
Combined Metabolic Activators Decrease Liver Steatosis by Activating Mitochondrial Metabolism in Hamsters Fed with a High-Fat Diet

 , ,
, ,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
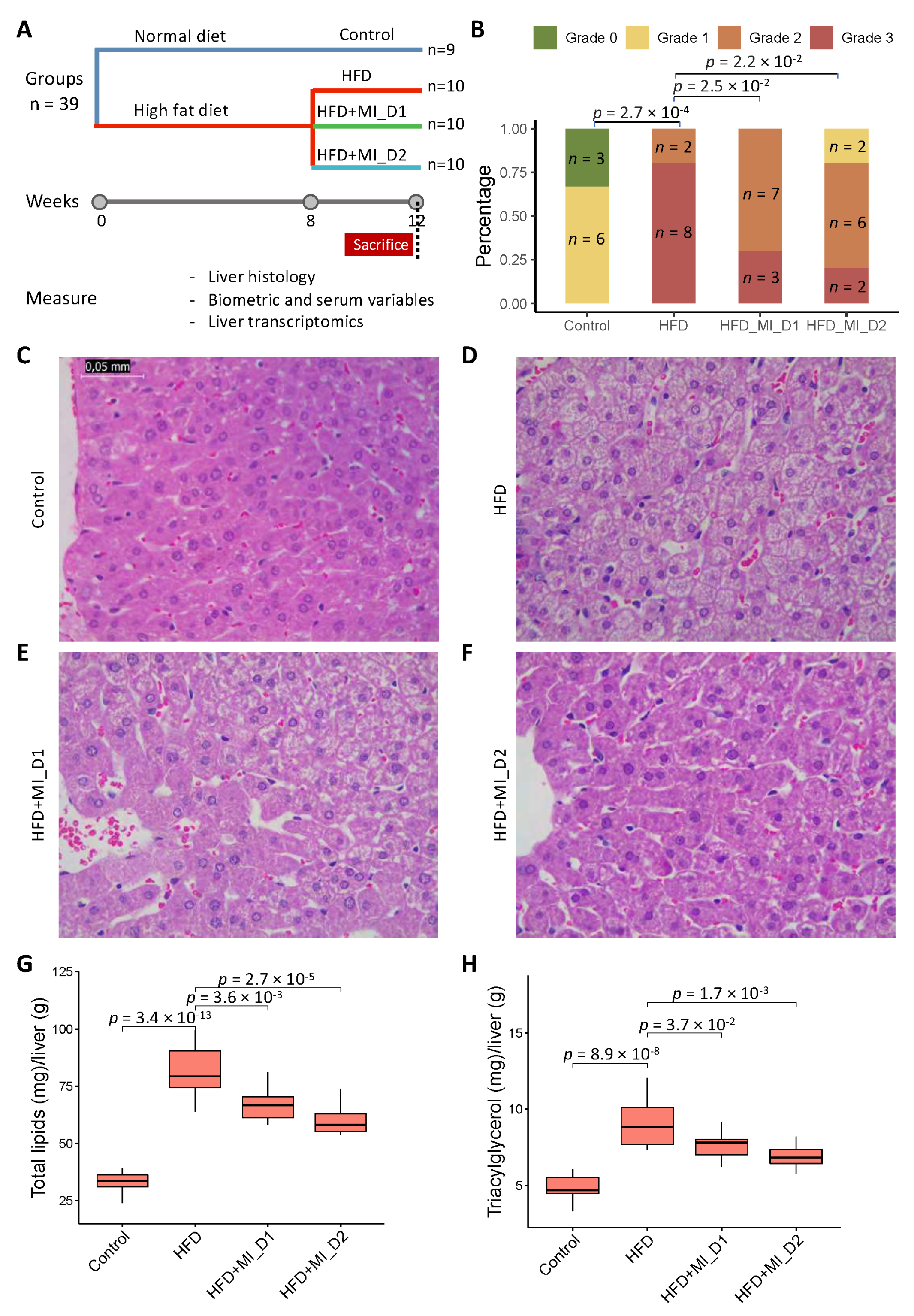
2.2. Histological Evaluation
2.3. Hepatic Lipid Extraction and Quantification
2.4. Body Composition Analyses
2.5. Serum Analysis
2.6. 1H NMR Analyses for NAD+, Sarcosine and Ascorbate Determination
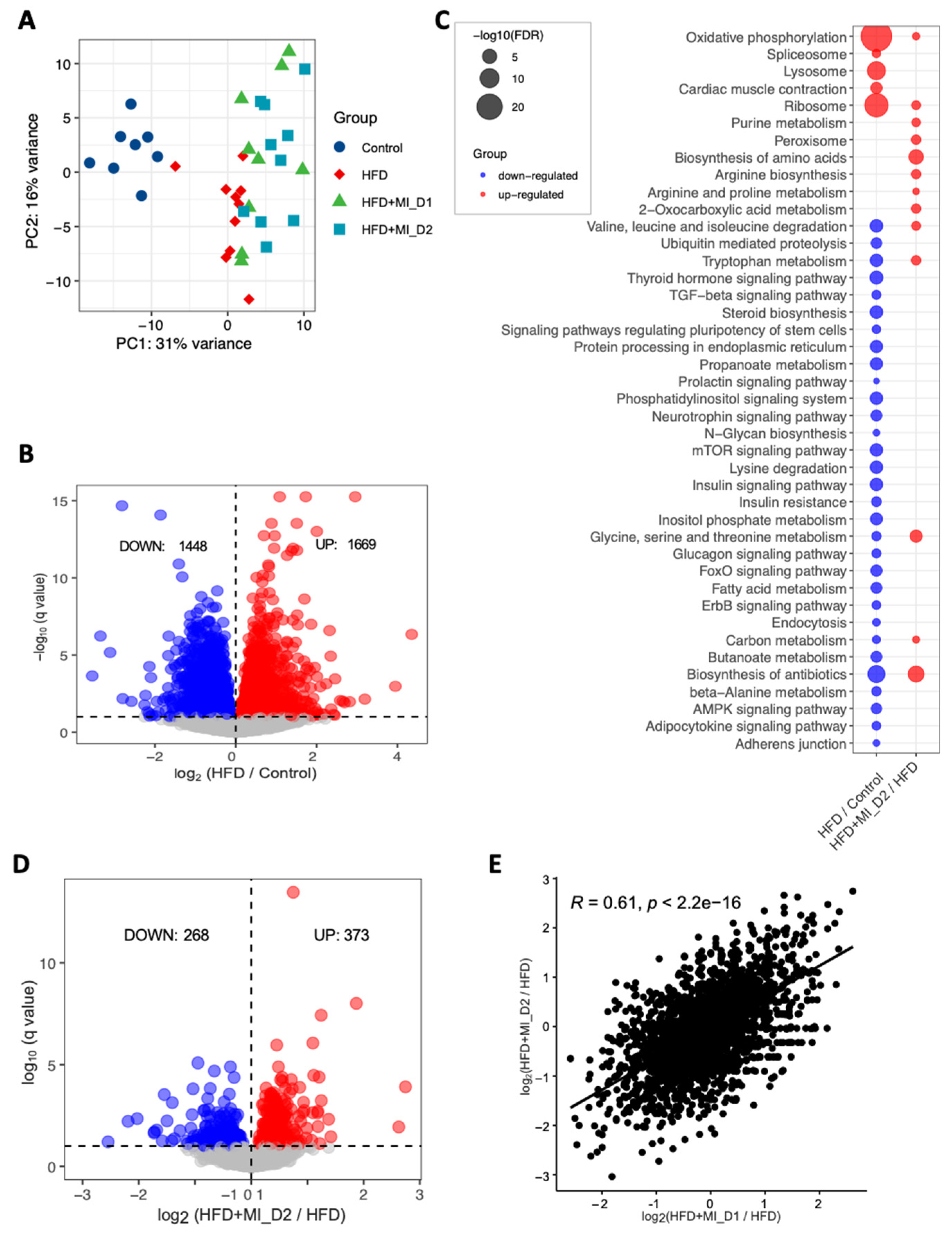
2.7. Transcriptomics and Gene Set Enrichment Analysis
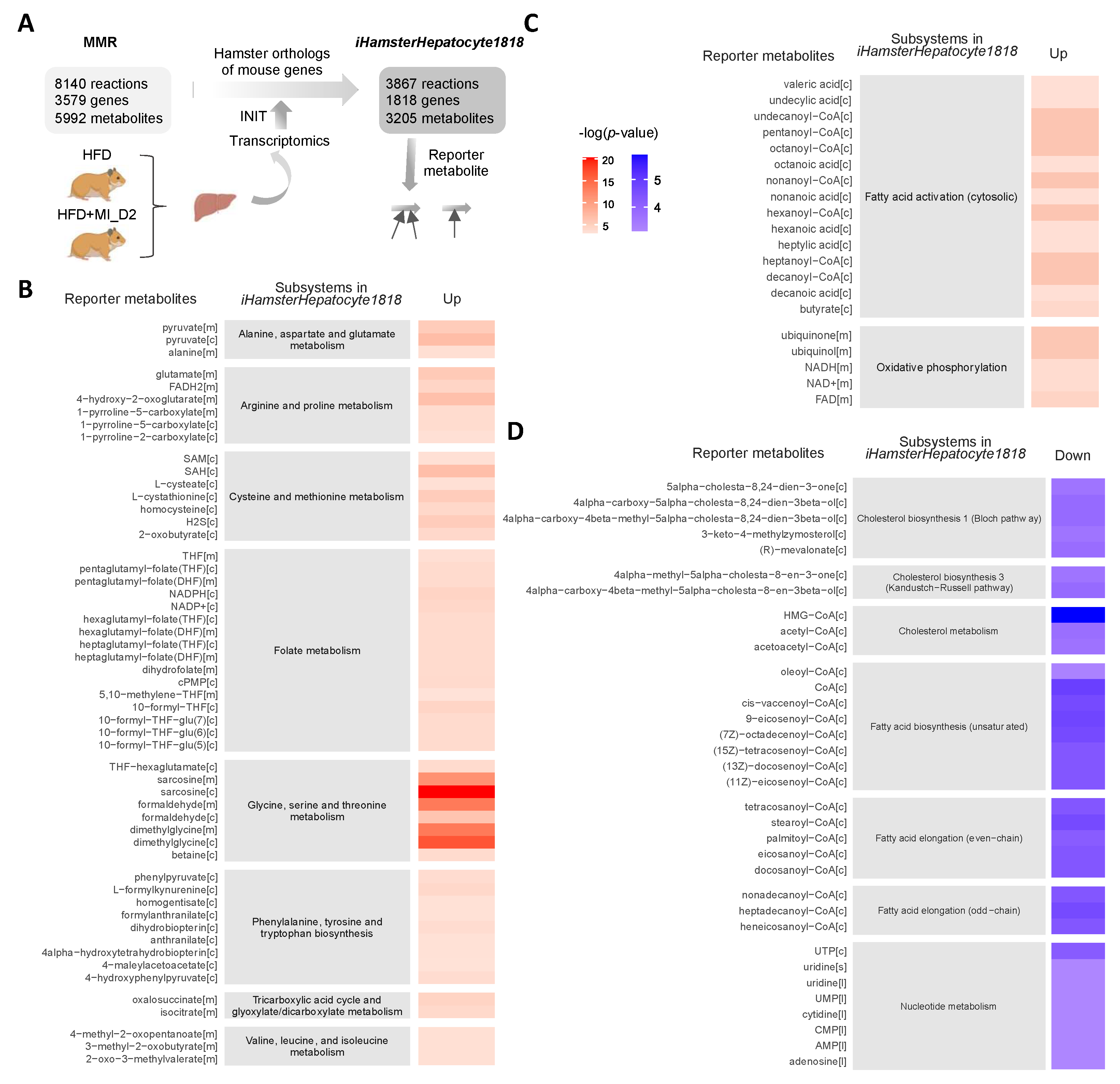
2.8. Metabolic Model Reconstruction and Reporter Metabolite Analysis
2.9. Statistical Analysis
3. Results
3.1. CMA Attenuated HFD-Induced HS in a Golden Syrian Hamster NAFLD Model
3.2. Transcriptomics Alteration with HFD and CMA Treatment
3.3. Reporter Metabolites through the Global Analysis of Transcriptomics Data
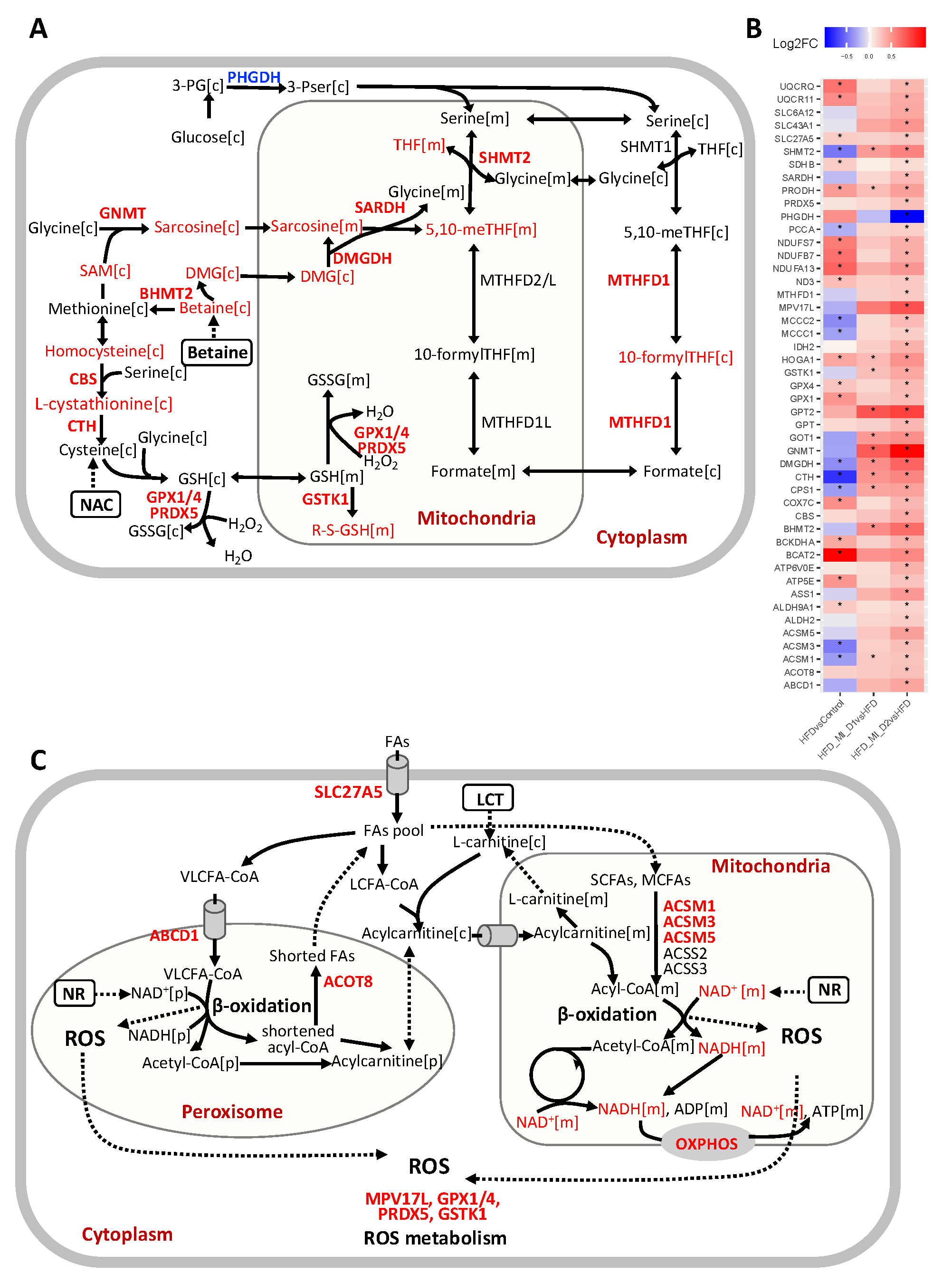
3.4. CMA Boosted Hepatic Metabolism to Attenuate HS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abeysekera, K.W.M.; Fernandes, G.S.; Hammerton, G.; Portal, A.J.; Gordon, F.H.; Heron, J.; Hickman, M. Prevalence of steatosis and fibrosis in young adults in the UK: A population-based study. Lancet Gastroenterol. Hepatol. 2020, 5, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 223–238. [Google Scholar] [CrossRef]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Lazarus, J.V.; Colombo, M.; Cortez-Pinto, H.; Huang, T.T.; Miller, V.; Ninburg, M.; Schattenberg, J.M.; Seim, L.; Wong, V.W.S.; Zelber-Sagi, S. NAFLD–sounding the alarm on a silent epidemic. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 377–379. [Google Scholar] [CrossRef]
- Suarez, M.; Boque, N.; Del Bas, J.M.; Mayneris-Perxachs, J.; Arola, L.; Caimari, A. Mediterranean Diet and Multi-Ingredient-Based Interventions for the Management of Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Agroudy, N.N.; Kurzbach, A.; Rodionov, R.N.; O’Sullivan, J.; Roden, M.; Birkenfeld, A.L.; Pesta, D.H. Are Lifestyle Therapies Effective for NAFLD Treatment? Trends Endocrinol. Metab. 2019, 30, 701–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra-Vargas, M.; Rodriguez-Echevarria, R.; Jimenez-Chillaron, J.C. Nutritional Approaches for the Management of Nonalcoholic Fatty Liver Disease: An Evidence-Based Review. Nutrients 2020, 12, 3860. [Google Scholar] [CrossRef]
- Parekh, S.; Anania, F.A. Abnormal lipid and glucose metabolism in obesity: Implications for nonalcoholic fatty liver disease. Gastroenterology 2007, 132, 2191–2207. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Soderlund, S.; Stahlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD(+) and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Sugimoto, M.; Honma, M.; Mori, M.; Igarashi, K.; Kashikura, K.; Ikeda, S.; Hirayama, A.; Yamamoto, T.; Yoshida, H.; et al. Serum metabolomics reveals gamma-glutamyl dipeptides as biomarkers for discrimination among different forms of liver disease. J. Hepatol. 2011, 55, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Rasanen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietilainen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e555. [Google Scholar] [CrossRef] [Green Version]
- Mardinoglu, A.; Uhlen, M.; Boren, J. Broad Views of Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Ural, D.; Zeybel, M.; Yuksel, H.H.; Uhlen, M.; Boren, J. The Potential Use of Metabolic Cofactors in Treatment of NAFLD. Nutrients 2019, 11, 1578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Bjornson, E.; Arif, M.; Tebani, A.; Lovric, A.; Benfeitas, R.; Ozcan, M.; Juszczak, K.; Kim, W.; Kim, J.T.; et al. The acute effect of metabolic cofactor supplementation: A potential therapeutic strategy against non-alcoholic fatty liver disease. Mol. Syst. Biol. 2020, 16, e9495. [Google Scholar] [CrossRef]
- Bravo, E.; Cantafora, A.; Calcabrini, A.; Ortu, G. Why prefer the golden Syrian hamster (Mesocricetus auratus) to the Wistar rat in experimental studies on plasma lipoprotein metabolism? Comp. Biochem. Physiol. Part B Comp. Biochem. 1994, 107, 347–355. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, H.; Jiao, R.; Peng, C.; Wong, Y.M.; Yeung, V.S.; Huang, Y.; Chen, Z.Y. Choosing hamsters but not rats as a model for studying plasma cholesterol-lowering activity of functional foods. Mol. Nutr. Food Res. 2009, 53, 921–930. [Google Scholar] [CrossRef]
- Dillard, A.; Matthan, N.R.; Lichtenstein, A.H. Use of hamster as a model to study diet-induced atherosclerosis. Nutr. Metab. 2010, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Laos, S.; Caimari, A.; Crescenti, A.; Lakkis, J.; Puiggros, F.; Arola, L.; del Bas, J.M. Long-term intake of soyabean phytosterols lowers serum TAG and NEFA concentrations, increases bile acid synthesis and protects against fatty liver development in dyslipidaemic hamsters. Br. J. Nutr. 2014, 112, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Pasquali, P.; Cryosurgery, A. A Practical Manual; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Radin, N.S. Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem. 1978, 90, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Sureda, V.; Peinado-Onsurbe, J. A procedure for measuring triacylglyceride and cholesterol content using a small amount of tissue. Anal. Biochem. 2005, 343, 277–282. [Google Scholar] [CrossRef]
- Caimari, A.; Oliver, P.; Palou, A. Adipose triglyceride lipase expression and fasting regulation are differently affected by cold exposure in adipose tissues of lean and obese Zucker rats. J. Nutr. Biochem. 2012, 23, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Rodriguez, M.A.; Rull, A.; Beltran, R.; Blade, C.; Brezmes, J.; Canellas, N.; Joven, J.; Correig, X. Metabolomic assessment of the effect of dietary cholesterol in the progressive development of fatty liver disease. J. Proteome Res. 2010, 9, 2527–2538. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, X.; Sherman, B.T.; Huang da, W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardinoglu, A.; Shoaie, S.; Bergentall, M.; Ghaffari, P.; Zhang, C.; Larsson, E.; Backhed, F.; Nielsen, J. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 2015, 11, 834. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef]
- Bin, P.; Huang, R.; Zhou, X. Oxidation Resistance of the Sulfur Amino Acids: Methionine and Cysteine. Biomed. Res. Int. 2017, 2017, 9584932. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Lismont, C.; Walton, P. The Peroxisome-Mitochondria Connection: How and Why? Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Hunt, M.C.; Alexson, S.E. Novel functions of acyl-CoA thioesterases and acyltransferases as auxiliary enzymes in peroxisomal lipid metabolism. Prog. Lipid Res. 2008, 47, 405–421. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.J.; Kemp, S.; Braiterman, L.T.; Watkins, P.A. Role of very-long-chain acyl-coenzyme A synthetase in X-linked adrenoleukodystrophy. Ann. Neurol. 1999, 46, 409–412. [Google Scholar] [CrossRef]
- Doege, H.; Baillie, R.A.; Ortegon, A.M.; Tsang, B.; Wu, Q.; Punreddy, S.; Hirsch, D.; Watson, N.; Gimeno, R.E.; Stahl, A. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: Alterations in hepatic lipid homeostasis. Gastroenterology 2006, 130, 1245–1258. [Google Scholar] [CrossRef] [Green Version]
- Arauz, J.; Ramos-Tovar, E.; Muriel, P. Redox state and methods to evaluate oxidative stress in liver damage: From bench to bedside. Ann. Hepatol. 2016, 15, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Buchet, J.P.; Lauwerys, R. Role of thiols in the in-vitro methylation of inorganic arsenic by rat liver cytosol. Biochem. Pharmacol. 1988, 37, 3149–3153. [Google Scholar] [CrossRef]
- Aasheim, E.T.; Hofso, D.; Hjelmesaeth, J.; Birkeland, K.I.; Bohmer, T. Vitamin status in morbidly obese patients: A cross-sectional study. Am. J. Clin. Nutr. 2008, 87, 362–369. [Google Scholar] [CrossRef]
- Wrieden, W.L.; Hannah, M.K.; Bolton-Smith, C.; Tavendale, R.; Morrison, C.; Tunstall-Pedoe, H. Plasma vitamin C and food choice in the third Glasgow MONICA population survey. J. Epidemiol. Commun. Health 2000, 54, 355–360. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Faga, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef]
- Park, J.K.; Ki, M.R.; Lee, H.R.; Hong, I.H.; Ji, A.R.; Ishigami, A.; Park, S.I.; Kim, J.M.; Chung, H.Y.; Yoo, S.E.; et al. Vitamin C deficiency attenuates liver fibrosis by way of up-regulated peroxisome proliferator-activated receptor-gamma expression in senescence marker protein 30 knockout mice. Hepatology 2010, 51, 1766–1777. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, Y.; Xiong, Y.; Tao, J.; Pan, T.; Tan, S.; Gao, G.; Chen, Y.; Abbas, N.; Getachew, A.; et al. Ascorbate protects liver from metabolic disorder through inhibition of lipogenesis and suppressor of cytokine signaling 3 (SOCS3). Nutr. Metab. 2020, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Rana, S.V. Ascorbic acid improves mitochondrial function in liver of arsenic-treated rat. Toxicol. Ind. Health 2010, 26, 265–272. [Google Scholar] [CrossRef]
- Lerin, C.; Goldfine, A.B.; Boes, T.; Liu, M.; Kasif, S.; Dreyfuss, J.M.; De Sousa-Coelho, A.L.; Daher, G.; Manoli, I.; Sysol, J.R.; et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol. Metab. 2016, 5, 926–936. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, F.; Heeboll, S.; Marrone, G.; Montoliu, C.; Hamilton-Dutoit, S.; Ferrandez, A.; Andreola, F.; Rombouts, K.; Gronbaek, H.; Felipo, V.; et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, P.L.; Vilstrup, H.; Rigbolt, K.; Suppli, M.P.; Sorensen, M.; Heeboll, S.; Veidal, S.S.; Knop, F.K.; Thomsen, K.L. Non-alcoholic fatty liver disease alters expression of genes governing hepatic nitrogen conversion. Liver Int. 2019, 39, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2020, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.E.; Wu, Q.; Wang, X.; Deng, L.; Caudill, M.A.; Rozen, R. Steatosis in mice is associated with gender, folate intake, and expression of genes of one-carbon metabolism. J. Nutr. 2010, 140, 1736–1741. [Google Scholar] [CrossRef] [Green Version]
- Pacana, T.; Cazanave, S.; Verdianelli, A.; Patel, V.; Min, H.K.; Mirshahi, F.; Quinlivan, E.; Sanyal, A.J. Dysregulated Hepatic Methionine Metabolism Drives Homocysteine Elevation in Diet-Induced Nonalcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0136822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.G.; Reily, M.D.; Lehman-McKeeman, L.D.; Vaillancourt, R.R.; Cherrington, N.J. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids 2015, 47, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Kalavalapalli, S.; Bril, F.; Garrett, T.J.; Nautiyal, M.; Mathew, J.T.; Williams, C.M.; Cusi, K. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am. J. Physiol. Metab. 2015, 309, E311–E319. [Google Scholar] [CrossRef] [Green Version]
- Neinast, M.D.; Jang, C.; Hui, S.; Murashige, D.S.; Chu, Q.; Morscher, R.J.; Li, X.; Zhan, L.; White, E.; Anthony, T.G.; et al. Quantitative Analysis of the Whole-Body Metabolic Fate of Branched-Chain Amino Acids. Cell Metab. 2019, 29, 417–429.e414. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.; Samson, S.L.; Reddy, V.T.; Gonzalez, E.V.; Sekhar, R.V. Impaired mitochondrial fatty acid oxidation and insulin resistance in aging: Novel protective role of glutathione. Aging Cell 2013, 12, 415–425. [Google Scholar] [CrossRef]
- Garcia, A.P.; Palou, M.; Sanchez, J.; Priego, T.; Palou, A.; Pico, C. Moderate caloric restriction during gestation in rats alters adipose tissue sympathetic innervation and later adiposity in offspring. PLoS ONE 2011, 6, e17313. [Google Scholar] [CrossRef] [Green Version]
- Avila, M.A.; Berasain, C.; Torres, L.; Martin-Duce, A.; Corrales, F.J.; Yang, H.; Prieto, J.; Lu, S.C.; Caballeria, J.; Rodes, J.; et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J. Hepatol. 2000, 33, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Varela-Rey, M.; Martinez-Lopez, N.; Fernandez-Ramos, D.; Embade, N.; Calvisi, D.F.; Woodhoo, A.; Rodriguez, J.; Fraga, M.F.; Julve, J.; Rodriguez-Millan, E.; et al. Fatty liver and fibrosis in glycine N-methyltransferase knockout mice is prevented by nicotinamide. Hepatology 2010, 52, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Una, M.; Varela-Rey, M.; Mestre, D.; Fernandez-Ares, L.; Fresnedo, O.; Fernandez-Ramos, D.; Gutierrez-de Juan, V.; Martin-Guerrero, I.; Garcia-Orad, A.; Luka, Z.; et al. S-Adenosylmethionine increases circulating very-low density lipoprotein clearance in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, M.; Wang, T.J.; Clish, C.; Engstrom, G.; Nilsson, P.; Gerszten, R.E.; Melander, O. Dimethylglycine Deficiency and the Development of Diabetes. Diabetes 2015, 64, 3010–3016. [Google Scholar] [CrossRef] [Green Version]
- Bravo, E.; Palleschi, S.; Aspichueta, P.; Buque, X.; Rossi, B.; Cano, A.; Napolitano, M.; Ochoa, B.; Botham, K.M. High fat diet-induced non alcoholic fatty liver disease in rats is associated with hyperhomocysteinemia caused by down regulation of the transsulphuration pathway. Lipids Health Dis. 2011, 10, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarna, L.K.; Siow, Y.L.; O, K. The CBS/CSE system: A potential therapeutic target in NAFLD? Can. J. Physiol. Pharmacol. 2015, 93, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maria Del Bas, J.; Rodriguez, B.; Puiggros, F.; Marine, S.; Rodriguez, M.A.; Morina, D.; Armengol, L.; Caimari, A.; Arola, L. Hepatic accumulation of S-adenosylmethionine in hamsters with non-alcoholic fatty liver disease associated with metabolic syndrome under selenium and vitamin E deficiency. Clin. Sci. 2019, 133, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Go, Y.; Kim, D.Y.; Lee, S.H.; Kim, O.H.; Jeon, Y.H.; Kwon, T.K.; Bae, J.H.; Song, D.K.; Rhyu, I.J.; et al. Correction: Isocitrate dehydrogenase 2 protects mice from high-fat diet-induced metabolic stress by limiting oxidative damage to the mitochondria from brown adipose tissue. Exp. Mol. Med. 2020, 52, 988. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Gao, L.; Ding, T. IDH2 protects against nonalcoholic steatohepatitis by alleviating dyslipidemia regulated by oxidative stress. Biochem. Biophys. Res. Commun. 2019, 514, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Bonekamp, N.A.; Volkl, A.; Fahimi, H.D.; Schrader, M. Reactive oxygen species and peroxisomes: Struggling for balance. Biofactors 2009, 35, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Shi, S.; Ju, W.; Faul, C.; Tsai, S.Y.; Mundel, P.; Bottinger, E.P. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc. Natl. Acad. Sci. USA 2008, 105, 14106–14111. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, A.C.; Coggan, M.; Shield, A.J.; Cappello, J.; Theodoratos, A.; Murray, T.P.; Rooke, M.; Larter, C.Z.; Koina, M.E.; Dahlstrom, J.E.; et al. Glutathione transferase kappa deficiency causes glomerular nephropathy without overt oxidative stress. Lab. Investig. 2011, 91, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Leach, N.V.; Dronca, E.; Vesa, S.C.; Sampelean, D.P.; Craciun, E.C.; Lupsor, M.; Crisan, D.; Tarau, R.; Rusu, R.; Para, I.; et al. Serum homocysteine levels, oxidative stress and cardiovascular risk in non-alcoholic steatohepatitis. Eur. J. Intern. Med. 2014, 25, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Zhang, G.; Zheng, Y.; Yang, Y.; Chen, C.; Xia, J.; Liang, L.; Lei, C.; Hu, Y.; Cai, X.; et al. SLC27A5 deficiency activates NRF2/TXNRD1 pathway by increased lipid peroxidation in HCC. Cell Death Differ. 2020, 27, 1086–1104. [Google Scholar] [CrossRef]
- Enooku, K.; Tsutsumi, T.; Kondo, M.; Fujiwara, N.; Sasako, T.; Shibahara, J.; Kado, A.; Okushin, K.; Fujinaga, H.; Nakagomi, R.; et al. Hepatic FATP5 expression is associated with histological progression and loss of hepatic fat in NAFLD patients. J. Gastroenterol. 2020, 55, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Moser, A.E.; Goldfischer, S.; Moser, H.W. Lignoceric acid is oxidized in the peroxisome: Implications for the Zellweger cerebro-hepato-renal syndrome and adrenoleukodystrophy. Proc. Natl. Acad. Sci. USA 1984, 81, 4203–4207. [Google Scholar] [CrossRef] [Green Version]
- Baarine, M.; Beeson, C.; Singh, A.; Singh, I. ABCD1 deletion-induced mitochondrial dysfunction is corrected by SAHA: Implication for adrenoleukodystrophy. J. Neurochem. 2015, 133, 380–396. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [Green Version]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Tanaka, M.; Okada, H.; Hashimoto, Y.; Kumagai, M.; Nishimura, H.; Oda, Y.; Fukui, M. Relationship between nonalcoholic fatty liver disease and muscle quality as well as quantity evaluated by computed tomography. Liver Int. 2020, 40, 120–130. [Google Scholar] [CrossRef]
- Aller, R.; Fernandez-Rodriguez, C.; Lo Iacono, O.; Banares, R.; Abad, J.; Carrion, J.A.; Garcia-Monzon, C.; Caballeria, J.; Berenguer, M.; Rodriguez-Peralvarez, M.; et al. Consensus document. Management of non-alcoholic fatty liver disease (NAFLD). Clinical practice guideline. Gastroenterol Hepatol. 2018, 41, 328–349. [Google Scholar] [CrossRef]
- Torres-Fuentes, C.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. A natural solution for obesity: Bioactives for the prevention and treatment of weight gain. A review. Nutr. Neurosci. 2015, 18, 49–65. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Zeybel, M.; Altay, O.; Arif, M.; Li, X.; Yang, H.; Fredolini, C.; Akyildiz, M.; Saglam, B.; Gonenli, M.G.; Ural, D. Combined Metabolic Activators Reduces Liver Fat in Nonalcoholic Fatty Liver Disease Patients. medRxiv 2021. [Google Scholar]
- Altay, O.; Arif, M.; Li, X.; Yang, H.; Aydin, M.; Alkurt, G.; Kim, W.; Akyol, D.; Zhang, C.; Dinler-Doganay, G.; et al. Combined Metabolic Activators Accelerates Recovery in Mild-to-Moderate COVID-19. Adv. Sci. 2021, 8, e2101222. [Google Scholar] [CrossRef] [PubMed]
- Yulug, B.; Altay, O.; Li, X.; Hanoglu, L.; Cankaya, S.; Lam, S.; Yang, H.; Coskun, E.; İdil, E.; Nogaylar, R. Combined Metabolic Activators Improve Cognitive Functions without Altering Motor Scores in Parkinson’s Disease. medRxiv 2021. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
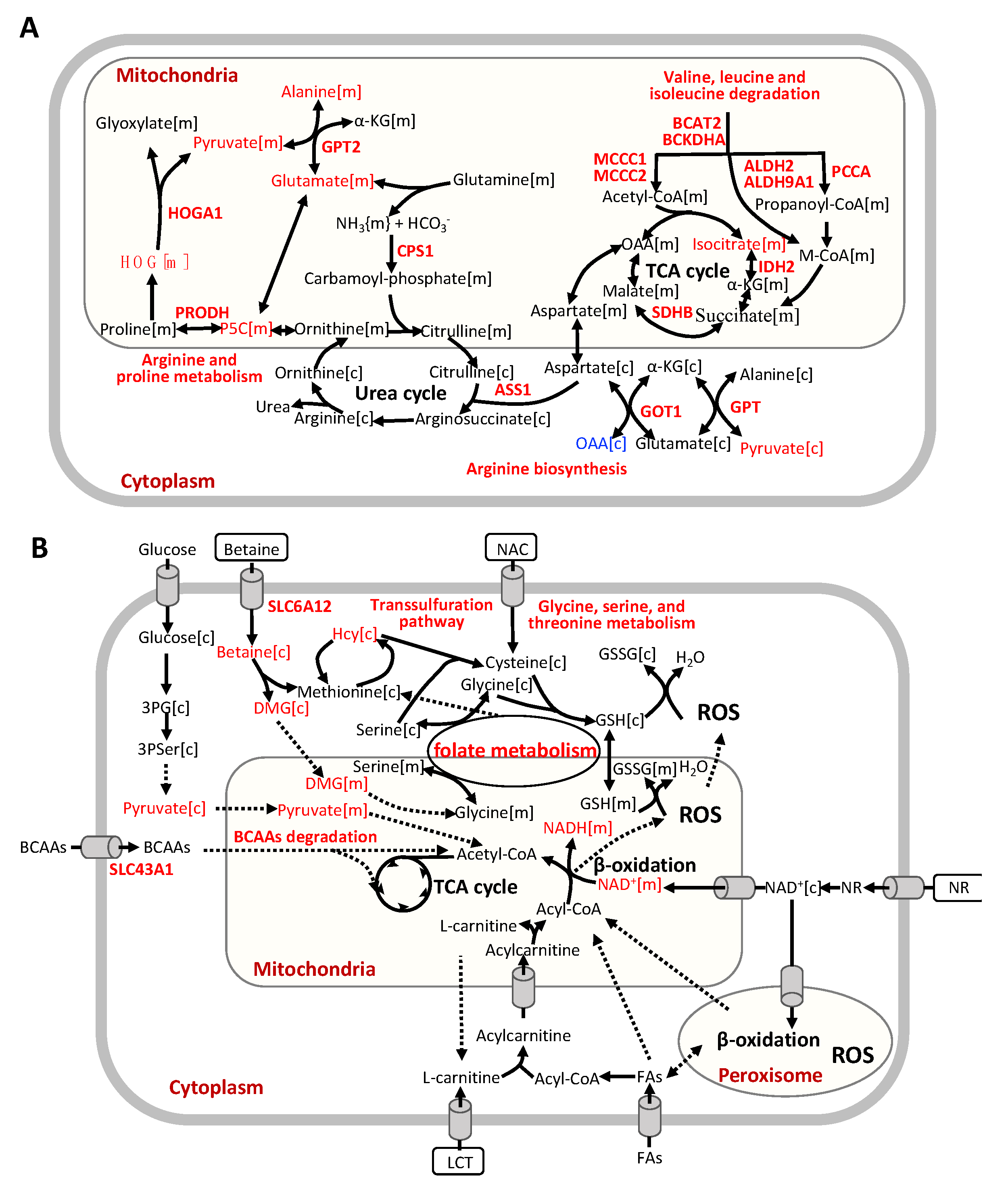{kind=link}
| Control (n = 9) | HFD (n = 30) | |
|---|---|---|
| Cumulative Food Intake (kcal) | 205.45 ± 14.08 | 194.73 ± 15.73 |
| Biometric variables | ||
| Body weight (g) | 123.05 ± 5.14 | 121.16 ± 7.16 |
| Fat mass (%) | 11.94 ± 3.20 | 14.10 ± 2.42 |
| Lean mass (%) | 84.89 ± 3.26 | 84.12 ± 2.34 |
| Lean/fat ratio | 7.83 ± 3.22 | 6.19 ± 1.41 |
| Serum parameters | ||
| CHOL (mM) | 3.90 ± 0.61 | 6.16 ± 0.91$ |
| Control (n = 9) | HFD (n = 10) | HFD + MI_D1 (n = 10) | HFD + MI_D2 (n =10) | |
|---|---|---|---|---|
| Cumulative food intake (kcal) * | 126.8 ± 14.4 | 111.1 ± 13.2 | 104.2 ± 15.9 | 95.2 ± 10.7 |
| Biometric variables | ||||
| Body weight (g) | 122.20 ± 5.62 | 126.57 ± 8.19 | 120.08 ± 10.32 | 116.28 ± 9.86 |
| Body weight gain (g) * | −0.85 ± 4.17 | 5.50 ± 6.61 | 0.27 ± 5.26 | −6.27± 5.38 b |
| Liver weight (g) * | 4.23 ± 0.30 | 5.10 ± 0.68 a | 4.69 ± 0.60 | 4.62 ± 0.61 |
| Liver weight (%) * | 3.49 ± 0.25 | 4.02 ± 0.30 a | 3.92 ± 0.43 | 3.98 ± 0.29 |
| MWAT (%) * | 0.88 ± 0.24 | 1.21 ± 0.24 a | 1.16 ± 0.29 | 1.14 ± 0.19 |
| MUS | 0.27 ± 0.03 | 0.25 ± 0.03 | 0.24 ± 0.03 | 0.24 ± 0.03 |
| Fat mass (%) | 11.28 ± 2.59 | 13.14 ± 2.78 | 14.84 ± 4.18 | 13.10 ± 2.87 |
| Lean mass (%) | 85.42 ± 2.39 | 85.74 ± 3.03 | 83.95 ± 4.04 | 85.22 ± 3.09 |
| Lean/fat mass ratio | 8.00 ± 2.13 | 6.84 ± 1.67 | 6.20 ± 2.19 | 6.83 ± 1.70 |
| Serum variables | ||||
| Glucose (mM) | 7.29 ± 1.50 | 9.42 ± 2.01 | 8.93 ± 1.42 | 9.27 ± 2.01 |
| Insulin (mIU/L) | 16.69 ± 2.92 | 16.26 ± 3.61 | 16.17 ± 1.34 | 16.78 ± 3.43 |
| CHOL (mM) * | 2.87 ± 0.21 | 4.03 ± 0.65 a | 3.77 ± 0.60 | 4.44 ± 0.85 |
| HDL-C (mM) * | 2.57 ± 0.36 | 3.15 ± 0.50 | 2.87 ± 0.52 | 3.32 ± 0.60 |
| LDL-C (mM) * | 0.90 ± 0.20 | 1.38 ± 0.32 a | 1.22 ± 0.24 | 1.34 ± 0.28 |
| TG (mM) | 1.07 ± 0.30 | 1.15 ± 0.53 | 0.96 ± 0.31 | 0.82 ± 0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Mayneris-Perxachs, J.; Boqué, N.; del Bas, J.M.; Arola, L.; Yuan, M.; Türkez, H.; Uhlén, M.; Borén, J.; Zhang, C.; et al. Combined Metabolic Activators Decrease Liver Steatosis by Activating Mitochondrial Metabolism in Hamsters Fed with a High-Fat Diet. Biomedicines 2021, 9, 1440. https://doi.org/10.3390/biomedicines9101440
Yang H, Mayneris-Perxachs J, Boqué N, del Bas JM, Arola L, Yuan M, Türkez H, Uhlén M, Borén J, Zhang C, et al. Combined Metabolic Activators Decrease Liver Steatosis by Activating Mitochondrial Metabolism in Hamsters Fed with a High-Fat Diet. Biomedicines. 2021; 9(10):1440. https://doi.org/10.3390/biomedicines9101440
Chicago/Turabian StyleYang, Hong, Jordi Mayneris-Perxachs, Noemí Boqué, Josep M. del Bas, Lluís Arola, Meng Yuan, Hasan Türkez, Mathias Uhlén, Jan Borén, Cheng Zhang, and et al. 2021. "Combined Metabolic Activators Decrease Liver Steatosis by Activating Mitochondrial Metabolism in Hamsters Fed with a High-Fat Diet" Biomedicines 9, no. 10: 1440. https://doi.org/10.3390/biomedicines9101440
APA StyleYang, H., Mayneris-Perxachs, J., Boqué, N., del Bas, J. M., Arola, L., Yuan, M., Türkez, H., Uhlén, M., Borén, J., Zhang, C., Mardinoglu, A., & Caimari, A. (2021). Combined Metabolic Activators Decrease Liver Steatosis by Activating Mitochondrial Metabolism in Hamsters Fed with a High-Fat Diet. Biomedicines, 9(10), 1440. https://doi.org/10.3390/biomedicines9101440