Changes in Resurgent Sodium Current Contribute to the Hyperexcitability of Muscles in Patients with Paramyotonia Congenita

Abstract
:1. Introduction
2. Experimental Section
2.1. Patients and Genetic Analysis
2.2. Preparation of cDNA Constructs for Mutagenesis
2.3. Cell Cultures and cDNA Transfection
2.4. Electrophysiological Recordings
2.5. Homology Modeling
2.6. Measurement of the Fast Steady-State of Activation and Inactivation Curves
2.7. Data Analysis
3. Results
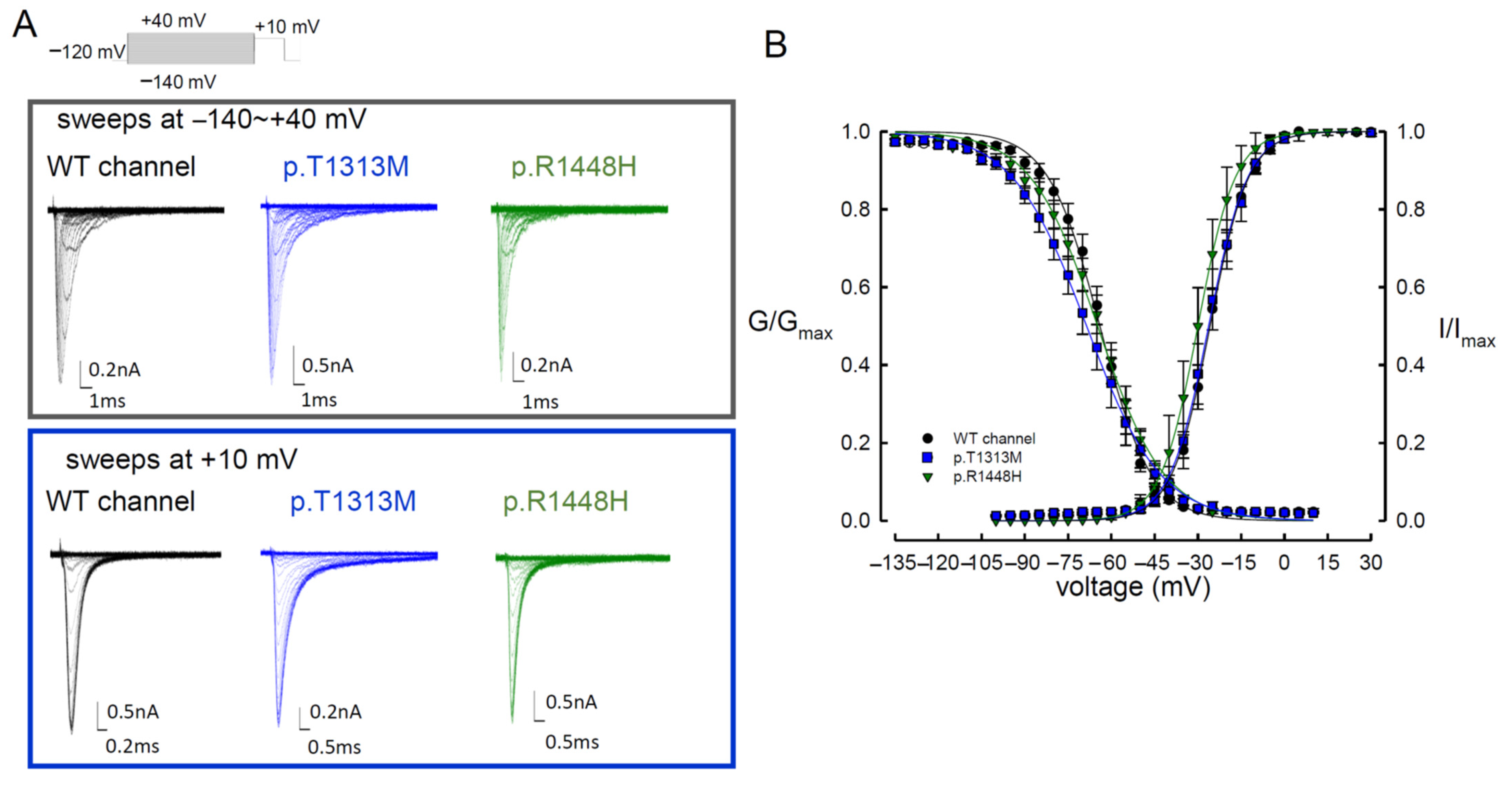
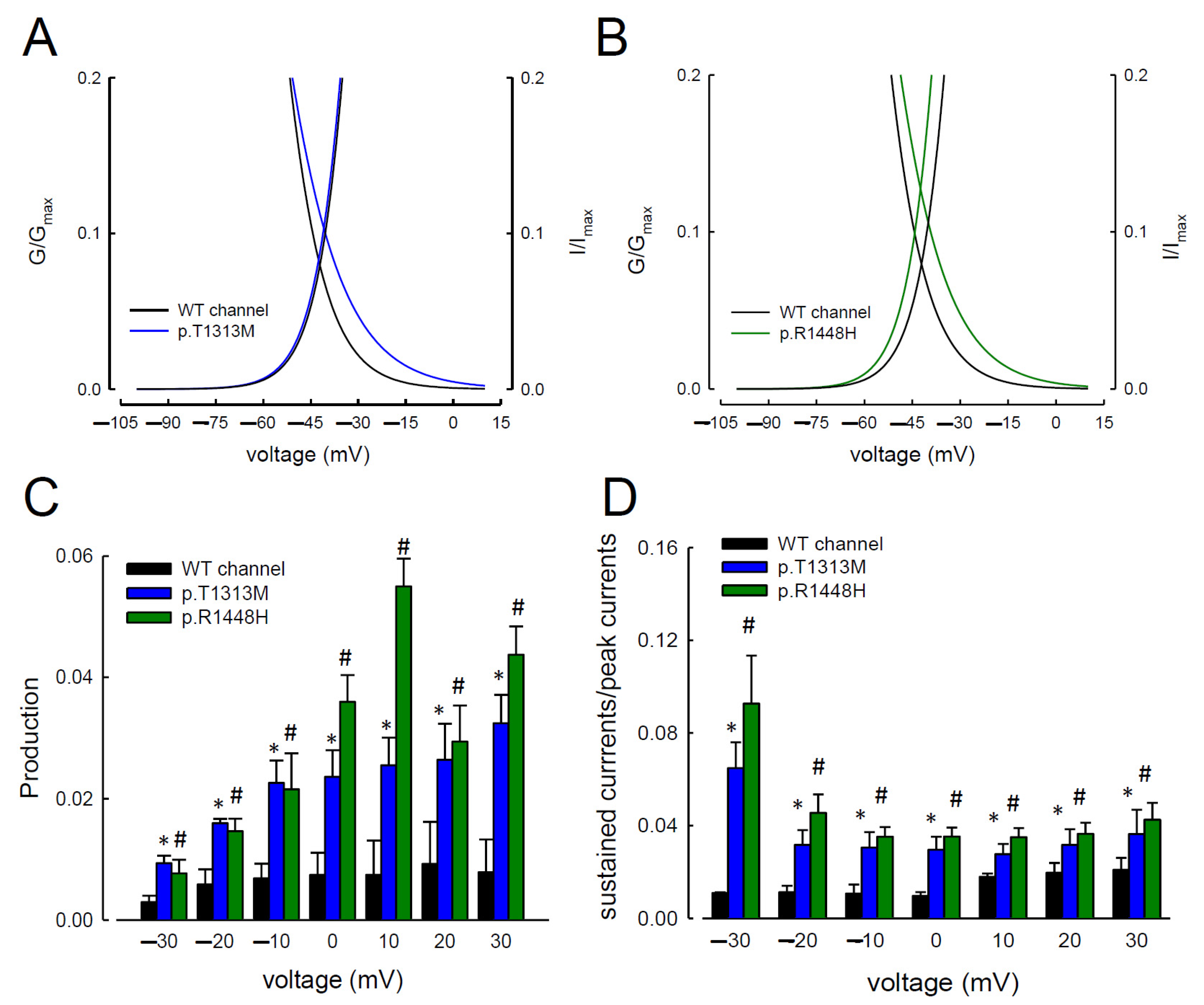
3.1. The Sustained Na+ Currents in the p.T1313M and p.R1448H Mutant Channels Were Larger than Those in the WT Nav1.4 Channel
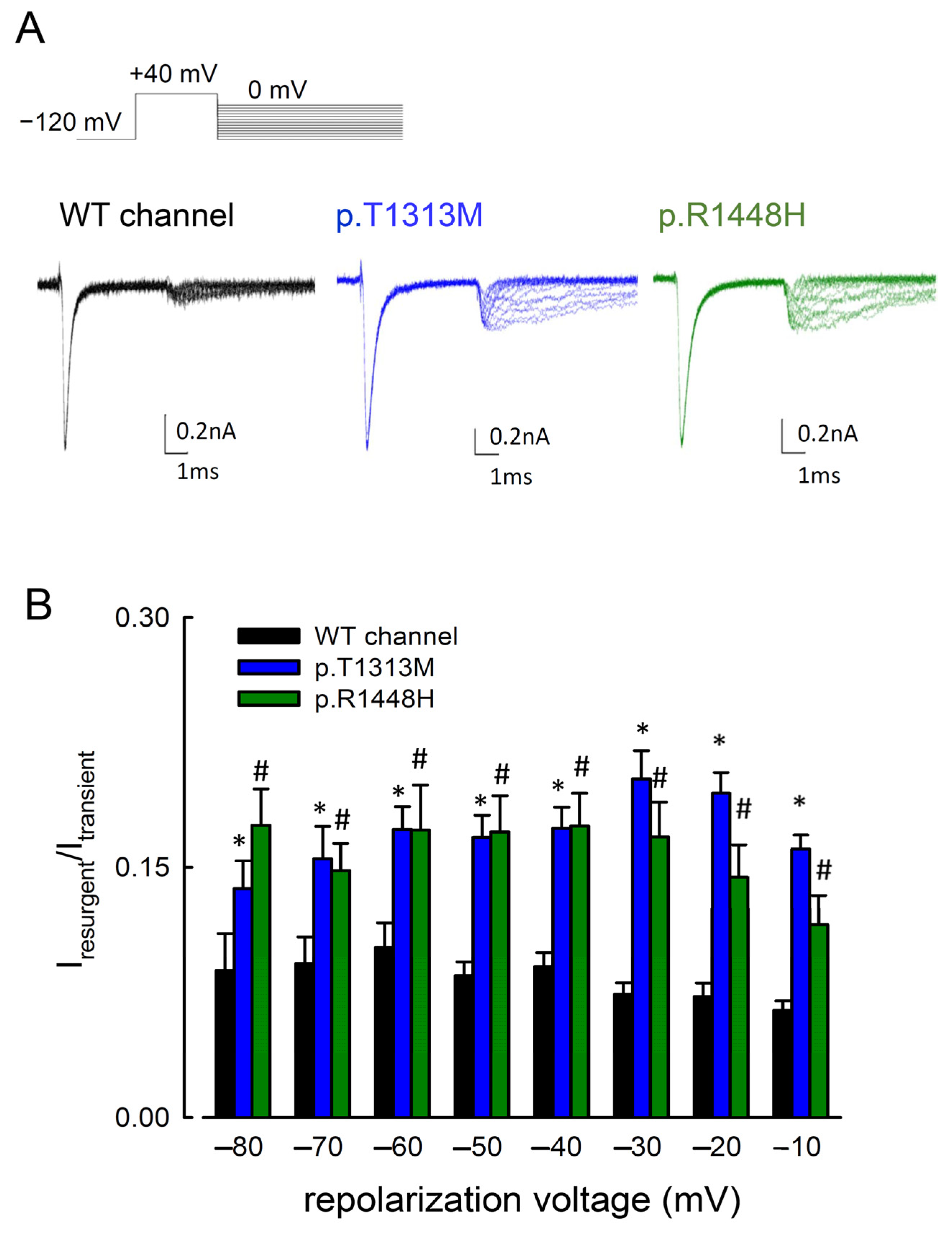
3.2. The Maximal Resurgent Na+ Currents in the p.T1313M and p.R1448H Mutant Channels Were between −80 and −10 mV
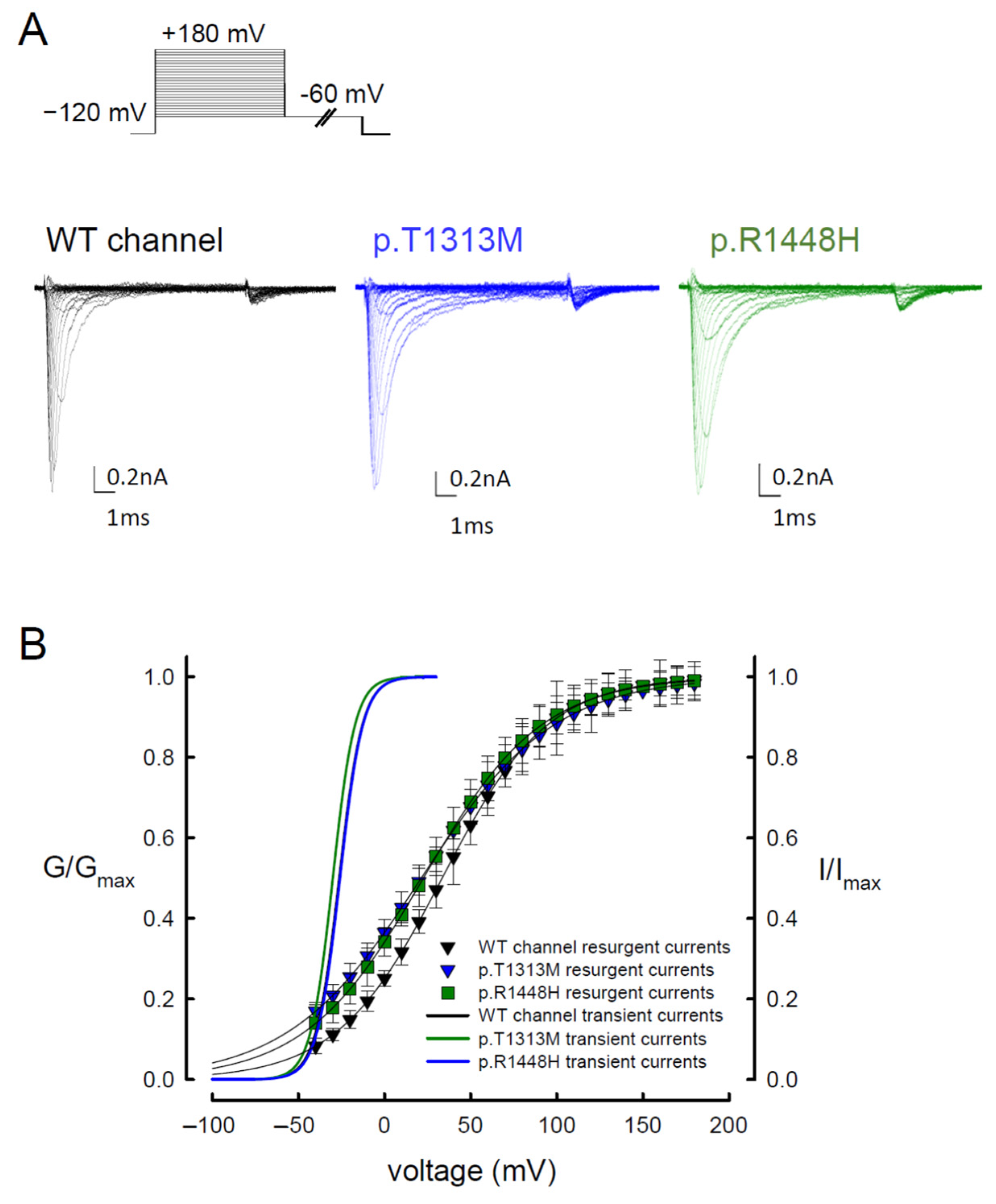
3.3. The WT Nav1.4 Channel Exhibits Two Different Activation Curves for Transient Na+ and Resurgent Na+ Currents
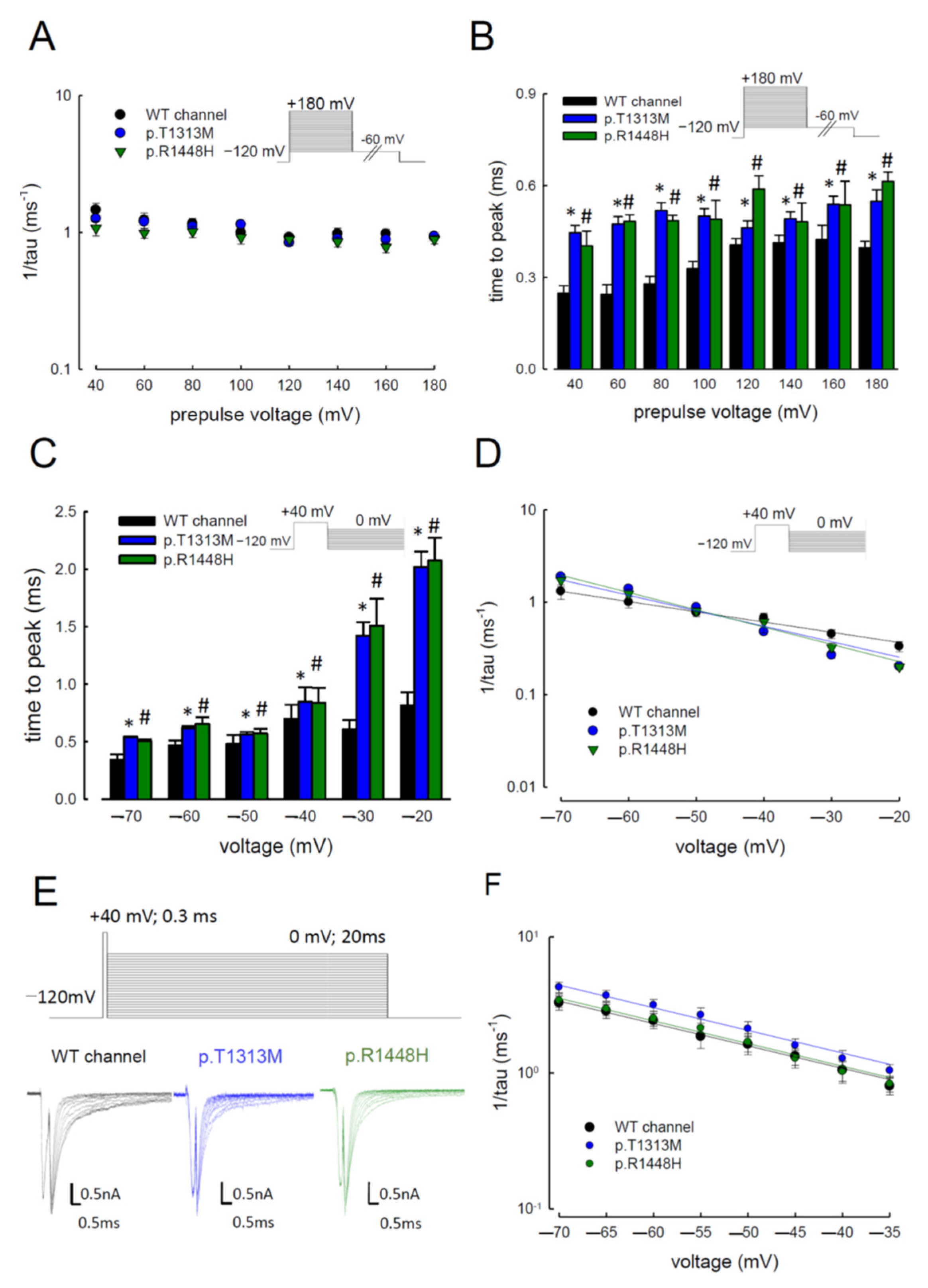
3.4. Delayed Times to Peak of Resurgent Na+ Currents in the p.T1313M and p.R1448H Mutant Channels than in the WT Nav1.4 Channel
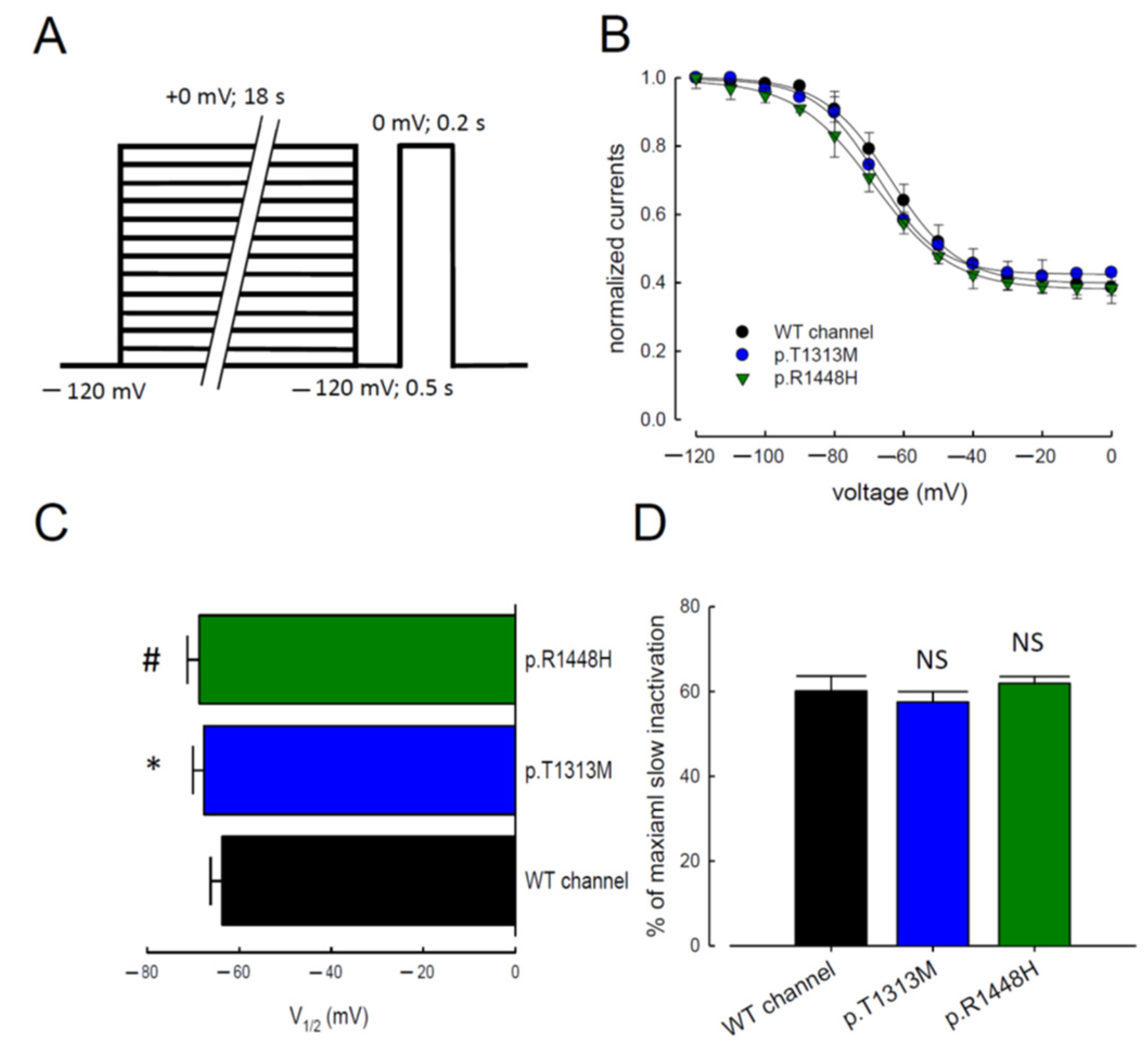
3.5. Slow Inactivation in WT Nav1.4, p.T1313M, and p.R1448H Mutant Channels
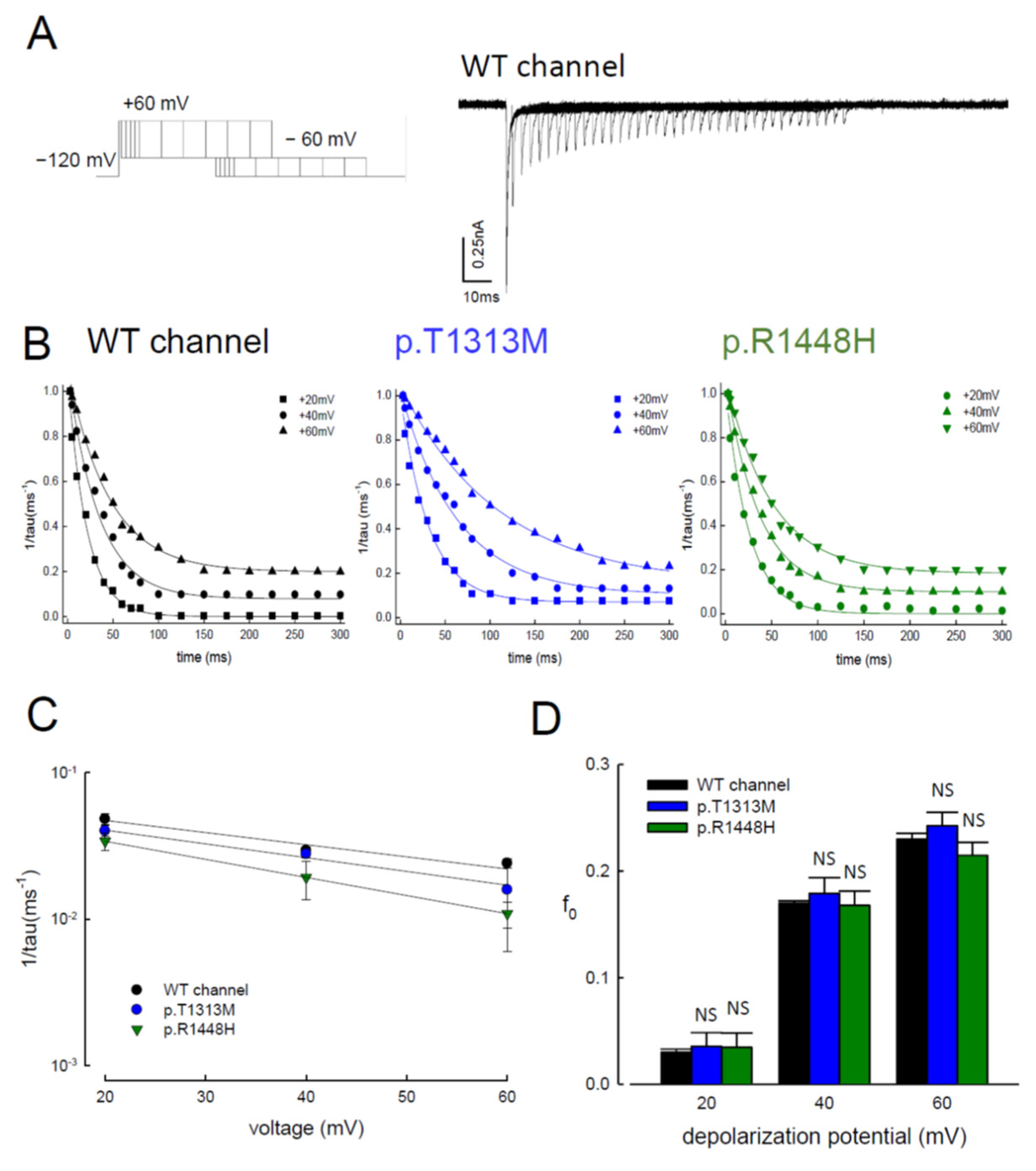
3.6. Reduced Magnitudes of the Resurgent Currents by Prolonging of the Depolarization Prepulses
3.7. Topological Changes in the p.T1313M and p.R1448H Mutant Channels Were Shown by Homology Modeling
4. Discussion
4.1. Sodium Channels Are Associated with Excitation-Contraction Coupling in the Skeletal Muscle
4.2. Molecular and Biophysical Basis of Resurgent Na+ Currents
4.3. Factors Contributed to the Hyperexcitability of Affected Muscles
4.4. Alteration in Fast Inactivation and Increase of Persistent Sodium Current in p.T1313 and p.R1448 Mutant Channels Causing for Cellular Hyperexcitability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mazón, M.J.; Barros, F.; De La Peña, P.; Quesada, J.F.; Escudero, A.; Cobo, A.M.; Pascual-Pascual, S.I.; Gutiérrez-Rivas, E.; Guillén, E.; Arpa, J.; et al. Screening for mutations in Spanish families with myotonia. Functional analysis of novel mutations in CLCN1 gene. Neuromuscul. Disord. 2012, 22, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, F.; Rüdel, R. Hereditary nondystrophic myotonias and periodic paralyses. Curr. Opin. Neurol. 1995, 8, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Lossin, C.; George, A.L., Jr. Myotonia congenita. Adv. Genet. 2008, 63, 25–55. [Google Scholar] [PubMed]
- Kol, S.; Turrell, B.R.; De Keyzer, J.; Van der Laan, M.; Nouwen, N.; Driessen, A.J. Yidc-mediated membrane insertion of assembly mutants of subunit c of the f1f0 atpase. J. Biol. Chem. 2006, 281, 29762–29768. [Google Scholar] [CrossRef] [Green Version]
- David, M.; Martinez-Marmol, R.; Gonzalez, T.; Felipe, A.; Valenzuela, C. Differential regulation of na(v)beta subunits during myogenesis. Biochem. Biophys. Res. Commun. 2008, 368, 761–766. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Kuo, C.-C. The Position of the Fourth Segment of Domain 4 Determines Status of the Inactivation Gate in Na+ Channels. J. Neurosci. 2003, 23, 4922–4930. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Zhang, W.; Chang, X.; Guo, J. Overlap of periodic paralysis and paramyotonia congenita caused by SCN4A gene mutations two family reports and literature review. Channels 2019, 13, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Holzherr, B.; Lehmann-Horn, F.; Kuzmenkina, E.; Fan, C.; Jurkat-Rott, K. A gating model for wildtype and R1448H Nav1.4 channels in paramyotonia. Acta Myol. 2014, 33, 22–33. [Google Scholar]
- Hsu, W.-C.; Huang, Y.-C.; Wang, C.-W.; Hsueh, C.-H.; Lai, L.-P.; Yeh, J.-H. Paralysis Periodica Paramyotonica Caused by SCN4A Arg1448Cys Mutation. J. Formos. Med. Assoc. 2006, 105, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Ke, Q.; Ye, J.; Tang, S.; Wang, J.; Luo, B.; Jin, W.; Zhang, X.; Yuezhou, L.; Cheng, X.; Li, Y. N1366S mutation of human skeletal muscle sodium channel causes paramyotonia congenita. J. Physiol. 2017, 595, 6837–6850. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; George, A.L.; Kyle, J.W.; Makielski, J.C. Two human paramyotonia congenita mutations have opposite effects on lidocaine block of Na+ channels expressed in a mammalian cell line. J. Physiol. 1996, 496, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouhours, M.; Sternberg, D.; Davoine, C.-S.; Xavier, F.; Willer, J.C.; Fontaine, B.; Tabti, N. Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans. J. Physiol. 2004, 554, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Dice, M.S.; Abbruzzese, J.L.; Wheeler, J.T.; Groome, J.R.; Fujimoto, E.; Ruben, P.C. Temperature-sensitive defects in paramyotonia congenita mutants R1448C and T1313M. Muscle Nerve 2004, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Richmond, J.E.; Featherstone, D.E.; Ruben, P.C. Human Na+ channel fast and slow inactivation in paramyotonia congenita mutants expressed in Xenopus laevis oocytes. J. Physiol. 1997, 499, 589–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Ji, S.; Zhou, M.; Ptacek, L.J.; Barchi, R.L.; Horn, R.; George, A.L. Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc. Natl. Acad. Sci. USA 1994, 91, 12785–12789. [Google Scholar] [CrossRef] [Green Version]
- Trimmer, J.S. Analysis of K+ channel biosynthesis and assembly in transfected mammalian cells. Methods Enzymol. 1998, 293, 32–49. [Google Scholar] [CrossRef]
- George, A.L.; Komisarof, J.; Kallen, R.G.; Barchi, R.L. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Ann. Neurol. 1992, 31, 131–137. [Google Scholar] [CrossRef]
- Huang, C.-W.; Lai, H.-J.; Huang, P.-Y.; Lee, M.-J.; Kuo, C.-C. The Biophysical Basis Underlying Gating Changes in the p.V1316A Mutant Nav1.7 Channel and the Molecular Pathogenesis of Inherited Erythromelalgia. PLoS Biol. 2016, 14, e1002561. [Google Scholar] [CrossRef]
- Huang, C.-W.; Lai, H.-J.; Huang, P.-Y.; Lee, M.-J.; Kuo, C.-C. Anomalous enhancement of resurgent Na+ currents at high temperatures by SCN9A mutations underlies the episodic heat-enhanced pain in inherited erythromelalgia. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-W.; Lai, H.-J.; Lin, P.-C.; Lee, M.-J. Changes of Resurgent Na+ Currents in the Nav1.4 Channel Resulting from an SCN4A Mutation Contributing to Sodium Channel Myotonia. Int. J. Mol. Sci. 2020, 21, 2593. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1. Science 2018, 362, eaau2486. [Google Scholar] [CrossRef] [PubMed]
- Farinato, A.; Altamura, C.; Imbrici, P.; Maggi, L.; Bernasconi, P.; Mantegazza, R.; Pasquali, L.; Siciliano, G.; Monaco, M.L.; Vial, C.; et al. Pharmacogenetics of myotonic hNav1.4 sodium channel variants situated near the fast inactivation gate. Pharmacol. Res. 2019, 141, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.; Sigworth, F. Impaired slow inactivation in mutant sodium channels. Biophys. J. 1996, 71, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Green, D.S.; Hayward, L.J.; George, A.L., Jr.; Cannon, S.C. A proposed mutation, val781ile, associated with hyperkalemic periodic paralysis and cardiac dysrhythmia is a benign polymorphism. Ann. Neurol. 1997, 42, 253–256. [Google Scholar] [CrossRef] [PubMed]
- DiFranco, M.; Vergara, J.L. The Na conductance in the sarcolemma and the transverse tubular system membranes of mammalian skeletal muscle fibers. J. Gen. Physiol. 2011, 138, 393–419. [Google Scholar] [CrossRef] [Green Version]
- Jaimovich, E.; Venosa, R.A.; Shrager, P.; Horowicz, P. Density and distribution of tetrodotoxin receptors in normal and detubulated frog sartorius muscle. J. Gen. Physiol. 1976, 67, 399–416. [Google Scholar] [CrossRef]
- Caldwell, J.H.; Campbell, D.T.; Beam, K.G. Na channel distribution in vertebrate skeletal muscle. J. Gen. Physiol. 1986, 87, 907–932. [Google Scholar] [CrossRef]
- Jong, D.S.; Stroffekova, K.; Heiny, J.A. A surface potential change in the membranes of frog skeletal muscle is associated with excitation-contraction coupling. J. Physiol. 1997, 499, 787–808. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Anderson, K.; Shirokov, R.; Levis, R.; Gonzalez, A.; Karhanek, M.; Hosey, M.M.; Meissner, G.; Ríos, E. Effects of perchlorate on the molecules of excitation-contraction coupling of skeletal and cardiac muscle. J. Gen. Physiol. 1993, 102, 423–448. [Google Scholar] [CrossRef] [Green Version]
- Cannon, S.C. Sodium channelopathies of skeletal muscle. Handb. Exp. Pharmacol. 2018, 246, 309–330. [Google Scholar]
- Lossin, C.; Nam, T.-S.; Shahangian, S.; Rogawski, M.A.; Choi, S.-Y.; Kim, M.-K.; Sunwoo, I.-N. Altered fast and slow inactivation of the N440K Nav1.4 mutant in a periodic paralysis syndrome. Neurology 2012, 79, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kinoshita, M.; Sasaki, R.; Aoike, F.; Takahashi, M.P.; Sakoda, S.; Hirose, K. New mutation of the Na channel in the severe form of potassium-aggravated myotonia. Muscle Nerve 2009, 39, 666–673. [Google Scholar] [CrossRef] [PubMed]
- El-Bizri, N.; Kahlig, K.M.; Shyrock, J.C.; George, J.A.L.; Belardinelli, L.; Rajamani, S. Ranolazine block of human Na v 1.4 sodium channels and paramyotonia congenita mutants. Channels 2011, 5, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferriby, D.; Stojkovic, T.; Sternberg, D.; Hurtevent, J.-F.; Hurtevent, J.-P.; Vermersch, P. A new case of autosomal dominant myotonia associated with the V1589M missense mutation in the muscle sodium channel gene and its phenotypic classification. Neuromuscul. Disord. 2006, 16, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Kim, E.J.; Jung, D.S.; Park, K.H.; Kim, I.J.; Kwak, K.Y.; Kim, C.M.; Ko, H.Y. A Korean Family with Arg1448Cys Mutation of SCN4A Channel Causing Paramyotonia Congenita: Electrophysiologic, Histopathologic, and Molecular Genetic Studies. J. Korean Med. Sci. 2002, 17, 856–860. [Google Scholar] [CrossRef] [Green Version]
- Nurputra, D.K.; Nakagawa, T.; Takeshima, Y.; Harahap, I.S.; Morikawa, S.; Sakaeda, T.; Lai, P.S.; Matsuo, M.; Takaoka, Y.; Nishio, H. Paramyotonia congenita: From clinical diagnosis to in silico protein modeling analysis. Pediatr. Int. 2012, 54, 602–612. [Google Scholar] [CrossRef]
- Yoshinaga, H.; Sakoda, S.; Good, J.-M.; Takahashi, M.P.; Kubota, T.; Arikawa-Hirasawa, E.; Nakata, T.; Ohno, K.; Kitamura, T.; Kobayashi, K.; et al. A novel mutation in SCN4A causes severe myotonia and school-age-onset paralytic episodes. J. Neurol. Sci. 2012, 315, 15–19. [Google Scholar] [CrossRef]
- Mohammadi, B.; Mitrovic, N.; Lehmann-Horn, F.; Dengler, R.; Bufler, J. Mechanisms of cold sensitivity of paramyotonia congenita mutation r1448h and overlap syndrome mutation m1360v. J. Physiol. 2003, 547, 691–698. [Google Scholar] [CrossRef]
- Lewis, A.H.; Raman, I.M. Resurgent current of voltage-gated na(+) channels. J. Physiol. 2014, 592, 4825–4838. [Google Scholar] [CrossRef]
- Raman, I.M.; Bean, B.P. Resurgent Sodium Current and Action Potential Formation in Dissociated Cerebellar Purkinje Neurons. J. Neurosci. 1997, 17, 4517–4526. [Google Scholar] [CrossRef] [Green Version]
- Raman, I.M.; Bean, B.P. Inactivation and Recovery of Sodium Currents in Cerebellar Purkinje Neurons: Evidence for Two Mechanisms. Biophys. J. 2001, 80, 729–737. [Google Scholar] [CrossRef] [Green Version]
- Raman, I.M.; Sprunger, L.K.; Meisler, M.H.; Bean, B.P. Altered Subthreshold Sodium Currents and Disrupted Firing Patterns in Purkinje Neurons of Scn8a Mutant Mice. Neuron 1997, 19, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Groome, J.R.; Lehmann-Horn, F.; Fan, C.; Wolf, M.; Winston, V.; Merlini, L.; Jurkat-Rott, K. NaV1.4 mutations cause hypokalaemic periodic paralysis by disrupting IIIS4 movement during recovery. Brain 2014, 137, 998–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, M.T.H.; Bean, B.P. Sodium Currents in Subthalamic Nucleus Neurons From Nav1.6-Null Mice. J. Neurophysiol. 2004, 92, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Do, M.T.H.; Bean, B.P. Subthreshold sodium currents and pacemaking of subthalamic neurons: Modulation by slow inactivation. Neuron 2003, 39, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Afshari, F.S.; Ptak, K.; Khaliq, Z.M.; Grieco, T.M.; Slater, N.T.; McCrimmon, D.R.; Raman, I.M. Resurgent Na Currents in Four Classes of Neurons of the Cerebellum. J. Neurophysiol. 2004, 92, 2831–2843. [Google Scholar] [CrossRef] [Green Version]
- Gittis, A.H.; Du Lac, S. Similar properties of transient, persistent, and resurgent na currents in gabaergic and non-gabaergic vestibular nucleus neurons. J. Neurophysiol. 2008, 99, 2060–2065. [Google Scholar] [CrossRef] [Green Version]
- Jarecki, B.W.; Piekarz, A.D.; Jackson, J.O.; Cummins, T.R. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J. Clin. Investig. 2010, 120, 369–378. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Bezanilla, F. Inactivation of the sodium channel. Ii. Gating current experiments. J. Gen. Physiol. 1977, 70, 567–590. [Google Scholar] [CrossRef]
- Kuo, C.-C.; Bean, B.P. Na+ channels must deactivate to recover from inactivation. Neuron 1994, 12, 819–829. [Google Scholar] [CrossRef]
- Grieco, T.M.; Malhotra, J.D.; Chen, C.; Isom, L.L.; Raman, I.M. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 2005, 45, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.P.; Cannon, S.C. Enhanced Slow Inactivation by V445M: A Sodium Channel Mutation Associated with Myotonia. Biophys. J. 1999, 76, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, G. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys. J. 1997, 72, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Männikkö, R.; Wong, L.; Tester, D.J.; Thor, M.G.; Sud, R.; Kullmann, D.M.; Sweeney, M.G.; Leu, C.; Sisodiya, S.M.; Fitzpatrick, D.R.; et al. Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: A case-control study. Lancet 2018, 391, 1483–1492. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT Channel | p.T1313M | p.R1448H | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Activation | Inactivation | Activation | Inactivation | Activation | Inactivation | ||||||
| Vh (mV) | k | Vh (mV) | k | Vh (mV) | k | Vh (mV) | k | Vh (mV) | k | Vh (mV) | k |
| −24.7 ± 1.5 | 6.9 ± 0.3 | −61.9 ± 0.9 | −8.9 ± 0.3 | −61.9 ± 0.9 | −8.9 ± 0.3 | −68.6 ± 0.3 | −12.8 ± 0.2 | −30.0 ± 1.5 | 6.4 ± 0.5 | −64.78 ± 0.5 | −11.6 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-W.; Lai, H.-J.; Lin, P.-C.; Lee, M.-J. Changes in Resurgent Sodium Current Contribute to the Hyperexcitability of Muscles in Patients with Paramyotonia Congenita. Biomedicines 2021, 9, 51. https://doi.org/10.3390/biomedicines9010051
Huang C-W, Lai H-J, Lin P-C, Lee M-J. Changes in Resurgent Sodium Current Contribute to the Hyperexcitability of Muscles in Patients with Paramyotonia Congenita. Biomedicines. 2021; 9(1):51. https://doi.org/10.3390/biomedicines9010051
Chicago/Turabian StyleHuang, Chiung-Wei, Hsing-Jung Lai, Pi-Chen Lin, and Ming-Jen Lee. 2021. "Changes in Resurgent Sodium Current Contribute to the Hyperexcitability of Muscles in Patients with Paramyotonia Congenita" Biomedicines 9, no. 1: 51. https://doi.org/10.3390/biomedicines9010051
APA StyleHuang, C. -W., Lai, H. -J., Lin, P. -C., & Lee, M. -J. (2021). Changes in Resurgent Sodium Current Contribute to the Hyperexcitability of Muscles in Patients with Paramyotonia Congenita. Biomedicines, 9(1), 51. https://doi.org/10.3390/biomedicines9010051