 352, 420–426. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Kuo, L.M.; Chen, P.J.; Sung, P.J.; Chang, Y.C.; Ho, C.T.; Wu, Y.H.; Hwang, T.L. The Bioactive Extract of Pinnigorgia sp. Induces Apoptosis of Hepatic Stellate Cells via ROS-ERK/JNK-Caspase-3 Signaling. Mar. Drugs 2018, 16, 19. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nature reviews. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Wobst, J.; Kessler, T.; Dang, T.A.; Erdmann, J.; Schunkert, H. Role of sGC-dependent NO signalling and myocardial infarction risk. J. Mol. Med. 2015, 93, 383–394. [Google Scholar] [CrossRef]
- Hollas, M.A.; Ben Aissa, M.; Lee, S.H.; Gordon-Blake, J.M.; Thatcher, G.R.J. Pharmacological manipulation of cGMP and NO/cGMP in CNS drug discovery. Nitric Oxide 2019, 82, 59–74. [Google Scholar] [CrossRef]
- Hu, L.; Wang, Z.; Yi, R.; Yi, H.; Xiao, S.; Chen, Z.; Hu, G.; Li, Q. Soluble Guanylate Cyclase: A New Therapeutic Target for Fibrotic Diseases. Curr. Med. Chem. 2017, 24, 3203–3215. [Google Scholar] [CrossRef] [PubMed]
- Priviero, F.B.; Webb, R.C. Heme-dependent and independent soluble guanylate cyclase activators and vasodilation. J. Cardiovasc. Pharmacol. 2010, 56, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Li, Q.; Hu, L.; Yu, Z.; Yang, J.; Chang, Q.; Chen, Z.; Hu, G. Soluble Guanylate Cyclase Stimulators and Activators: Where are We and Where to Go? Mini Rev. Med. Chem. 2019, 19, 1544–1557. [Google Scholar] [CrossRef] [PubMed]
- Perri, R.E.; Langer, D.A.; Chatterjee, S.; Gibbons, S.J.; Gadgil, J.; Cao, S.; Farrugia, G.; Shah, V.H. Defects in cGMP-PKG pathway contribute to impaired NO-dependent responses in hepatic stellate cells upon activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G535–G542. [Google Scholar] [CrossRef]
- Sandner, P.; Stasch, J.P. Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Respir. Med. 2017, 122 (Suppl. S1), S1–S9. [Google Scholar] [CrossRef] [Green Version]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, I.; Caballero-Diaz, D. Transforming Growth Factor-beta-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; Consortium, I.-L. TGF-beta signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [Green Version]
- Carthy, J.M. TGFbeta signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J. Cell. Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Konigsrainer, A.; Weng, H.; Dooley, S.; ten Dijke, P. Transforming growth factor-beta (TGF-beta)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [Green Version]
- Colak, Y.; Senates, E.; Coskunpinar, E.; Oltulu, Y.M.; Zemheri, E.; Ozturk, O.; Doganay, L.; Mesci, B.; Yilmaz, Y.; Enc, F.Y.; et al. Concentrations of connective tissue growth factor in patients with nonalcoholic fatty liver disease: Association with liver fibrosis. Dis. Markers 2012, 33, 77–83. [Google Scholar] [CrossRef]
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef]
- Huang, G.; Brigstock, D.R. Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 2012, 17, 2495–2507. [Google Scholar]
- Wang, Y.; Kramer, S.; Loof, T.; Martini, S.; Kron, S.; Kawachi, H.; Shimizu, F.; Neumayer, H.H.; Peters, H. Enhancing cGMP in experimental progressive renal fibrosis: Soluble guanylate cyclase stimulation vs. phosphodiesterase inhibition. Am. J. Physiol. Ren. Physiol. 2006, 290, F167–F176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuyama, H.; Tsuruda, T.; Sekita, Y.; Hatakeyama, K.; Imamura, T.; Kato, J.; Asada, Y.; Stasch, J.P.; Kitamura, K. Pressure-independent effects of pharmacological stimulation of soluble guanylate cyclase on fibrosis in pressure-overloaded rat heart. Hypertens. Res. 2009, 32, 597–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, C.; Zenzmaier, C.; Palumbo-Zerr, K.; Mancuso, R.; Distler, A.; Dees, C.; Zerr, P.; Huang, J.; Maier, C.; Pachowsky, M.L.; et al. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFbeta signalling. Ann. Rheum. Dis. 2015, 74, 1408–1416. [Google Scholar] [CrossRef] [Green Version]
- Kadoya, H.; Satoh, M.; Nagasu, H.; Sasaki, T.; Kashihara, N. Deficiency of endothelial nitric oxide signaling pathway exacerbates peritoneal fibrosis in mice. Clin. Exp. Nephrol. 2015, 19, 567–575. [Google Scholar] [CrossRef]
- Matei, A.E.; Beyer, C.; Gyorfi, A.H.; Soare, A.; Chen, C.W.; Dees, C.; Bergmann, C.; Ramming, A.; Friebe, A.; Hofmann, F.; et al. Protein kinases G are essential downstream mediators of the antifibrotic effects of sGC stimulators. Ann. Rheum. Dis. 2018, 77, 459. [Google Scholar] [CrossRef]
- Lambers, C.; Boehm, P.M.; Karabacak, Y.; Samaha, E.; Benazzo, A.; Jaksch, P.; Roth, M. Combined Activation of Guanylate Cyclase and Cyclic AMP in Lung Fibroblasts as a Novel Therapeutic Concept for Lung Fibrosis. Biomed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Chen, C.H.; Kuo, L.M.; Chang, Y.; Wu, W.; Goldbach, C.; Ross, M.A.; Stolz, D.B.; Chen, L.; Fung, J.J.; Lu, L.; et al. In Vivo immune modulatory activity of hepatic stellate cells in mice. Hepatology 2006, 44, 1171–1181. [Google Scholar] [CrossRef]
- Yu, M.C.; Chen, C.H.; Liang, X.; Wang, L.; Gandhi, C.R.; Fung, J.J.; Lu, L.; Qian, S. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology 2004, 40, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gaca, M.D.; Swenson, E.S.; Vellucci, V.F.; Reiss, M.; Wells, R.G. Smads 2 and 3 are differentially activated by transforming growth factor-beta (TGF-beta) in quiescent and activated hepatic stellate cells. Constitutive nuclear localization of Smads in activated cells is TGF-beta-independent. J. Biol. Chem. 2003, 278, 11721–11728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugnier, C.; Meyer, A.; Talha, S.; Geny, B. Cyclic nucleotide phosphodiesterases: New targets in the metabolic syndrome? Pharmacol. Ther. 2020, 208, 107475. [Google Scholar] [CrossRef] [PubMed]
- Son, G.; Hines, I.N.; Lindquist, J.; Schrum, L.W.; Rippe, R.A. Inhibition of phosphatidylinositol 3-kinase signaling in hepatic stellate cells blocks the progression of hepatic fibrosis. Hepatology 2009, 50, 1512–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.K.; Ryu, Y.L.; Jung, K.H.; Lee, H.; Lee, H.S.; Yan, H.H.; Park, H.J.; Ryu, J.K.; Suh, J.K.; Hong, S.; et al. HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis. Sci. Rep. 2013, 3, 3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Wang, F.; Guo, Q.; Li, M.; Wang, L.; Zhang, Z.; Jiang, S.; Jin, H.; Chen, A.; Tan, S.; et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 2018, 19, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Gao, R.; Brigstock, D.R. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J. Biol. Chem. 2004, 279, 8848–8855. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Charrier, A.L.; Leask, A.; French, S.W.; Brigstock, D.R. Ethanol-stimulated differentiated functions of human or mouse hepatic stellate cells are mediated by connective tissue growth factor. J. Hepatol. 2011, 55, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.Y.; Lee, S.H.; Lee, J.H.; Kang, Y.N.; Hwang, J.S.; Park, K.G.; Kim, M.K.; Jang, B.K. Src Inhibition Attenuates Liver Fibrosis by Preventing Hepatic Stellate Cell Activation and Decreasing Connetive Tissue Growth Factor. Cells 2020, 9, 558. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Zheng, S. Curcumin inhibits connective tissue growth factor gene expression in activated hepatic stellate cells in vitro by blocking NF-kappaB and ERK signalling. Br. J. Pharmacol. 2008, 153, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, C.; Xie, Y.; Peng, M.; Ma, L.; Zhou, Y.; Zhang, Y.; Kang, W.; Wang, J.; Bai, X.; Wang, P.; et al. Inhibition of connective tissue growth factor suppresses hepatic stellate cell activation In Vitro and prevents liver fibrosis In Vivo. Clin. Exp. Med. 2014, 14, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Berger, P.; Zenzmaier, C. The Potential of sGC Modulators for the Treatment of Age-Related Fibrosis: A Mini-Review. Gerontology 2017, 63, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Kawada, N.; Kuroki, T.; Uoya, M.; Inoue, M.; Kobayashi, K. Smooth muscle alpha-actin expression in rat hepatic stellate cell is regulated by nitric oxide and cGMP production. Biochem. Biophys. Res. Commun. 1996, 229, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Failli, P.; De, F.R.; Caligiuri, A.; Gentilini, A.; Romanelli, R.G.; Marra, F.; Batignani, G.; Guerra, C.T.; Laffi, G.; Gentilini, P.; et al. Nitrovasodilators inhibit platelet-derived growth factor-induced proliferation and migration of activated human hepatic stellate cells. Gastroenterology 2000, 119, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Thirunavukkarasu, C.; Watkins, S.C.; Gandhi, C.R. Mechanisms of endotoxin-induced NO, IL-6, and TNF-alpha production in activated rat hepatic stellate cells: Role of p38 MAPK. Hepatology 2006, 44, 389–398. [Google Scholar] [CrossRef]
- Uemura, T.; Gandhi, C.R. Inhibition of DNA synthesis in cultured hepatocytes by endotoxin-conditioned medium of activated stellate cells is transforming growth factor-beta and nitric oxide-independent. Br. J. Pharmacol. 2001, 133, 1125–1133. [Google Scholar] [CrossRef] [Green Version]
- Urtasun, R.; Cubero, F.J.; Vera, M.; Nieto, N. Reactive nitrogen species switch on early extracellular matrix remodeling via induction of MMP1 and TNFalpha. Gastroenterology 2009, 136, 1410–1422. [Google Scholar] [CrossRef]
- Wang, P.G.; Xian, M.; Tang, X.; Wu, X.; Wen, Z.; Cai, T.; Janczuk, A.J. Nitric oxide donors: Chemical activities and biological applications. Chem. Rev. 2002, 102, 1091–1134. [Google Scholar] [CrossRef]
- Hall, K.C.; Bernier, S.G.; Jacobson, S.; Liu, G.; Zhang, P.Y.; Sarno, R.; Catanzano, V.; Currie, M.G.; Masferrer, J.L. sGC stimulator praliciguat suppresses stellate cell fibrotic transformation and inhibits fibrosis and inflammation in models of NASH. Proc. Natl. Acad. Sci. USA 2019, 116, 11057–11062. [Google Scholar] [CrossRef] [Green Version]
- Hwang, T.L.; Tang, M.C.; Kuo, L.M.; Chang, W.D.; Chung, P.J.; Chang, Y.W.; Fang, Y.C. YC-1 potentiates cAMP-induced CREB activation and nitric oxide production in alveolar macrophages. Toxicol. Appl. Pharmacol. 2012, 260, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Breitenstein, S.; Roessig, L.; Sandner, P.; Lewis, K.S. Novel sGC Stimulators and sGC Activators for the Treatment of Heart Failure. Handb. Exp. Pharmacol. 2017, 243, 225–247. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Zimmer, D.P.; Milne, G.T.; Follmann, M.; Hobbs, A.; Stasch, J.P. Soluble Guanylate Cyclase Stimulators and Activators. Handb. Exp. Pharmacol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Abdelaziz, N.; Colombo, F.; Mercier, I.; Calderone, A. Nitric oxide attenuates the expression of transforming growth factor-beta(3) mRNA in rat cardiac fibroblasts via destabilization. Hypertension 2001, 38, 261–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitson, T.D.; Martic, M.; Darby, I.A.; Kelynack, K.J.; Bisucci, T.; Tait, M.G.; Becker, G.J. Intracellular cyclic nucleotide analogues inhibit in vitro mitogenesis and activation of fibroblasts derived from obstructed rat kidneys. Nephron. Exp. Nephrol. 2004, 96, e59–e66. [Google Scholar] [CrossRef]
- Frey, R.; Becker, C.; Saleh, S.; Unger, S.; van der Mey, D.; Muck, W. Clinical Pharmacokinetic and Pharmacodynamic Profile of Riociguat. Clin. Pharmacokinet. 2018, 57, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Schwabl, P.; Brusilovskaya, K.; Supper, P.; Bauer, D.; Konigshofer, P.; Riedl, F.; Hayden, H.; Fuchs, C.D.; Stift, J.; Oberhuber, G.; et al. The soluble guanylate cyclase stimulator riociguat reduces fibrogenesis and portal pressure in cirrhotic rats. Sci. Rep. 2018, 8, 9372. [Google Scholar] [CrossRef]
- Flores-Costa, R.; Alcaraz-Quiles, J.; Titos, E.; Lopez-Vicario, C.; Casulleras, M.; Duran-Guell, M.; Rius, B.; Diaz, A.; Hall, K.; Shea, C.; et al. The soluble guanylate cyclase stimulator IW-1973 prevents inflammation and fibrosis in experimental non-alcoholic steatohepatitis. Br. J. Pharmacol. 2018, 175, 953–967. [Google Scholar] [CrossRef]
- Knorr, A.; Hirth-Dietrich, C.; Alonso-Alija, C.; Harter, M.; Hahn, M.; Keim, Y.; Wunder, F.; Stasch, J.P. Nitric oxide-independent activation of soluble guanylate cyclase by BAY 60-2770 in experimental liver fibrosis. Arzneimittel-Forschung 2008, 58, 71–80. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, Q.L.; Shen, L.; Tao, Y.Y.; Liu, C.H. MicroRNA-101 suppresses liver fibrosis by downregulating PI3K/Akt/mTOR signaling pathway. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 575–584. [Google Scholar] [CrossRef]
- Lao, Y.; Li, Y.; Zhang, P.; Shao, Q.; Lin, W.; Qiu, B.; Lv, Y.; Tang, L.; Su, S.; Zhang, H.; et al. Targeting Endothelial Erk1/2-Akt Axis as a Regeneration Strategy to Bypass Fibrosis during Chronic Liver Injury in Mice. Mol. Ther. 2018, 26, 2779–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.; Bai, R.; Wang, L.; Gao, J.; Zhang, H. Artesunate may inhibit liver fibrosis via the FAK/Akt/beta-catenin pathway in LX-2 cells. BMC Pharmacol. Toxicol. 2018, 19, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.-J.; Kuo, L.-M.; Wu, Y.-H.; Chang, Y.-C.; Lai, K.-H.; Hwang, T.-L. BAY 41-2272 Attenuates CTGF Expression via sGC/cGMP-Independent Pathway in TGFβ1-Activated Hepatic Stellate Cells. Biomedicines 2020, 8, 330. https://doi.org/10.3390/biomedicines8090330
Chen P-J, Kuo L-M, Wu Y-H, Chang Y-C, Lai K-H, Hwang T-L. BAY 41-2272 Attenuates CTGF Expression via sGC/cGMP-Independent Pathway in TGFβ1-Activated Hepatic Stellate Cells. Biomedicines. 2020; 8(9):330. https://doi.org/10.3390/biomedicines8090330
Chicago/Turabian StyleChen, Po-Jen, Liang-Mou Kuo, Yi-Hsiu Wu, Yu-Chia Chang, Kuei-Hung Lai, and Tsong-Long Hwang. 2020. "BAY 41-2272 Attenuates CTGF Expression via sGC/cGMP-Independent Pathway in TGFβ1-Activated Hepatic Stellate Cells" Biomedicines 8, no. 9: 330. https://doi.org/10.3390/biomedicines8090330
APA StyleChen, P. -J., Kuo, L. -M., Wu, Y. -H., Chang, Y. -C., Lai, K. -H., & Hwang, T. -L. (2020). BAY 41-2272 Attenuates CTGF Expression via sGC/cGMP-Independent Pathway in TGFβ1-Activated Hepatic Stellate Cells. Biomedicines, 8(9), 330. https://doi.org/10.3390/biomedicines8090330