
Mental Health, Mitochondria, and the Battle of the Sexes



{kind=link}
{kind=link}
Abstract
:Honour thy father and thy mother.—Exodus 20:12
1. Female versus Male
2. Autism versus Psychosis
3. Expressing versus Silencing
3.1. Mitochondria Enable Epigenetics
3.2. Epigenetic Battles
4. (Too Much) Mother versus Father
4.1. Beckwith–Wiedemann Syndrome versus Silver–Russell Syndrome
4.2. Angelman Syndrome versus Prader–Willi Syndrome
4.3. Rett Syndrome versus PPM-X Syndrome
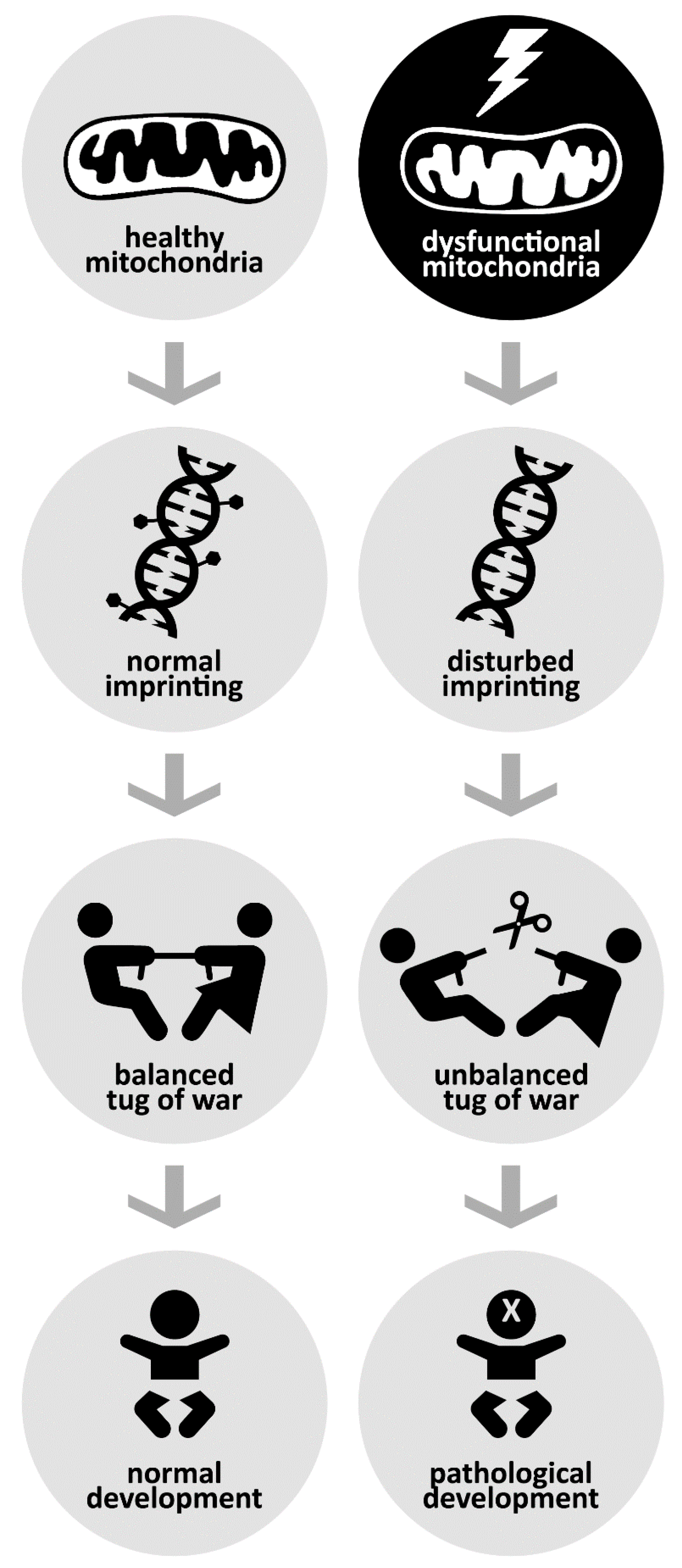
4.4. Mitochondria in the Mix
5. Coda: the Inevitable Instability of Mental Health
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trivers, R. Social Evolution; Benjamin/Cummings: Menlo Park, CA, USA, 1985; ISBN 080538507X. [Google Scholar]
- Xirocostas, Z.A.; Everingham, S.E.; Moles, A.T. The sex with the reduced sex chromosome dies earlier: A comparison across the tree of life. Biol. Lett. 2020, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geary, D.C. Evolutionary perspective on sex differences in the expression of neurological diseases. Prog. Neurobiol. 2019, 176, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Badcock, C.R. The Imprinted Brain: How Genes Set the Balance between Autism and Psychosis; Jessica Kingsley Publishers: Philadelphia, PA, USA, 2009; ISBN 1849050236. [Google Scholar]
- Badcock, C. The Diametric Mind: New Insights into AI, IQ, the Self, and Society; TLU Press: Tallinn, Estonia, 2019; ISBN 978-9985-58-866-6. [Google Scholar]
- Herlitz, A.; Nilsson, L.-G.; Bäckman, L. Gender differences in episodic memory. Mem. Cogn. 1997, 25, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.A. Life Unfolding; Oxford University Press: Oxford, UK, 2014; ISBN 9780199673537. [Google Scholar]
- Lombardo, M.V.; Ashwin, E.; Auyeung, B.; Chakrabarti, B.; Taylor, K.; Hackett, G.; Bullmore, E.T.; Baron-Cohen, S. Fetal testosterone influences sexually dimorphic gray matter in the human brain. J. Neurosci. 2012, 32, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Puts, D.A.; McDaniel, M.A.; Jordan, C.L.; Breedlove, S.M. Spatial ability and prenatal androgens: Meta-analyses of congenital adrenal hyperplasia and digit ratio (2D:4D) studies. Arch. Sex. Behav. 2008, 37, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Collaer, M.L.; Hines, M. Human behavioral sex differences: A role for gonadal hormones during early development. Psychol. Bull. 1995, 118, 55. [Google Scholar] [CrossRef]
- Pol, H.E.H.; Cohen-Kettenis, P.T.; Van Haren, N.E.M.; Peper, J.S.; Brans, R.G.H.; Cahn, W.; Schnack, H.G.; Gooren, L.J.G.; Kahn, R.S. Changing your sex changes your brain: Influences of testosterone and estrogen on adult human brain structure. Eur. J. Endocrinol. 2006, 155, S107–S114. [Google Scholar] [CrossRef] [Green Version]
- Nottebohm, F. Testosterone triggers growth of brain vocal control nuclei in adult female canaries. Brain Res. 1980, 189, 429–436. [Google Scholar] [CrossRef]
- Laws, K.R.; Irvine, K.; Gale, T.M. Sex differences in cognitive impairment in Alzheimer’s disease. World J. Psychiatry 2016, 6, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Gale, S.D.; Baxter, L.; Thompson, J. Greater memory impairment in dementing females than males relative to sex-matched healthy controls. J. Clin. Exp. Neuropsychol. 2016, 38, 527–533. [Google Scholar] [CrossRef]
- Fengler, S.; Roeske, S.; Heber, I.; Reetz, K.; Schulz, J.B.; Riedel, O.; Wittchen, H.U.; Storch, A.; Linse, K.; Baudrexel, S.; et al. Verbal memory declines more in female patients with Parkinson’s disease: The importance of gender-corrected normative data. Psychol. Med. 2016, 46, 2275–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, J.R.; Deskin, K.A.; Josephson, K.T.; Wichmann, R.L. Sex differences in tracking performance in patients with Parkinson’s disease. Arch. Phys. Med. Rehabil. 2002, 83, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, L.L.; Rinne, J.O.; Puukka, P.J.; Laine, H.K.; Ahtiluoto, S.E.; Sulkava, R.O.; Viitanen, M.H.; Jula, A.M. Insulin resistance is associated with poorer verbal fluency performance in women. Diabetologia 2015, 58, 2545–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, P.; Bressan, P. Mitochondria inspire a lifestyle. Adv. Anat. Embryol. Cell Biol. 2019, 231, 105–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bora, E.; Köse, S. Meta-analysis of theory of mind in anorexia nervosa and bulimia nervosa: A specific impairment of cognitive perspective taking in anorexia nervosa? Int. J. Eat. Disord. 2016, 49, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Goddard, E.; Carral-Fernández, L.; Denneny, E.; Campbell, I.C.; Treasure, J. Cognitive flexibility, central coherence and social emotional processing in males with an eating disorder. World J. Biol. Psychiatry 2014, 15, 317–326. [Google Scholar] [CrossRef]
- Asperger, H. Die "autistischen Psychopathen" im Kindesalter. Arch. Psychiatr. Nervenkr. Z. Gesamte Neurol. Psychiatr. 1944, 117, 76–136. [Google Scholar] [CrossRef]
- Baron-Cohen, S. The extreme male brain theory of autism. Trends Cogn. Sci. 2002, 6, 248–254. [Google Scholar] [CrossRef]
- Dammann, G. Borderline personality disorder and theory of mind: An evolutionary perspective. In The Social Brain: Evolution and Pathology; Brüne, M., Ribbert, H., Schiefenhövel, W., Eds.; John Wiley & Sons: Chichester, UK, 2003; pp. 373–417. ISBN 0470849606. [Google Scholar]
- Dinsdale, N.; Mokkonen, M.; Crespi, B. The ‘extreme female brain’: Increased cognitive empathy as a dimension of psychopathology. Evol. Hum. Behav. 2016, 37, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An overview of autism spectrum disorder, heterogeneity and treatment options. Neurosci. Bull. 2017, 33, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Carruthers, S.P.; Van Rheenen, T.E.; Gurvich, C.; Sumner, P.J.; Rossell, S.L. Characterising the structure of cognitive heterogeneity in schizophrenia spectrum disorders. A systematic review and narrative synthesis. Neurosci. Biobehav. Rev. 2019, 107, 252–278. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B.; Badcock, C. Psychosis and autism as diametrical disorders of the social brain. Behav. Brain Sci. 2008, 31, 241–261. [Google Scholar] [CrossRef] [PubMed]
- Badcock, C.; Crespi, B. Battle of the sexes may set the brain. Nature 2008, 454, 1054–1055. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, M.; Angeleri, R.; Brizio, A.; Elena, M.R. The evolution of autistic-like and schizotypal traits: A sexual selection hypothesis. Front. Psychol. 2010, 1, 41. [Google Scholar] [CrossRef] [Green Version]
- Del Giudice, M.; Klimczuk, A.C.; Traficonte, D.M.; Maestripieri, D. Autistic-like and schizotypal traits in a life history perspective: Diametrical associations with impulsivity, sensation seeking, and sociosexual behavior. Evol. Hum. Behav. 2014, 35, 415–424. [Google Scholar] [CrossRef]
- Crespi, B. Oxytocin, testosterone, and human social cognition. Biol. Rev. 2016, 91, 390–408. [Google Scholar] [CrossRef]
- Mokkonen, M.; Koskela, E.; Procyshyn, T.; Crespi, B. Socio-reproductive conflicts and the father’s curse dilemma. Am. Nat. 2018, 192, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Loomes, R.; Hull, L.; Mandy, W. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Raine, A. Sex differences in schizotypal personality in a nonclinical population. J. Abnorm. Psychol. 1992, 101, 361–364. [Google Scholar] [CrossRef]
- Goldstein, J.M.; Santangelo, S.L.; Simpson, J.C.; Tsuang, M.T. The role of gender in identifying subtypes of schizophrenia: A latent class analytic approach. Schizophr. Bull. 1990, 16, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Skuse, D.H. Imprinting, the X-chromosome, and the male brain: Explaining sex differences in the liability to autism. Pediatr. Res. 2000, 47, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespi, B.; Summers, K.; Dorus, S. Genomic sister-disorders of neurodevelopment: An evolutionary approach. Evol. Appl. 2009, 2, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Power, Sex, Suicide: Mitochondria and the Meaning of Life; Oxford University Press: Oxford, UK, 2005; ISBN 0192804812. [Google Scholar]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. Acta 2013, 1833, 1979–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beekman, M.; Dowling, D.K.; Aanen, D.K. The costs of being male: Are there sex-specific effects of uniparental mitochondrial inheritance? Philos. Trans. R. Soc. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innocenti, P.; Morrow, E.H.; Dowling, D.K. Experimental evidence supports a sex-specific selective sieve in mitochondrial genome evolution. Science 2011, 332, 845–848. [Google Scholar] [CrossRef]
- Kramer, P.; Bressan, P. Our (mother’s) mitochondria and our mind. Perspect. Psychol. Sci. 2018, 13, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Vaught, R.C.; Dowling, D.K. Maternal inheritance of mitochondria: Implications for male fertility? Reproduction 2018, 155, R159–R168. [Google Scholar] [CrossRef] [Green Version]
- Zeh, J.A.; Zeh, D.W. Maternal inheritance, sexual conflict and the maladapted male. Trends Genet. 2005, 21, 281–286. [Google Scholar] [CrossRef]
- Hurst, L.D. Embryonic growth and the evolution of the mammalian Y chromosome. I. The Y as an attractor for selfish growth factors. Heredity 1994, 73, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Boklage, C. The epigenetic environment: Secondary sex ratio depends on differential survival in embryogenesis. Hum. Reprod. 2005, 20, 583–587. [Google Scholar] [CrossRef] [Green Version]
- Castegna, A.; Iacobazzi, V.; Infantino, V. The mitochondrial side of epigenetics. Physiol. Genom. 2015, 47, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.F.C. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenetics 2020, 12, 182. [Google Scholar] [CrossRef] [PubMed]
- Forés-Martos, J.; Catalá-López, F.; Sánchez-Valle, J.; Ibáñez, K.; Tejero, H.; Palma-Gudiel, H.; Climent, J.; Pancaldi, V.; Fañanás, L.; Arango, C.; et al. Transcriptomic metaanalyses of autistic brains reveals shared gene expression and biological pathway abnormalities with cancer. Mol. Autism 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, H.-L.; Liu, C.-J.; Hu, Y.-W.; Chen, S.-C.; Hu, L.-Y.; Shen, C.-C.; Yeh, C.-M.; Chen, T.-J.; Gau, S.S.-F. Risk of cancer in children, adolescents, and young adults with autistic disorder. J. Pediatr. 2015, 166, 418–423.e1. [Google Scholar] [CrossRef]
- Catts, V.S.; Catts, S.V.; O’Toole, B.I.; Frost, A.D.J. Cancer incidence in patients with schizophrenia and their first-degree relatives—A meta-analysis. Acta Psychiatr. Scand. 2008, 117, 323–336. [Google Scholar] [CrossRef]
- Crespi, B. The evolutionary biology of child health. Proc. Royal Soc. B 2011, 278, 1441–1449. [Google Scholar] [CrossRef] [Green Version]
- Gilman, S.R.; Chang, J.; Xu, B.; Bawa, T.S.; Gogos, J.A.; Karayiorgou, M.; Vitkup, D. Diverse types of genetic variation converge on functional gene networks involved in schizophrenia. Nat. Neurosci. 2012, 15, 1723–1728. [Google Scholar] [CrossRef]
- Susser, E.; St Clair, D.; He, L. Latent effects of prenatal malnutrition on adult health: The example of schizophrenia. Ann. N. Y. Acad. Sci. 2008, 1136, 185–192. [Google Scholar] [CrossRef]
- Chiarotti, F.; Venerosi, A. Epidemiology of Autism Spectrum Disorders: A review of worldwide prevalence estimates since 2014. Brain Sci. 2020, 10, 274. [Google Scholar] [CrossRef]
- Loenen, W.A.M. S-Adenosylmethionine: Jack of all trades and master of everything? Biochem. Soc. Trans. 2006, 34, 330–333. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019, 20, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdge, G.C.; Slater-Jefferies, J.; Torrens, C.; Phillips, E.S.; Hanson, M.A.; Lillycrop, K.A. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br. J. Nutr. 2007, 97, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.H. Mitochondria signaling to the epigenome: A novel role for an old organelle. Free Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef] [Green Version]
- Smiraglia, D.J.; Kulawiec, M.; Bistulfi, G.L.; Gupta, S.G.; Singh, K.K. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol. Ther. 2008, 7, 1182–1190. [Google Scholar] [CrossRef] [Green Version]
- Lozoya, O.A.; Xu, F.; Grenet, D.; Wang, T.; Grimm, S.A.; Godfrey, V.; Waidyanatha, S.; Woychik, R.P.; Santos, J.H. Single nucleotide resolution analysis reveals pervasive, long-lasting DNA methylation changes by developmental exposure to a mitochondrial toxicant. Cell Rep. 2020, 32, 108131. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting Group. Genomic imprinting and physiological processes in mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.; Haig, D. Genomic imprinting in mammalian development: A parental tug-of-war. Trends. Genet. 1991, 7, 45–49. [Google Scholar] [CrossRef]
- Haig, D. Transfers and transitions: Parent-offspring conflict, genomic imprinting, and the evolution of human life history. Proc. Natl. Acad. Sci. USA 2010, 107, 1731–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haig, D. Frugal fat or munificent muscle: Genomic imprinting and metabolism. BMC Biol. 2014, 12, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespi, B. Why and how imprinted genes drive fetal programming. Front. Endocrinol. 2020, 10, 940. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Hivert, M.-F.; Perron, P.; Poirier, P.; Guay, S.-P.; Brisson, D.; Bouchard, L. IGF2 DNA methylation is a modulator of newborn’s fetal growth and development. Epigenetics 2012, 7, 1125–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haig, D. Genetic conflicts in human pregnancy. Q. Rev. Biol. 1993, 68, 495–532. [Google Scholar] [CrossRef]
- World Health Organization. Trends in Maternal Mortality 2000 to 2017: Estimates By Who, Unicef, Unfpa, World Bank Group and the United Nations Population Division; World Health Organization: Geneva, Switzerland, 2019; ISBN 9241516488. [Google Scholar]
- Say, L.; Chou, D.; Gemmill, A.; Tunçalp, Ö.; Moller, A.-B.; Daniels, J.; Gülmezoglu, A.M.; Temmerman, M.; Alkema, L. Global causes of maternal death: A WHO systematic analysis. Lancet Glob. Health 2014, 2, e323–e333. [Google Scholar] [CrossRef] [Green Version]
- Úbeda, F. Evolution of genomic imprinting with biparental care: Implications for Prader-Willi and Angelman syndromes. PLoS Biol. 2008, 6, e208. [Google Scholar] [CrossRef] [PubMed]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef]
- Soellner, L.; Begemann, M.; Mackay, D.J.G.; Grønskov, K.; Tümer, Z.; Maher, E.R.; Temple, I.K.; Monk, D.; Riccio, A.; Linglart, A.; et al. Recent advances in imprinting disorders. Clin. Genet. 2017, 91, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.M.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver–Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef]
- Perrone, S.; Lotti, F.; Geronzi, U.; Guidoni, E.; Longini, M.; Buonocore, G. Oxidative stress in cancer-prone genetic diseases in pediatric age: The role of mitochondrial dysfunction. Oxid. Med. Cell. Longev. 2016, 2016, 4782426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalish, J.M.; Doros, L.; Helman, L.J.; Hennekam, R.C.; Kuiper, R.P.; Maas, S.M.; Maher, E.R.; Nichols, K.E.; Plon, S.E.; Porter, C.C.; et al. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin. Cancer Res. 2017, 23, e115–e122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, L.; Bowdin, S.; Kirby, G.A.; Cooper, W.N.; Maher, E.R. Beckwith Weidemann syndrome: A behavioral phenotype-genotype study. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147B, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Salminen, I.I.; Crespi, B.J.; Mokkonen, M. Baby food and bedtime: Evidence for opposite phenotypes from different genetic and epigenetic alterations in Prader-Willi and Angelman syndromes. SAGE Open Med. 2019, 7, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 2010, 38, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespi, B. Genomic imprinting in the development and evolution of psychotic spectrum conditions. Biol. Rev. 2008, 83, 441–493. [Google Scholar] [CrossRef]
- Krefft, M.; Frydecka, D.; Adamowski, T.; Misiak, B. From Prader–Willi syndrome to psychosis: Translating parent-of-origin effects into schizophrenia research. Epigenomics 2014, 6, 677–688. [Google Scholar] [CrossRef]
- Matijevic, T.; Knezevic, J.; Slavica, M.; Pavelic, J. Rett syndrome: From the gene to the disease. Eur. Neurol. 2009, 61, 3–10. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef]
- Trappe, R.; Laccone, F.; Cobilanschi, J.; Meins, M.; Huppke, P.; Hanefeld, F.; Engel, W. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 2001, 68, 1093–1101. [Google Scholar] [CrossRef] [Green Version]
- Sandweiss, A.J.; Brandt, V.L.; Zoghbi, H.Y. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: Prospects for future therapies. Lancet Neurol. 2020, 19, 689–698. [Google Scholar] [CrossRef]
- Percy, A.K. Rett syndrome: Exploring the autism link. Arch. Neurol. 2011, 68, 985–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, S.; Splitt, M.; Edney, S.; Berney, T.P.; Knight, S.J.L.; Davies, K.E.; O’Brien, O.; Gale, M.; Burn, J. PPM-X: A new X-linked mental retardation syndrome with psychosis, pyramidal signs, and macroorchidism maps to Xq28. Am. J. Hum. Genet. 1996, 58, 1120–1126. [Google Scholar] [PubMed]
- Klauck, S.M.; Lindsay, S.; Beyer, K.S.; Splitt, M.; Burn, J.; Poustka, A. A mutation hot spot for nonspecific X-linked mental retardation in the MECP2 gene causes the PPM-X syndrome. Am. J. Hum. Genet. 2002, 70, 1034–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorini, C.; De Felice, C.; Leoncini, S.; Møller, R.S.; Zollo, G.; Buoni, S.; Cortelazzo, A.; Guerranti, R.; Durand, T.; Ciccoli, L.; et al. MECP2 duplication syndrome: Evidence of enhanced oxidative stress. A comparison with Rett syndrome. PLoS ONE 2016, 11, e0150101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicaloni, V.; Pecorelli, A.; Tinti, L.; Rossi, M.; Benedusi, M.; Cervellati, C.; Spiga, O.; Santucci, A.; Hayek, J.; Salvini, L.; et al. Proteomic profiling reveals mitochondrial alterations in Rett syndrome. Free. Radic. Biol. Med. 2020, 155, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial dysfunction in the pathogenesis of Rett Syndrome: Implications for mitochondria-targeted therapies. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- De Felice, C.; Signorini, C.; Durand, T.; Ciccoli, L.; Leoncini, S.; D’Esposito, M.; Filosa, S.; Oger, C.; Guy, A.; Bultel-Poncé, V.; et al. Partial rescue of Rett syndrome by ω-3 polyunsaturated fatty acids (PUFAs) oil. Genes Nutr. 2012, 7, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Aldosary, M.; Al-Bakheet, A.; Al-Dhalaan, H.; Almass, R.; Alsagob, M.; Al-Younes, B.; AlQuait, L.; Mustafa, O.M.; Bulbul, M.; Rahbeeni, Z.; et al. Rett Syndrome, a neurodevelopmental disorder, whole-transcriptome, and mitochondrial genome multiomics analyses identify novel variations and disease pathways. OMICS 2020, 24, 160–171. [Google Scholar] [CrossRef]
- Panov, J.; Simchi, L.; Feuermann, Y.; Kaphzan, H. Bioinformatics analyses of the transcriptome reveal Ube3a-dependent effects on mitochondrial-related pathways. Int. J. Mol. Sci. 2020, 21, 4156. [Google Scholar] [CrossRef]
- Simchi, L.; Panov, J.; Morsy, O.; Feuermann, Y.; Kaphzan, H. Novel insights into the role of UBE3A in regulating apoptosis and proliferation. J. Clin. Med. 2020, 9, 1573. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Yasuda, T.; Shiraishi, C.; Fujiwara, K.; Przedborski, S.; Mochizuki, H.; Yoshikawa, K. Promotion of mitochondrial biogenesis by necdin protects neurons against mitochondrial insults. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castora, F.J. Mitochondrial function and abnormalities implicated in the pathogenesis of ASD. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 83–108. [Google Scholar] [CrossRef] [PubMed]
- Hollis, F.; Kanellopoulos, A.K.; Bagni, C. Mitochondrial dysfunction in Autism Spectrum Disorder: Clinical features and perspectives. Curr. Opin. Neurobiol. 2017, 45, 178–187. [Google Scholar] [CrossRef]
- Gonçalves, V.F.; Andreazza, A.C.; Kennedy, J.L. Mitochondrial dysfunction in schizophrenia: An evolutionary perspective. Hum. Genet 2015, 134, 13–21. [Google Scholar] [CrossRef]
- Ni, P.; Chung, S. Mitochondrial dysfunction in schizophrenia. BioEssays 2020, 42, e1900202. [Google Scholar] [CrossRef]
- Pei, L.; Wallace, D.C. Mitochondrial etiology of neuropsychiatric disorders. Biol. Psychiatry 2018, 83, 722–730. [Google Scholar] [CrossRef]
- Picard, M.; McEwen, B.S. Psychological stress and mitochondria: A systematic review. Psychosom. Med. 2018, 80, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.; Maes, M. Mitochondria and immunity in chronic fatigue syndrome. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 103. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-Junior, H.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction, oxidative stress, and neuroinflammation: Intertwined roads to neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef]
- Anglin, R.E.; Tarnopolsky, M.A.; Mazurek, M.F.; Rosebush, P.I. The psychiatric presentation of mitochondrial disorders in adults. J. Neuropsychiatry Clin. Neurosci. 2012, 24, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.C.P.; Ferrasa, A.; Muotri, A.R.; Herai, R.H. Frequency and association of mitochondrial genetic variants with neurological disorders. Mitochondrion 2019, 46, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Gong, J.-S.; Zhang, J.; Yamada, Y.; Borgeld, H.-J.; Yagi, K. Mitochondrial genotype associated with longevity and its inhibitory effect on mutagenesis. Mech. Ageing Dev. 2000, 116, 65–76. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial etiology of neuropsychiatric disorders. JAMA Psychiat. 2017, 74, 863–864. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bressan, P.; Kramer, P. Mental Health, Mitochondria, and the Battle of the Sexes. Biomedicines 2021, 9, 116. https://doi.org/10.3390/biomedicines9020116
Bressan P, Kramer P. Mental Health, Mitochondria, and the Battle of the Sexes. Biomedicines. 2021; 9(2):116. https://doi.org/10.3390/biomedicines9020116
Chicago/Turabian StyleBressan, Paola, and Peter Kramer. 2021. "Mental Health, Mitochondria, and the Battle of the Sexes" Biomedicines 9, no. 2: 116. https://doi.org/10.3390/biomedicines9020116
APA StyleBressan, P., & Kramer, P. (2021). Mental Health, Mitochondria, and the Battle of the Sexes. Biomedicines, 9(2), 116. https://doi.org/10.3390/biomedicines9020116