Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future?

 , , ,
, , ,
Abstract
:1. Introduction
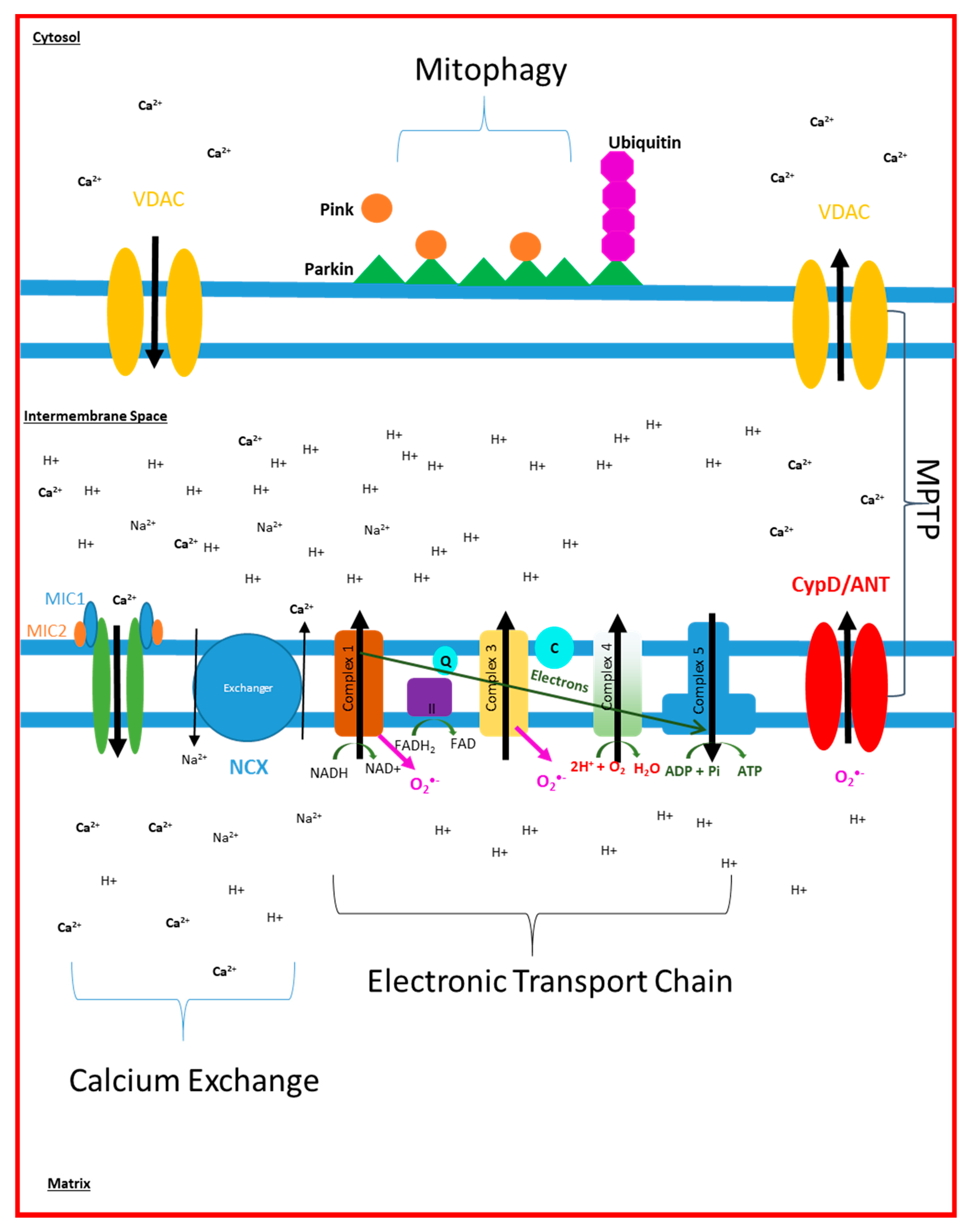
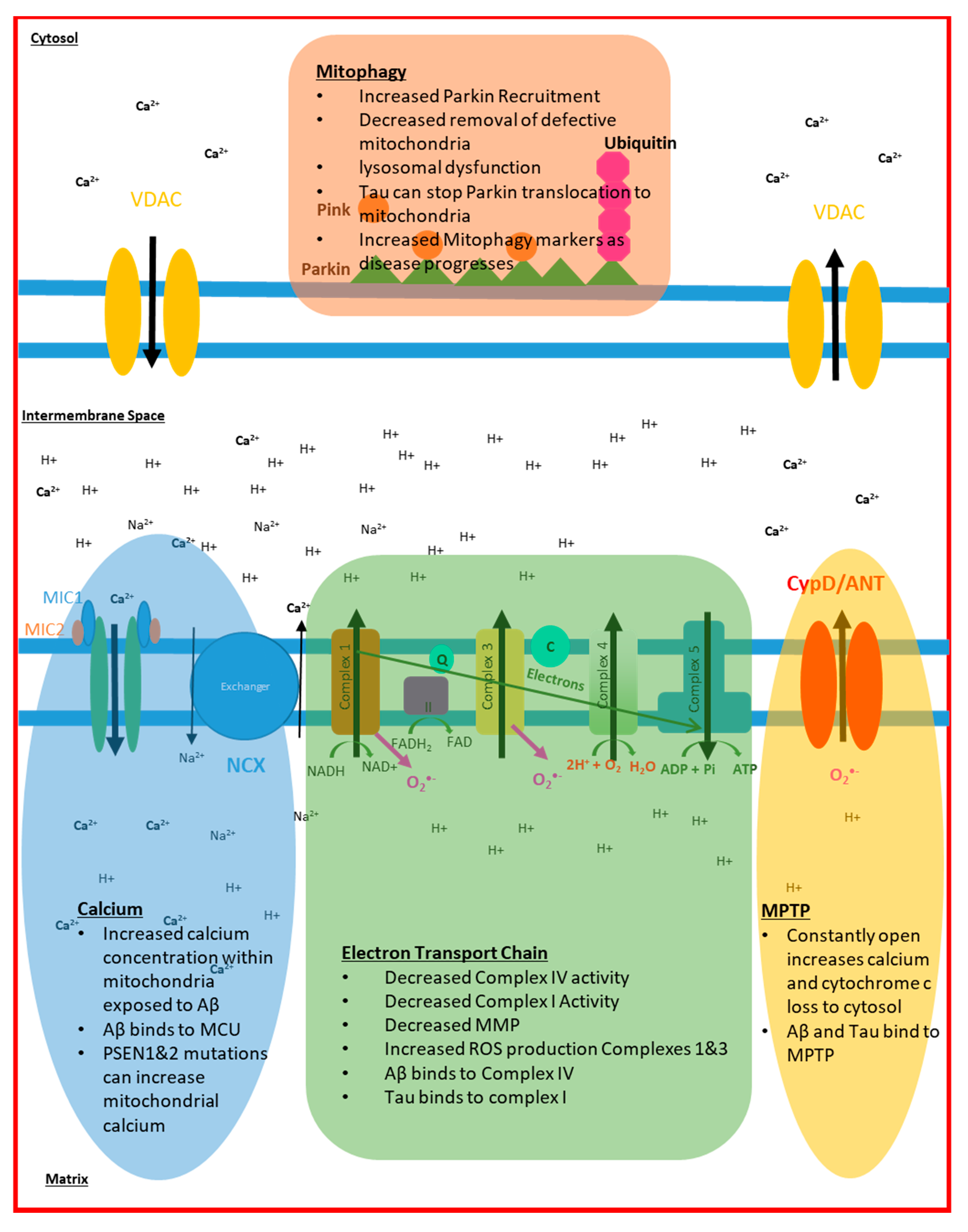
2. Electron Transport Chain Disruption in AD
3. Mitochondrial Dynamic Changes Seen in AD
4. Mitochondrial Calcium Signalling in AD
5. Mitochondrial ROS Production in AD
6. Mitophagy and Cell Death in AD
7. Mitochondrial Abnormalities in AD Summary
8. Mitochondrial Dysfunction and Its Current Clinical Imaging Applications
9. Mitochondrial Dysfunction: A Future Biomarker of AD?
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer‘s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer‘s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.G.; Mirra, S.S.; Pollock, N.J.; Binder, L.I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. USA 1986, 83, 4040–4043. [Google Scholar] [CrossRef] [Green Version]
- Binder, L.I.; Guillozet-Bongaarts, A.L.; Garcia-Sierra, F.; Berry, R.W. Tau, tangles, and Alzheimer‘s disease. Biochim. Biophys. Acta 2005, 1739, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Morgen, K.; Frolich, L. The metabolism hypothesis of Alzheimer‘s disease: From the concept of central insulin resistance and associated consequences to insulin therapy. J. Neural Transm. 2015, 122, 499–504. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A "mitochondrial cascade hypothesis" for sporadic Alzheimer’s disease. Med. Hypoth. 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer‘s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Hirai, K.; Aliev, G.; Takeda, A.; Chiba, S.; Smith, M.A. Neuronal RNA oxidation in Alzheimer’s disease and Down’s syndrome. Ann. N. Y. Acad. Sci. 1999, 893, 362–364. [Google Scholar] [CrossRef]
- Simpson, J.E.; Ince, P.G.; Haynes, L.J.; Theaker, R.; Gelsthorpe, C.; Baxter, L.; Forster, G.; Lace, G.L.; Shaw, P.J.; Matthews, F.E.; et al. Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol. Appl. Neurobiol. 2010, 36, 25–40. [Google Scholar] [CrossRef]
- Tayler, H.; Fraser, T.; Miners, J.S.; Kehoe, P.G.; Love, S. Oxidative balance in Alzheimer’s disease: Relationship to APOE, Braak tangle stage, and the concentrations of soluble and insoluble amyloid-β. J. Alzheimer’s Dis. 2010, 22, 1363–1373. [Google Scholar] [CrossRef]
- Wang, J.; Xiong, S.; Xie, C.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 2005, 93, 953–962. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.L. Steroid hormone synthesis in mitochondria. Mol. Cell Endocrinol. 2013, 379, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L. Role of mitochondria in steroidogenesis. Endocr. Dev. 2011, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect Biol. 2011, 3, a007559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [Green Version]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Borland, M.K. Differentiated Alzheimer’s disease transmitochondrial cybrid cell lines exhibit reduced organelle movement. Antioxid Redox. Signal. 2005, 7, 1101–1109. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [Green Version]
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimers. Dement. 2017, 13, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, K.; Giordano, T.; Brady, D.R.; Stoll, J.; Martin, L.J.; Rapoport, S.I. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res. 1994, 24, 336–340. [Google Scholar] [CrossRef]
- Hatanpää, K.; Chandrasekaran, K.; Brady, D.R.; Rapoport, S.I. No association between Alzheimer plaques and decreased levels of cytochrome oxidase subunit mRNA, a marker of neuronal energy metabolism. Mol. Brain Res. 1998, 59, 13–21. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, D.A.; Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Turnbull, D.M. The role of cytochrome c oxidase deficient hippocampal neurones in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2002, 28, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Simonian, N.A.; Hyman, B.T. Functional Alterations in Alzheimer’s Disease: Selective Loss of Mitochondrial-encoded Cytochrome Oxidase mRNA in the Hippocampal Formation. J. Neuropathol. Exp. Neurol. 1994, 53, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromol. Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Fukuyama, R.; Hatanpää, K.; Rapoport, S.I.; Chandrasekaran, K. Gene expression of ND4, a subunit of complex I of oxidative phosphorylation in mitochondria, is decreased in temporal cortex of brains of Alzheimer’s disease patients. Brain Res. 1996, 713, 290–293. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Luong, T.; Neely, T.R.; Robinson, D.; Wands, J.R. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab. Investig. 2000, 80, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.H.; Lin, R.; Wisniewski, H.M.; Hwang, Y.W.; Grundke-Iqbal, I.; Healy-Louie, G.; Iqbal, K. Detection of point mutations in codon 331 of mitochondrial NADH dehydrogenase subunit 2 in Alzheimer’s brains. Biochem. Biophys. Res. Commun. 1992, 182, 238–246. [Google Scholar] [CrossRef]
- Linnane, A.; Ozawa, T.; Marzuki, S.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 333, 642–645. [Google Scholar] [CrossRef]
- Griffiths, K.K.; Levy, R.J. Evidence of Mitochondrial Dysfunction in Autism: Biochemical Links, Genetic-Based Associations, and Non-Energy-Related Mechanisms. Oxid. Med. Cell. Long. 2017, 2017, 4314025. [Google Scholar] [CrossRef]
- Chagnon, P.; Bétard, C.; Robitaille, Y.; Cholette, A.; Gauvreau, D. Distribution of brain cytochrome oxidase activity in various neurodegenerative diseases. Neuroreport 1995, 6, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.; Filley, C.M.; Kleinschmidt-DeMasters, B.K. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090–1096. [Google Scholar] [CrossRef]
- Parker, W.D.; Parks, J.K. Cytochrome C Oxidase in Alzheimer’s Disease Brain. Purif. Character. 1995, 45, 482–486. [Google Scholar] [CrossRef]
- Wong-Riley, M.; Antuono, P.; Ho, K.-C.; Egan, R.; Hevner, R.; Liebl, W.; Huang, Z.; Rachel, R.; Jones, J. Cytochrome oxidase in Alzheimer’s disease: Biochemical, histochemical, and immunohistochemical analyses of the visual and other systems. Vis. Res. 1997, 37, 3593–3608. [Google Scholar] [CrossRef] [Green Version]
- Hevner, R.F.; Wong-Riley, M.T. Mitochondrial and nuclear gene expression for cytochrome oxidase subunits are disproportionately regulated by functional activity in neurons. J. Neurosci. 1993, 13, 1805–1819. [Google Scholar] [CrossRef] [Green Version]
- Wong-Riley, M.T. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989, 12, 94–101. [Google Scholar] [CrossRef]
- Wong-Riley, M.T.; Walsh, S.M.; Leake-Jones, P.A.; Merzenich, M.M. Maintenance of neuronal activity by electrical stimulation of unilaterally deafened cats demonstrable with cytochrome oxidase technique. Ann. Otol. Rhinol. Laryngol. 1981, 90, 30–32. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Hatanpää, K.; Brady, D.R.; Rapoport, S.I. Evidence for Physiological Down-regulation of Brain Oxidative Phosphorylation in Alzheimer’s Disease. Exp. Neurol. 1996, 142, 80–88. [Google Scholar] [CrossRef]
- Maynard, S.; Hejl, A.M.; Dinh, T.S.T.; Keijzers, G.; Hansen, A.M.; Desler, C.; Moreno-Villanueva, M.; Burkle, A.; Rasmussen, L.J.; Waldemar, G.; et al. Defective mitochondrial respiration, altered dNTP pools and reduced AP endonuclease 1 activity in peripheral blood mononuclear cells of Alzheimer’s disease patients. Aging 2015, 7, 793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuner, K.; Schulz, K.; Schütt, T.; Pantel, J.; Prvulovic, D.; Rhein, V.; Savaskan, E.; Czech, C.; Eckert, A.; Müller, W.E. Peripheral mitochondrial dysfunction in Alzheimer’s disease: Focus on lymphocytes. Mol. Neurobiol. 2012, 46, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Coskun, P.; Helguera, P.; Nemati, Z.; Bohannan, R.C.; Thomas, J.; Schriner, S.E.; Argueta, J.; Doran, E.; Wallace, D.C.; Lott, I.T.; et al. Metabolic and Growth Rate Alterations in Lymphoblastic Cell Lines Discriminate Between Down Syndrome and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Keohane, A.; Pidsley, R.; Newhouse, S.; Riddoch-Contreras, J.; Thubron, E.B.; Devall, M.; Soininen, H.; Koszewska, I.; Mecocci, P.; et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol. Aging 2017, 53, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Fisar, Z.; Hroudova, J.; Hansikova, H.; Spacilova, J.; Lelkova, P.; Wenchich, L.; Jirak, R.; Zverova, M.; Zeman, J.; Martasek, P.; et al. Mitochondrial Respiration in the Platelets of Patients with Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 930–941. [Google Scholar] [CrossRef]
- Talib, L.L.; Joaquim, H.P.; Forlenza, O.V. Platelet biomarkers in Alzheimer’s disease. World J. Psychiatr. 2012, 2, 95–101. [Google Scholar] [CrossRef]
- Casoli, T.; Di Stefano, G.; Giorgetti, B.; Grossi, Y.; Balietti, M.; Fattoretti, P.; Bertoni-Freddari, C. Release of beta-amyloid from high-density platelets: Implications for Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2007, 1096, 170–178. [Google Scholar] [CrossRef]
- Smith, C.C. Stimulated release of the beta-amyloid protein of Alzheimer’s disease by normal human platelets. Neurosci. Lett. 1997, 235, 157–159. [Google Scholar] [CrossRef]
- Prestia, F.A.; Galeano, P.; Martino Adami, P.V.; Do Carmo, S.; Castaño, E.M.; Cuello, A.C.; Morelli, L. Platelets Bioenergetics Screening Reflects the Impact of Brain Aβ Plaque Accumulation in a Rat Model of Alzheimer. Neurochem. Res. 2019, 44, 1375–1386. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 1990, 40, 1302–1303. [Google Scholar] [CrossRef]
- Valla, J.; Schneider, L.; Niedzielko, T.; Coon, K.D.; Caselli, R.; Sabbagh, M.N.; Ahern, G.L.; Baxter, L.; Alexander, G.; Walker, D.G.; et al. Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion 2006, 6, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citron, M.; Vigo-Pelfrey, C.; Teplow, D.B.; Miller, C.; Schenk, D.; Johnston, J.; Winblad, B.; Venizelos, N.; Lannfelt, L.; Selkoe, D.J. Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc. Natl Acad. Sci. USA 1994, 91, 11993–11997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, N.E.; Quinn, J.F. Alterations in mitochondrial number and function in Alzheimer’s disease fibroblasts. Metab. Brain Dis. 2015, 30, 1275–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, M.J.; Ponce, D.P.; Osorio-Fuentealba, C.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial Bioenergetics Is Altered in Fibroblasts from Patients with Sporadic Alzheimer’s Disease. Front. Neurosci. 2017, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Sorbi, S.; Piacentini, S.; Latorraca, S.; Piersanti, P.; Amaducci, L. Alterations in metabolic properties in fibroblasts in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1995, 9, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Barnes, K.; Clemmens, H.; Al-Rafiah, A.R.; Al-Ofi, E.A.; Leech, V.; Bandmann, O.; Shaw, P.J.; Blackburn, D.J.; Ferraiuolo, L.; et al. Ursodeoxycholic Acid Improves Mitochondrial Function and Redistributes Drp1 in Fibroblasts from Patients with Either Sporadic or Familial Alzheimer’s Disease. J. Mol. Biol. 2018, 430, 3942–3953. [Google Scholar] [CrossRef] [PubMed]
- Curti, D.; Rognoni, F.; Gasparini, L.; Cattaneo, A.; Paolillo, M.; Racchi, M.; Zani, L.; Bianchetti, A.; Trabucchi, M.; Bergamaschi, S.; et al. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer’s disease (AD) patients. Neurosci. Lett. 1997, 236, 13–16. [Google Scholar] [CrossRef]
- Sonntag, K.-C.; Ryu, W.-I.; Amirault, K.M.; Healy, R.A.; Siegel, A.J.; McPhie, D.L.; Forester, B.; Cohen, B.M. Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 2017, 7, 14038. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.M.; De Marco, M.; Barnes, K.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Mortiboys, H.; Venneri, A. Deficits in Mitochondrial Spare Respiratory Capacity Contribute to the Neuropsychological Changes of Alzheimer’s Disease. J. Pers. Med. 2020, 10, 32. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S265–S279. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.Q.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.L.; Shenoy, D.V.; Thomas, N.; Choudhary, P.K.; LaFerla, F.M.; Goodman, S.R.; Breen, G.A.M. Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J. Proteom. 2011, 74, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Schuldt, V.; Forler, S.; Zabel, C.; Klose, J.; Rohe, M. Presymptomatic Alterations in Energy Metabolism and Oxidative Stress in the APP23 Mouse Model of Alzheimer Disease. J. Proteome Res. 2012, 11, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Behbahani, H.; Shabalina, I.G.; Wiehager, B.; Concha, H.; Hultenby, K.; Petrovic, N.; Nedergaard, J.; Winblad, B.; Cowburn, R.F.; Ankarcrona, M. Differential role of Presenilin-1 and -2 on mitochondrial membrane potential and oxygen consumption in mouse embryonic fibroblasts. J. Neurosci. Res. 2006, 84, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Contino, S.; Porporato, P.E.; Bird, M.; Marinangeli, C.; Opsomer, R.; Sonveaux, P.; Bontemps, F.; Dewachter, I.; Octave, J.N.; Bertrand, L.; et al. Presenilin 2-Dependent Maintenance of Mitochondria! Oxidative Capacity and Morphology. Front. Physiol. 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Escobar-Khondiker, M.; Höllerhage, M.; Muriel, M.P.; Champy, P.; Bach, A.; Depienne, C.; Respondek, G.; Yamada, E.S.; Lannuzel, A.; Yagi, T.; et al. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J. Neurosci. 2007, 27, 7827–7837. [Google Scholar] [CrossRef]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Rhein, V.; Müller-Spahn, F.; Götz, J.; Müller, W.E. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neuro Degenerat. Dis. 2008, 5, 157–159. [Google Scholar] [CrossRef] [Green Version]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial dysfunction—The beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimers Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef]
- Sanchez, M.; Waugh, H.S.; Tsatsanis, A.; Wong, B.X.; Crowston, J.G.; Duce, J.A.; Trounce, I.A. Amyloid precursor protein drives down-regulation of mitochondrial oxidative phosphorylation independent of amyloid beta. Sci. Rep. 2017, 7, 10. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Santos, S.; Swerdlow, R.H.; Oliveira, C.R. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001, 15, 1439. [Google Scholar] [CrossRef] [PubMed]
- Fleck, D.; Phu, L.; Verschueren, E.; Hinkle, T.; Reichelt, M.; Bhangale, T.; Haley, B.; Wang, Y.; Graham, R.; Kirkpatrick, D.S.; et al. PTCD1 Is Required for Mitochondrial Oxidative-Phosphorylation: Possible Genetic Association with Alzheimer’s Disease. J. Neurosci. 2019, 39, 4636–4656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, X.Q.; Huang, S.B.; Wu, L.; Wang, Y.F.; Hu, G.; Li, G.Y.; Zhang, H.J.; Yu, H.Y.; Swerdlow, R.H.; Chen, J.X.; et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harbor Perspect. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Shen, Y.; Wang, X.; Wei, L.F.; Wang, P.; Yang, H.; Wang, C.F.; Xie, Z.H.; Bi, J.Z. Mitochondrial dynamics changes with age in an APPsw/PS1dE9 mouse model of Alzheimer’s disease. NeuroReport 2017. [Google Scholar] [CrossRef] [Green Version]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Djordjevic, J.; Chowdhury, S.R.; Snow, W.M.; Perez, C.; Cadonic, C.; Fernyhough, P.; Albensi, B.C. Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice. Cells 2020, 9, 1541. [Google Scholar] [CrossRef]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018. [Google Scholar] [CrossRef]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmukler, E.; Solomon, S.; Simonovitch, S.; Goldshmit, Y.; Wolfson, E.; Morris Michaelson, D.; Pinkas-Kramarski, R. Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis. 2020, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Paidi, R.K.; Nthenge-Ngumbau, D.N.; Singh, R.; Kankanala, T.; Mehta, H.; Mohanakumar, K.P. Mitochondrial Deficits Accompany Cognitive Decline Following Single Bilateral Intracerebroventricular Streptozotocin. Curr. Alzheimer Res. 2015, 12, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Lee, H.g.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Wang, S.; Song, J.; Tan, M.; Albers, K.M.; Jia, J. Mitochondrial fission proteins in peripheral blood lymphocytes are potential biomarkers for Alzheimer’s disease. Eur. J. Neurol. 2012. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.-H.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Bossy, B.; Petrilli, A.; Klinglmayr, E.; Chen, J.; Lütz-Meindl, U.; Knott, A.B.; Masliah, E.; Schwarzenbacher, R.; Bossy-Wetzel, E. S-nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20. [Google Scholar] [CrossRef] [Green Version]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Günther, C.; von Hadeln, K.; Müller-Thomsen, T.; Alberici, A.; Binetti, G.; Hock, C.; Nitsch, R.M.; Stoppe, G.; Reiss, J.; Gal, A.; et al. Possible association of mitochondrial transcription factor A (TFAM) genotype with sporadic Alzheimer disease. Neurosci. Lett. 2004, 369, 219–223. [Google Scholar] [CrossRef]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.X.; Dai, S.X.; Guo, Y.C.; Han, F.F.; Zheng, J.J.; Li, G.H.; Huang, J.F. Meta-Analysis of Parkinson’s Disease and Alzheimer’s Disease Revealed Commonly Impaired Pathways and Dysregulation of NRF2-Dependent Genes. J. Alzheimer’s Dis. 2017, 56, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Ylä-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Ylä-Herttuala, S.; Tanila, H.; Levonen, A.-L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Kandimalla, R.; Kuruva, C.S. Protective effects of a natural product, curcumin, against amyloid β induced mitochondrial and synaptic toxicities in Alzheimer’s disease. J. Investig. Med. 2016, 64, 1220–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Fink, A.; Doblhammer, G. Effect of pioglitazone medication on the incidence of dementia. Ann. Neurol. 2015, 78, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatr. 2005, 13, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.F.; Song, C.Y.; Wang, X.; Huang, L.Y.; Ding, M.; Yang, H.; Wang, P.; Xu, L.L.; Xie, Z.H.; Bi, J.Z. Protective effects of melatonin on mitochondrial biogenesis and mitochondrial structure and function in the HEK293-APPswe cell model of Alzheimer’s disease. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3542–3550. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Guo, Q.; Sopher, B.L.; Furukawa, K.; Pham, D.G.; Robinson, N.; Martin, G.M.; Mattson, M.P. Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: Involvement of calcium and oxyradicals. J. Neurosci. 1997, 17, 4212–4222. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Christakos, S.; Robinson, N.; Mattson, M.P. Calbindin D28k blocks the proapoptotic actions of mutant presenilin 1: Reduced oxidative stress and preserved mitochondrial function. Proc. Natl. Acad. Sci. USA 1998, 95, 3227–3232. [Google Scholar] [CrossRef] [Green Version]
- Sarasija, S.; Laboy, J.T.; Ashkavand, Z.; Bonner, J.; Tang, Y.; Norman, K.R. Presenilin mutations deregulate mitochondrial Ca(2+) homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. Elife 2018, 7, e33052. [Google Scholar] [CrossRef] [PubMed]
- Begley, J.G.; Duan, W.; Chan, S.; Duff, K.; Mattson, M.P. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J. Neurochem. 1999, 72, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)–mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepulveda-Falla, D.; Barrera-Ocampo, A.; Hagel, C.; Korwitz, A.; Vinueza-Veloz, M.F.; Zhou, K.; Schonewille, M.; Zhou, H.; Velazquez-Perez, L.; Rodriguez-Labrada, R.; et al. Familial Alzheimer’s disease–associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Investig. 2014, 124, 1552–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, D.H.W.; Paillusson, S.; Hartopp, N.; Rupawala, H.; Mórotz, G.M.; Gomez-Suaga, P.; Greig, J.; Troakes, C.; Noble, W.; Miller, C.C.J. Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 2020, 143, 105020. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-g.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta (BBA) 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Cecchi, C.; Fiorillo, C.; Sorbi, S.; Latorraca, S.; Nacmias, B.; Bagnoli, S.; Nassi, P.; Liguri, G. Oxidative stress and reduced antioxidant defenses in peripheral cells from familial Alzheimer’s patients. Free Rad. Biol. Med. 2002, 33, 1372–1379. [Google Scholar] [CrossRef]
- Moreira, P.I.; Harris, P.L.; Zhu, X.; Santos, M.S.; Oliveira, C.R.; Smith, M.A.; Perry, G. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheimer’s Dis. 2007, 12, 195–206. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odetti, P.; Angelini, G.; Dapino, D.; Zaccheo, D.; Garibaldi, S.; Dagna-Bricarelli, F.; Piombo, G.; Perry, G.; Smith, M.; Traverso, N.; et al. Early Glycoxidation Damage in Brains from Down’s Syndrome. Biochem. Biophys. Res. Commun. 1998, 243, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Santa-María, I.; Hernández, F.; Martín, C.P.; Avila, J.; Moreno, F.J. Quinones Facilitate the Self-Assembly of the Phosphorylated Tubulin Binding Region of Tau into Fibrillar Polymers. Biochemistry 2004, 43, 2888–2897. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.D.; Stern, D.M. Mitochondrial dysfunction and Alzheimer’s disease: Role of amyloid-beta peptide alcohol dehydrogenase (ABAD). Int. J. Exp. Pathol. 2005, 86, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, M.Y.; Tucker, H.M.; Nair, P.; Aksenova, M.V.; Butterfield, D.A.; Estus, S.; Markesbery, W.R. The expression of key oxidative stress-handling genes in different brain regions in Alzheimer’s disease. J. Mol. Neurosci. 1998, 11, 151–164. [Google Scholar] [CrossRef]
- Kontush, A.; Berndt, C.; Weber, W.; Akopyan, V.; Arlt, S.; Schippling, S.; Beisiegel, U. Amyloid-β is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Rad. Biol. Med. 2001, 30, 119–128. [Google Scholar] [CrossRef]
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909. [Google Scholar] [CrossRef] [Green Version]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 3270–3274. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Ciotti, M.T.; Natale, F.; Della Valle, F.; Borreca, A.; Manca, A.; Meli, G.; et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: Implications in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 3058–3081. [Google Scholar] [CrossRef] [Green Version]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Maestro, P.; Gargini, R.; A Sproul, A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy Failure in Fibroblasts and iPSC-Derived Neurons of Alzheimer’s Disease-Associated Presenilin 1 Mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro-Cardoso, V.F.; Oliveira, M.M.; Melo, T.; Domingues, M.R.; Moreira, P.I.; Ferreiro, E.; Peixoto, F.; Videira, R.A. Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 43, 1375–1392. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Siedlak, S.L.; Wang, X.; Santos, M.S.; Oliveira, C.R.; Tabaton, M.; Nunomura, A.; Szweda, L.I.; Aliev, G.; Smith, M.A.; et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 2007, 3, 614–615. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Peterhoff, C.M.; Schmidt, S.D.; Terio, N.B.; Duff, K.; Beard, M.; Mathews, P.M.; Nixon, R.A. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol. 2004, 63, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease. Redox Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.S.; Müllendorff, K.; Cheng, I.H.; Miranda, R.D.; Huang, Y.; Mahley, R.W. Reactivity of apolipoprotein E4 and amyloid beta peptide: Lysosomal stability and neurodegeneration. J. Biol. Chem. 2006, 281, 2683–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Partin, J.; Begley, J.G. Amyloid β-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998, 807, 167–176. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.; García, E.; Perry, G.; Avila, J. Slower Dynamics and Aged Mitochondria in Sporadic Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9302761. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, K.; Hatanpää, K.; Rapoport, S.I.; Brady, D.R. Decreased expression of nuclear and mitochondrial DNA-encoded genes of oxidative phosphorylation in association neocortex in Alzheimer disease. Brain Res. 1997, 44, 99–104. [Google Scholar] [CrossRef]
- Buckner, R.L.; Andrews-Hanna, J.R.; Schacter, D.L. The brain’s default network: Anatomy, function, and relevance to disease. Ann. N. Y. Acad. Sci. 2008, 1124, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Dolui, S.; Li, Z.; Nasrallah, I.M.; Detre, J.A.; Wolk, D.A. Arterial spin labeling versus (18)F-FDG-PET to identify mild cognitive impairment. NeuroImage Clin. 2020, 25, 102146. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Laviolette, P.S.; O’Keefe, K.; O’Brien, J.; Rentz, D.M.; Pihlajamaki, M.; Marshall, G.; Hyman, B.T.; Selkoe, D.J.; Hedden, T.; et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 2009, 63, 178–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria in Alzheimer brains. PET Project Shows Complex Chang. 2020, 94, 646–647. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur. J. Nuclear Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Chételat, G.; Desgranges, B.; de la Sayette, V.; Viader, F.; Eustache, F.; Baron, J.C. Mild cognitive impairment: Can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology 2003, 60, 1374–1377. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. Scaling of brain metabolism with a fixed energy budget per neuron: Implications for neuronal activity, plasticity and evolution. PLoS ONE 2011, 6, e17514. [Google Scholar] [CrossRef] [Green Version]
- Yanase, D.; Matsunari, I.; Yajima, K.; Chen, W.; Fujikawa, A.; Nishimura, S.; Matsuda, H.; Yamada, M. Brain FDG PET study of normal aging in Japanese: Effect of atrophy correction. Eur. J. Nuclear Med. Mol. Imaging 2005, 32, 794–805. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.R.; Pan, P.L.; Sheng, L.Q.; Dai, Z.Y.; Wang, G.D.; Luo, R.; Chen, J.H.; Xiao, P.R.; Zhong, J.G.; Shi, H.C. Aberrant pattern of regional cerebral blood flow in Alzheimer’s disease: A voxel-wise meta-analysis of arterial spin labeling MR imaging studies. Oncotarget 2017, 8, 93196–93208. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, S.C.; Adriaanse, S.M.; Binnewijzend, M.A.; Benedictus, M.R.; Ossenkoppele, R.; Wattjes, M.P.; Pijnenburg, Y.A.; van der Flier, W.M.; Lammertsma, A.A.; Kuijer, J.P.; et al. Cerebral perfusion and glucose metabolism in Alzheimer’s disease and frontotemporal dementia: Two sides of the same coin? Eur. Radiol. 2015, 25, 3050–3059. [Google Scholar] [CrossRef] [Green Version]
- Daulatzai, M.A. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 943–972. [Google Scholar] [CrossRef] [PubMed]
- Sheikh-Bahaei, N.; Sajjadi, S.A.; Manavaki, R.; McLean, M.; O’Brien, J.T.; Gillard, J.H. Positron emission tomography-guided magnetic resonance spectroscopy in Alzheimer disease. Ann. Neurol. 2018, 83, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.; Reiter, D.; Kapogiannis, D. Magnetic resonance spectroscopy reveals abnormalities of glucose metabolism in the Alzheimer’s brain. Ann. Clin. Transl. Neurol. 2018, 5, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Blamire, A.M.; Watson, R.; He, J.; Hayes, L.; O’Brien, J.T. Whole-brain patterns of (1)H-magnetic resonance spectroscopy imaging in Alzheimer’s disease and dementia with Lewy bodies. Transl. Psychiatr. 2016, 6, e877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, N.; Nishiyama, S.; Kanazawa, M.; Tsukada, H. Development of novel PET probes, [18F]BCPP-EF, [18F]BCPP-BF, and [11C]BCPP-EM for mitochondrial complex 1 imaging in the living brain. J. Label. Compounds Radiopharm. 2013, 56, 553–561. [Google Scholar] [CrossRef]
- Mansur, A.; Rabiner, E.A.; Comley, R.A.; Lewis, Y.; Middleton, L.T.; Huiban, M.; Passchier, J.; Tsukada, H.; Gunn, R.N. Characterization of 3 PET Tracers for Quantification of Mitochondrial and Synaptic Function in Healthy Human Brain: (18)F-BCPP-EF, (11)C-SA-4503, and (11)C-UCB-J. J. Nuclear Med. 2020, 61, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020, 94, e1592–e1604. [Google Scholar] [CrossRef]
- Ishii, K.; Kitagaki, H.; Kono, M.; Mori, E. Decreased medial temporal oxygen metabolism in Alzheimer’s disease shown by PET. J. Nuclear Med. 1996, 37, 1159–1165. [Google Scholar]
- Nagata, K.; Maruya, H.; Yuya, H.; Terashi, H.; Mito, Y.; Kato, H.; Sato, M.; Satoh, Y.; Watahiki, Y.; Hirata, Y.; et al. Can PET data differentiate Alzheimer’s disease from vascular dementia? Ann. N. Y. Acad. Sci. 2000, 903, 252–261. [Google Scholar] [CrossRef]
- Seeley, W.W.; Crawford, R.K.; Zhou, J.; Miller, B.L.; Greicius, M.D. Neurodegenerative diseases target large-scale human brain networks. Neuron 2009, 62, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Greicius, M.D.; Srivastava, G.; Reiss, A.L.; Menon, V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: Evidence from functional MRI. Proc. Natl. Acad. Sci. USA 2004, 101, 4637–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verber, N.S.; Shepheard, S.R.; Sassani, M.; McDonough, H.E.; Moore, S.A.; Alix, J.J.P.; Wilkinson, I.D.; Jenkins, T.M.; Shaw, P.J. Biomarkers in Motor Neuron Disease: A State of the Art Review. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassani, M.; Alix, J.; McDermott, C.; Baster, K.; Wild, J.; Mortiboys, H.; Shaw, P.J.; Wilkinson, I.; Jenkins, T. Magnetic resonance spectroscopy reveals mitochondrial dysfunction in amyotrophic lateral sclerosis. Brain J. Neurol. 2020, in press. [Google Scholar]
- Mayeux, R. Biomarkers: Potential uses and limitations. NeuroRx 2004, 1, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esterhuizen, K.; van der Westhuizen, F.H.; Louw, R. Metabolomics of mitochondrial disease. Mitochondrion 2017, 35, 97–110. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Ekman, U.; Ferreira, D.; Westman, E. The A/T/N biomarker scheme and patterns of brain atrophy assessed in mild cognitive impairment. Sci. Rep. 2018, 8, 8431. [Google Scholar] [CrossRef] [Green Version]
- Humpel, C. Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011, 29, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Bookheimer, S.; Burggren, A. APOE-4 genotype and neurophysiological vulnerability to Alzheimer’s and cognitive aging. Annu. Rev. Clin. Psychol. 2009, 5, 343–362. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Teipel, S.J.; Fuchsberger, T.; Andreasen, N.; Wiltfang, J.; Otto, M.; Shen, Y.; Dodel, R.; Du, Y.; Farlow, M.; et al. Value of CSF beta-amyloid1-42 and tau as predictors of Alzheimer’s disease in patients with mild cognitive impairment. Mol. Psychiatry 2004, 9, 705–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, I.; Vos, S.J.B.; Jansen, W.J.; Vandenberghe, R.; Gabel, S.; Estanga, A.; Ecay-Torres, M.; Tomassen, J.; den Braber, A.; Lleó, A.; et al. Amyloid-β, Tau, and Cognition in Cognitively Normal Older Individuals: Examining the Necessity to Adjust for Biomarker Status in Normative Data. Front. Aging Neurosci. 2018, 10, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, S.M.; Burgess, T.; Lee, J.; Blackburn, D.J.; Allen, S.P.; Mortiboys, H. Peripheral Glycolysis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8924. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mitochondrial Property | Organ/Cell Type | Change Seen to Mitochondrial Property | Reference |
|---|---|---|---|
| ETC mRNA/Protein Expression | Brain | ↑ OxPHOS protein expression ↓ mRNA subunits 1 and 2 Complex IV ↓ RNA of subunit 3 Complex IV ↑ Mitochondrial Subunits of Complex IV and III ↓ Mitochondrial Subunits of Complex I | [157] [158] [30] [33,35,159] [36] |
| RBC | ↓ OxPHOS genes | [54] | |
| Platelets | ↓ Complex IV subunits | [59,60,61] | |
| ETC Activity | Brain | ↓ Complex IV activity | [41,42,43,44,45,46,47,48,49] |
| Fibroblasts | ↓ Mitochondrial Spare Capacity ↓ OxPHOS, ↓NAD/NADH ratio ↓ Complex IV activity | [66] [68] [67] | |
| RBC | ↓ Oxygen Consumption rates | [106] | |
| Platelets | ↓ Oxygen consumption rates | [110] | |
| Mitochondrial Dynamics | Brain | ↓ and ↑ of both fission and fusion proteins ↑ SNO-Drp1 | [95,99] |
| Fibroblasts | ↓ and ↑ of both fission and fusion proteins and ↑Fis1 | [27,66] [97,98] | |
| Blood Cells | ↑ SNO-Drp1 in lymphocytes | [96] | |
| Calcium Homeostasis | Brain | ↑ IP3R3-VDAC ↑ efflux transporters ↓ influx transporters ↑ Mitochondrial Calcium ↑ MAM Contacts | [124] [124] [123] [116] [116] |
| Fibroblasts | ↑ MAM Contacts | [123] | |
| ROS Production | Brain | ↑ ROS production in areas with lower AP ↑ ROS around AP | [16,138,160] [27,138] |
| Fibroblasts | ↑ ROS production | [129,130] | |
| Bloods | ↑ ROS production | [137] | |
| Mitophagy | Brain | ↑ Parkin Recruitment to mitochondria ↑ Mitochondrial accumulation ↑ Lysosomal dysfunction | [140,141,142,143,144,145,146,147,148] [149,150,151] [150,152] |
| Fibroblasts | ↑ Parkin Recruitment to mitochondria | [146] |
| Imaging Technique | Advantages | Disadvantages |
|---|---|---|
| FDG-PET |
|
|
| 18 F-BCPP-EF |
|
|
| MRI Spectroscopy |
|
|
| MRI Phosphorous Spectroscopy |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. https://doi.org/10.3390/biomedicines9010063
Bell SM, Barnes K, De Marco M, Shaw PJ, Ferraiuolo L, Blackburn DJ, Venneri A, Mortiboys H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines. 2021; 9(1):63. https://doi.org/10.3390/biomedicines9010063
Chicago/Turabian StyleBell, Simon M., Katy Barnes, Matteo De Marco, Pamela J. Shaw, Laura Ferraiuolo, Daniel J. Blackburn, Annalena Venneri, and Heather Mortiboys. 2021. "Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future?" Biomedicines 9, no. 1: 63. https://doi.org/10.3390/biomedicines9010063
APA StyleBell, S. M., Barnes, K., De Marco, M., Shaw, P. J., Ferraiuolo, L., Blackburn, D. J., Venneri, A., & Mortiboys, H. (2021). Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines, 9(1), 63. https://doi.org/10.3390/biomedicines9010063