
A Comparative Analysis of Punicalagin Interaction with PDIA1 and PDIA3 by Biochemical and Computational Approaches

 ,
,  , , , and
, , , and 
Abstract
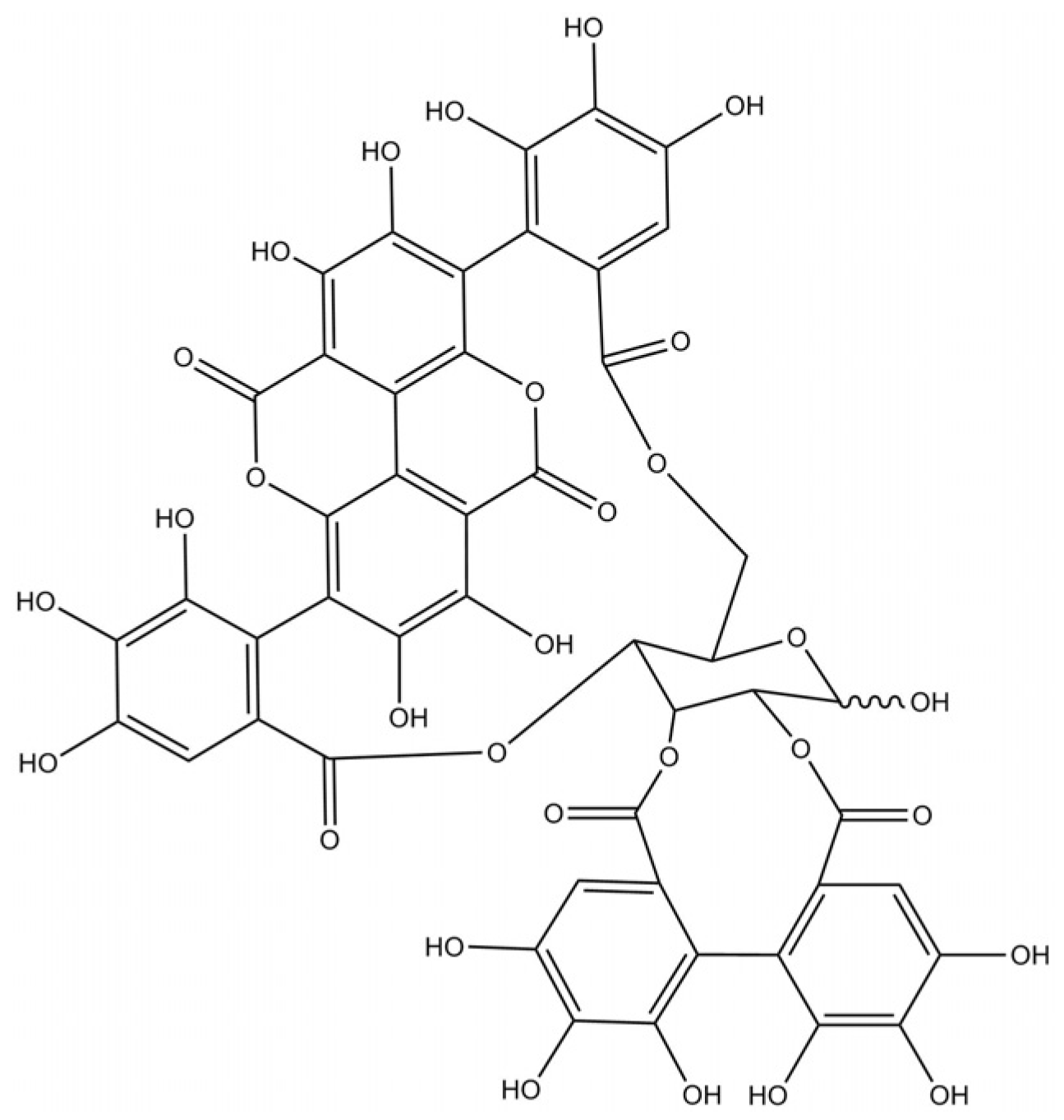
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Recombinant Proteins: Production and Purification
2.3. Measurements of Tryptophan Fluorescence Quenching
2.4. Isothermal Titration Calorimetry
2.5. Differential Scanning Calorimetry
2.6. Measurement of PDIAs Disulfide Reductase Activity
3. Results
3.1. Biochemical Studies
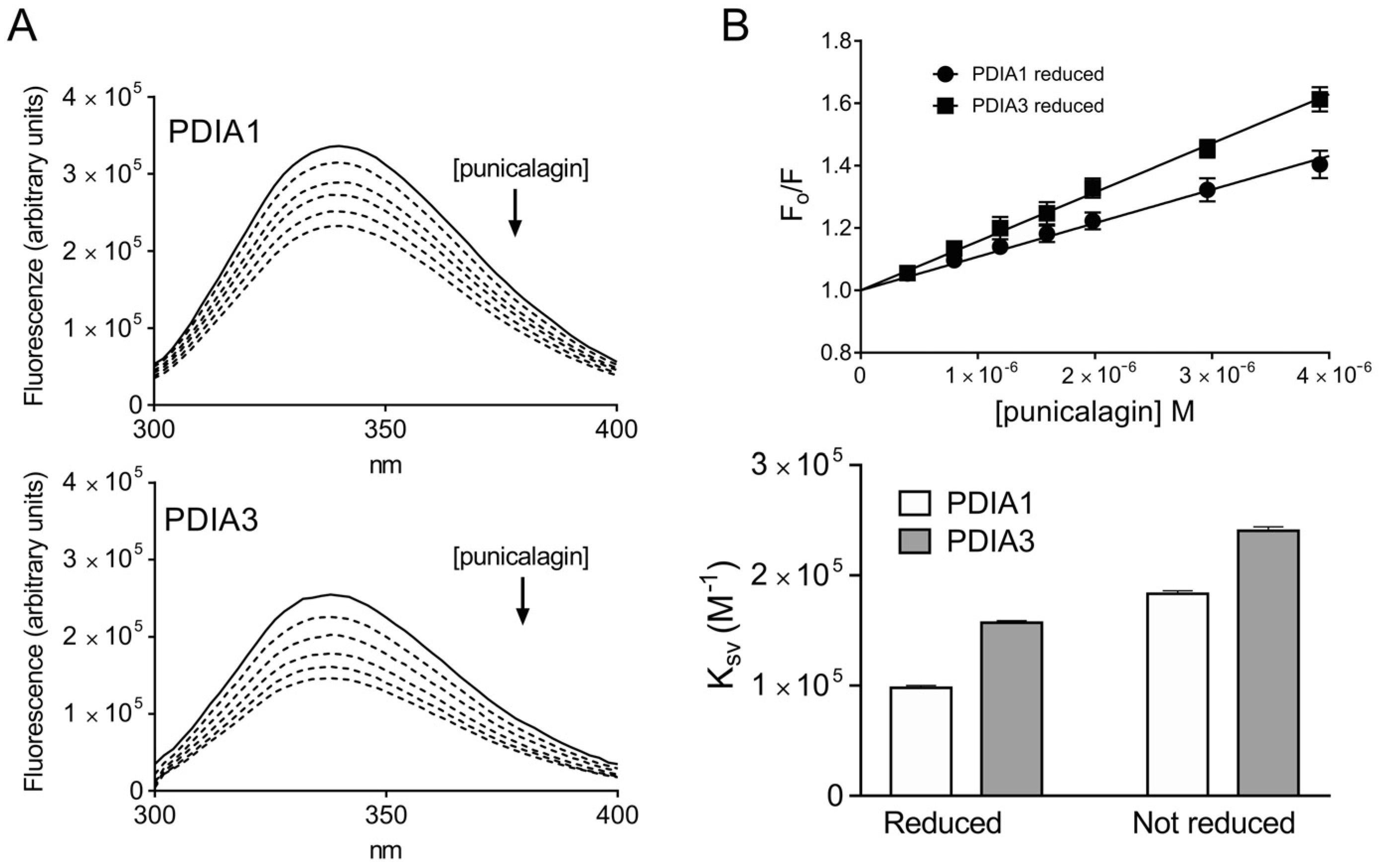
3.1.1. Assessment of PDIA-Punicalagin Interactions by Intrinsic Fluorescence Spectroscopy
3.1.2. Analysis of PDIA-Punicalagin Interactions by Isothermal Titration Calorimetry
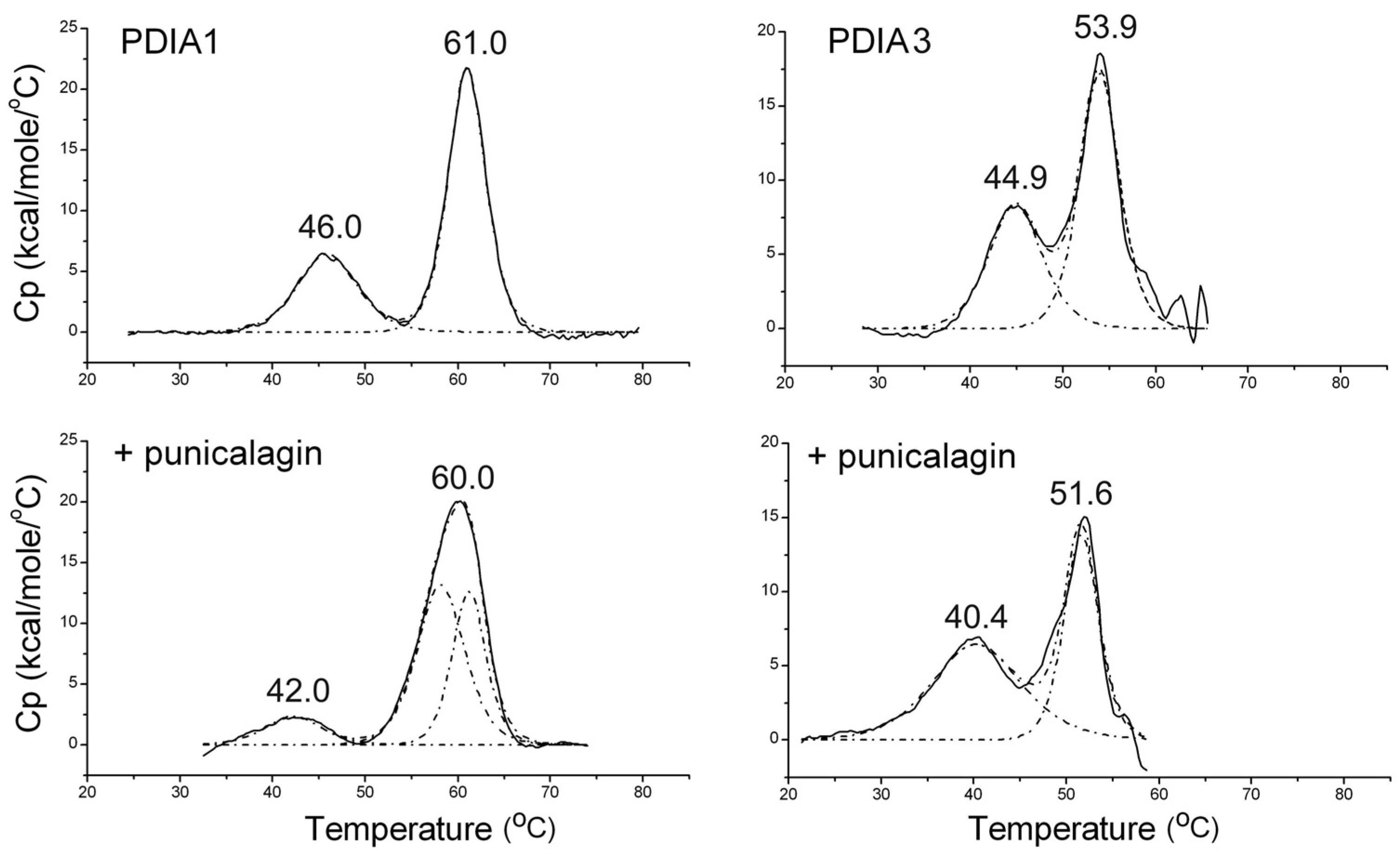
3.1.3. Thermally Induced Transitions of PDIAs upon Punicalagin Binding
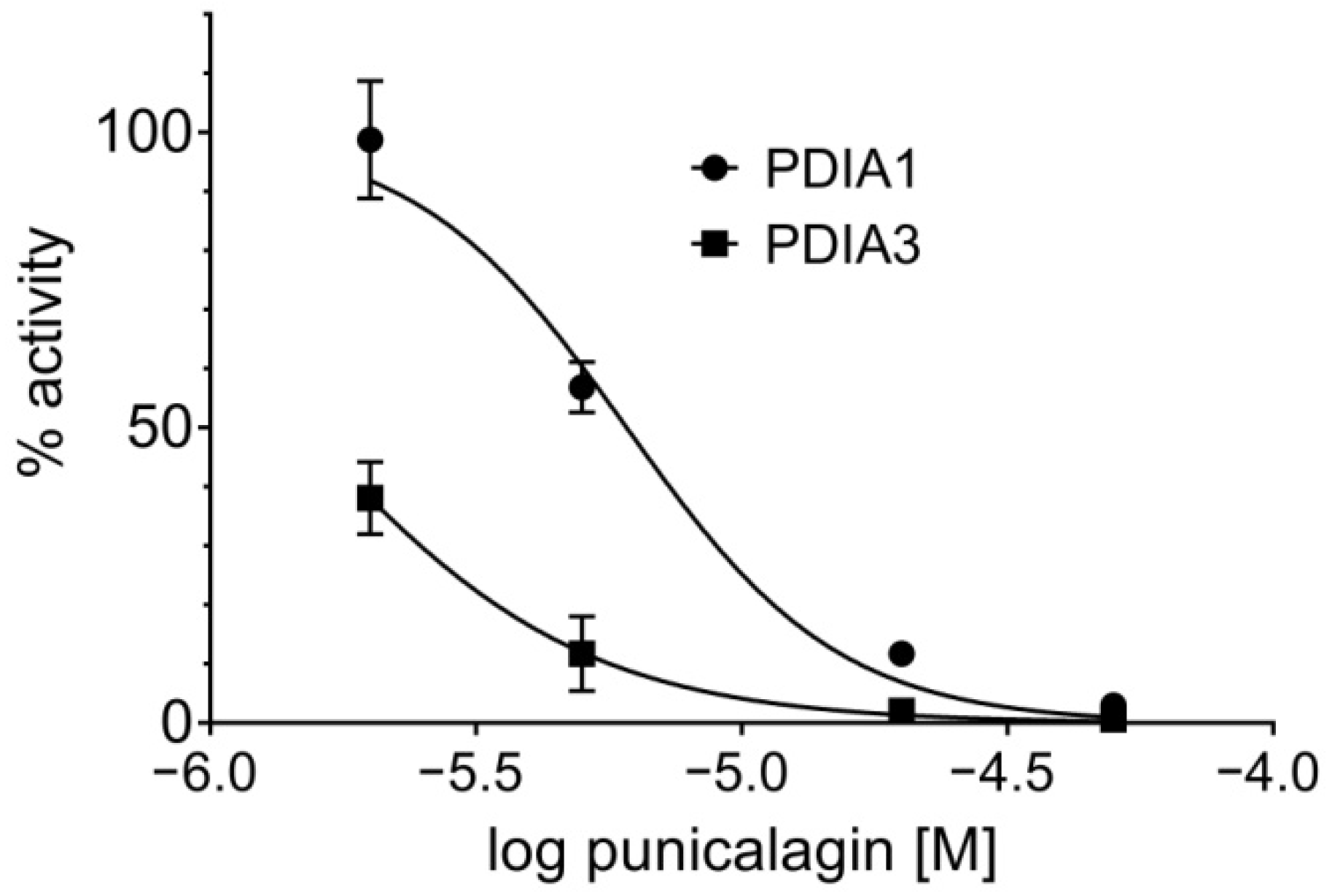
3.1.4. Punicalagin Effect on PDIA3 and PDIA1 Reductase Activity
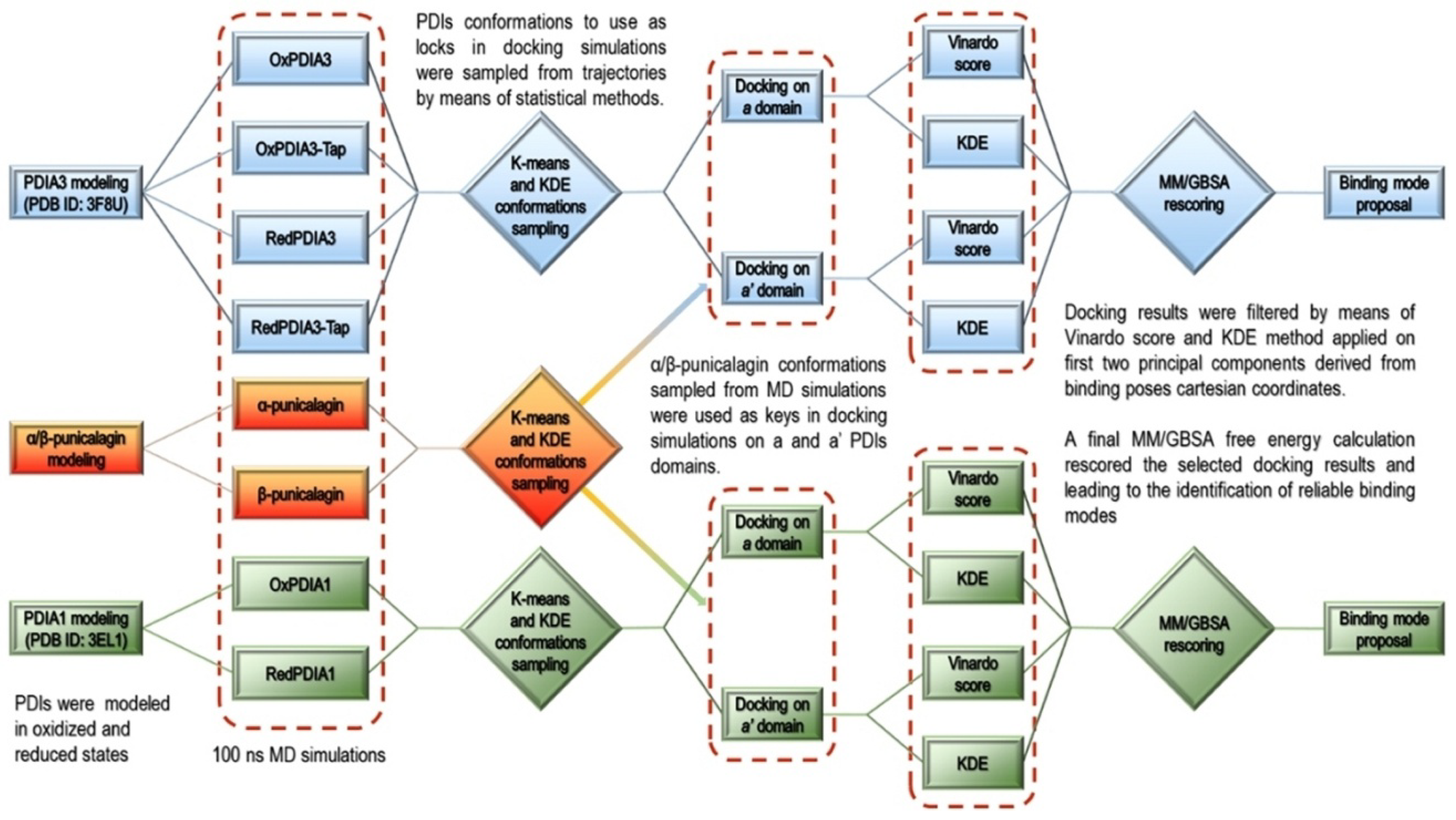
3.2. Computational Studies
- Modeling of the PDIAs crystal structures in oxidized and reduced states;
- MD simulations of the modeled PDIAs;
- Analysis of the MD trajectories and conformations’ sampling for the subsequent ensemble molecular docking (cross-docking) [39] simulations;
- α and β-punicalagin MD-based conformational analysis for the molecular docking into a and a’ domains of PDIAs sampled conformations;
- Analysis of the molecular docking results by means of statistical techniques and docking score ranking allowed binding poses selection for final rescoring with MM/GBSA [40].
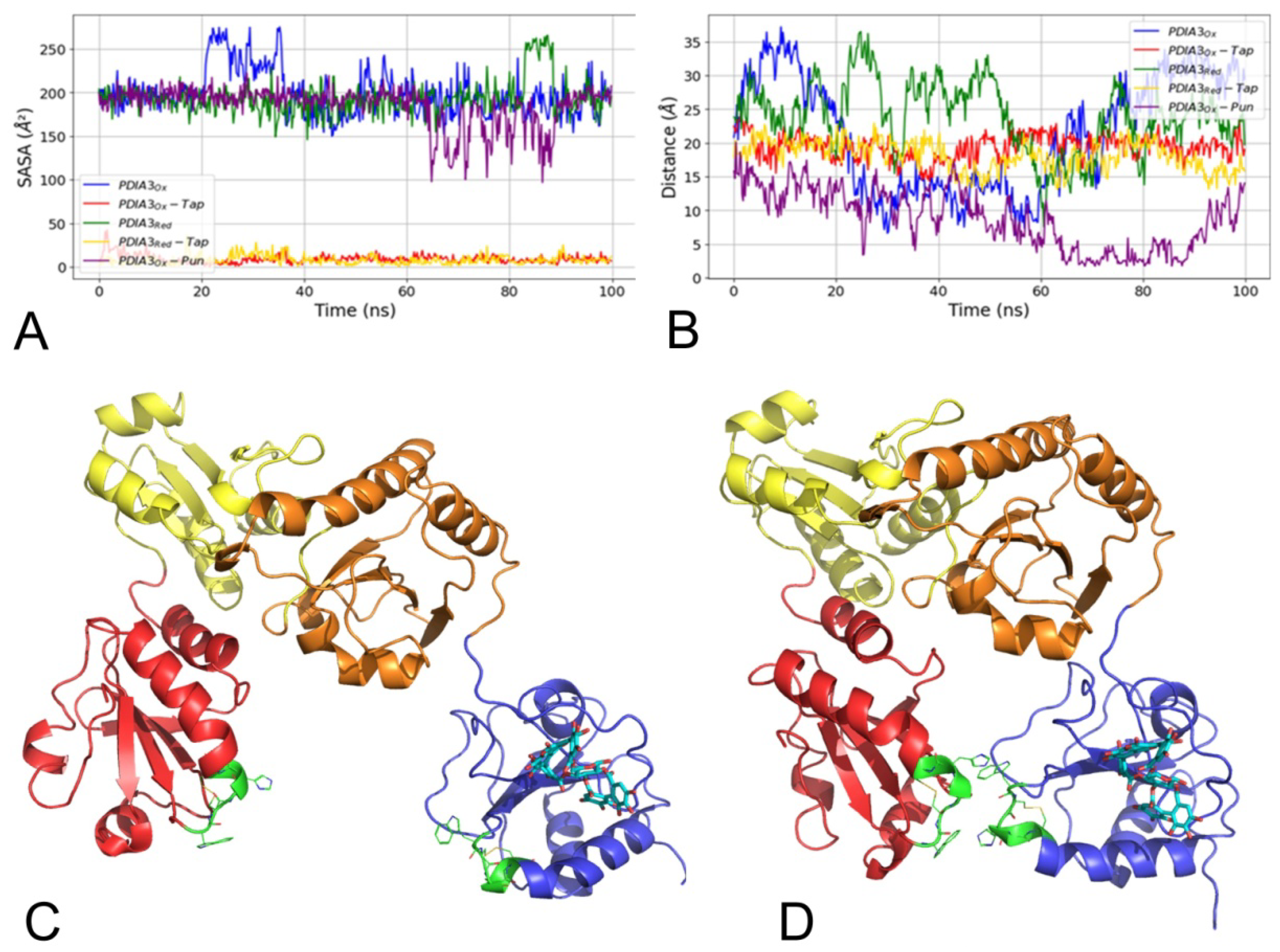
3.2.1. Molecular Dynamics Simulations
3.2.2. Molecular Docking Simulations
3.2.3. Punicalagin Binding Mode Selection by Free Energy Calculations
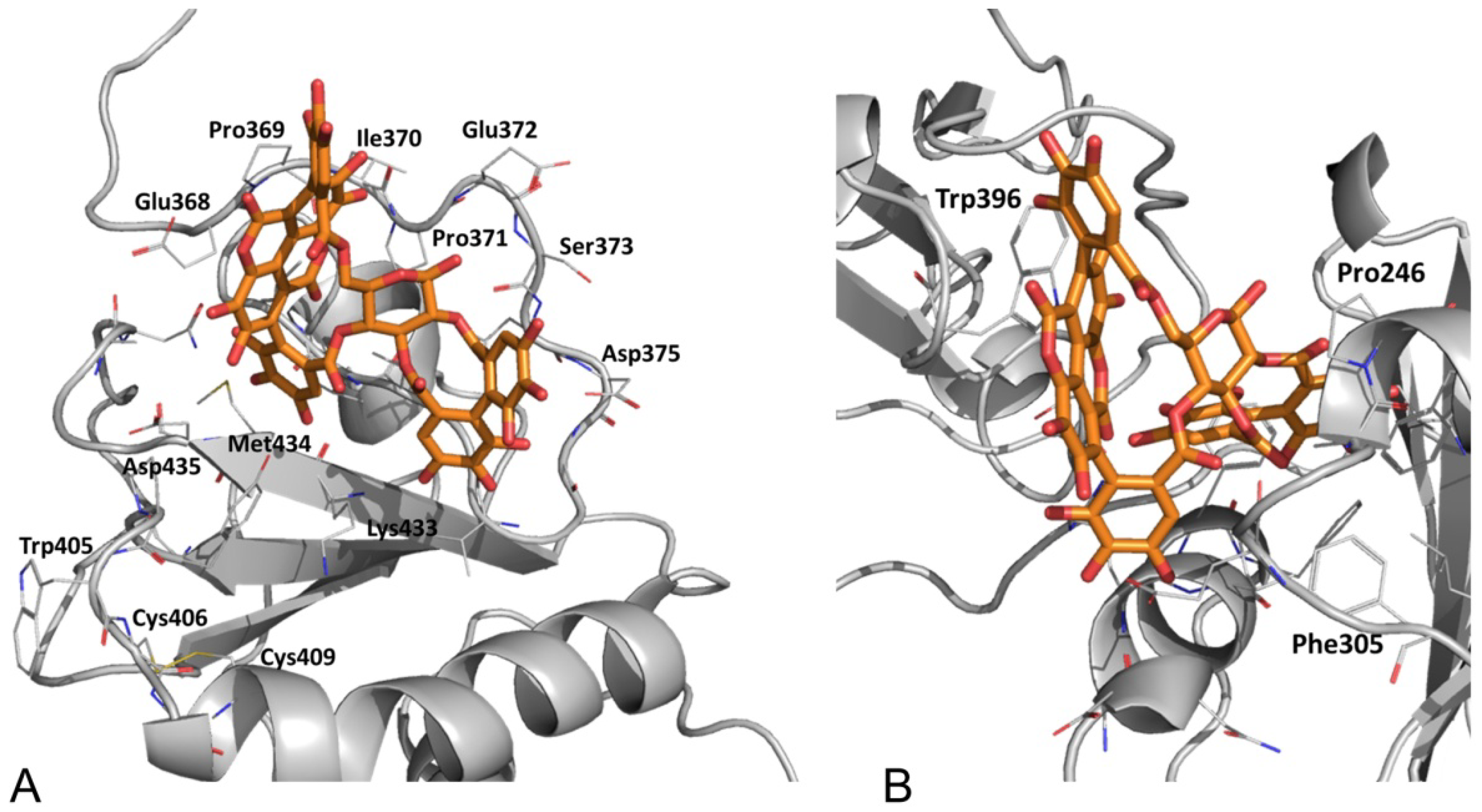
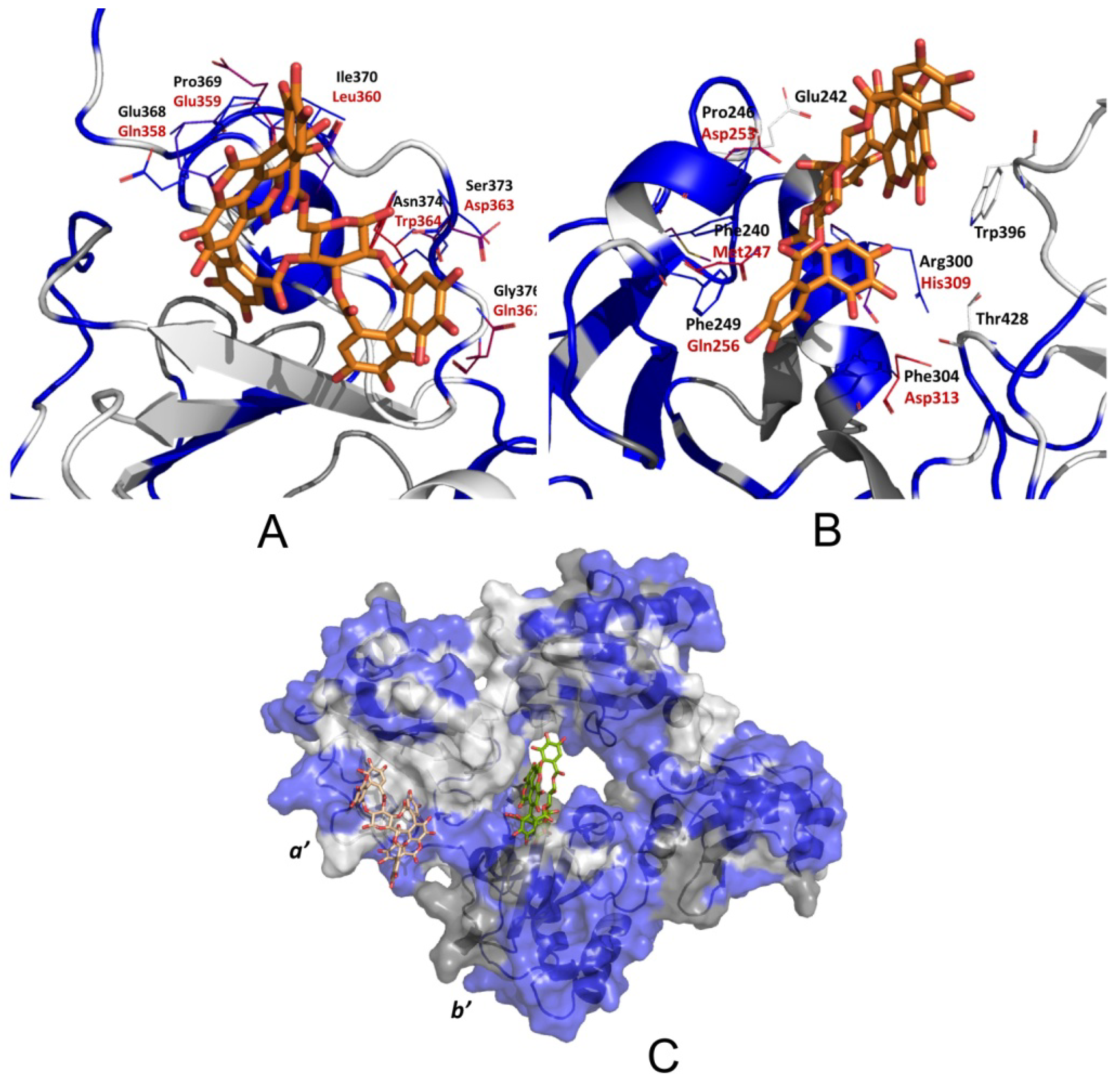
3.2.4. Punicalagin Binding Mode on PDIA3 Refinement by MD Simulation
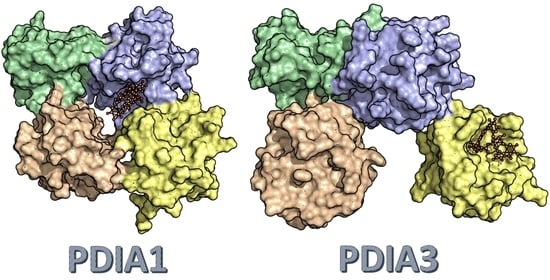
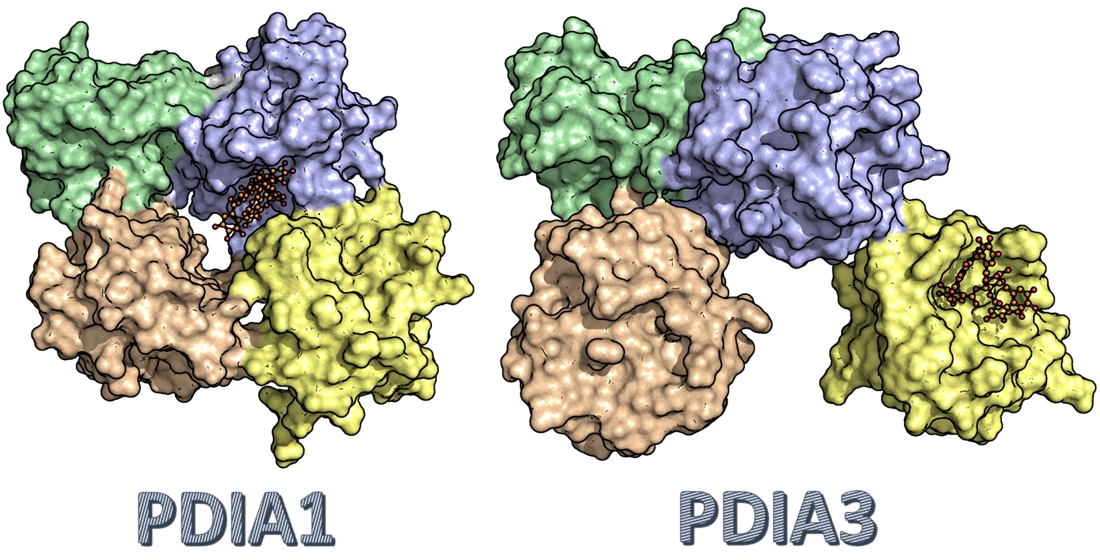
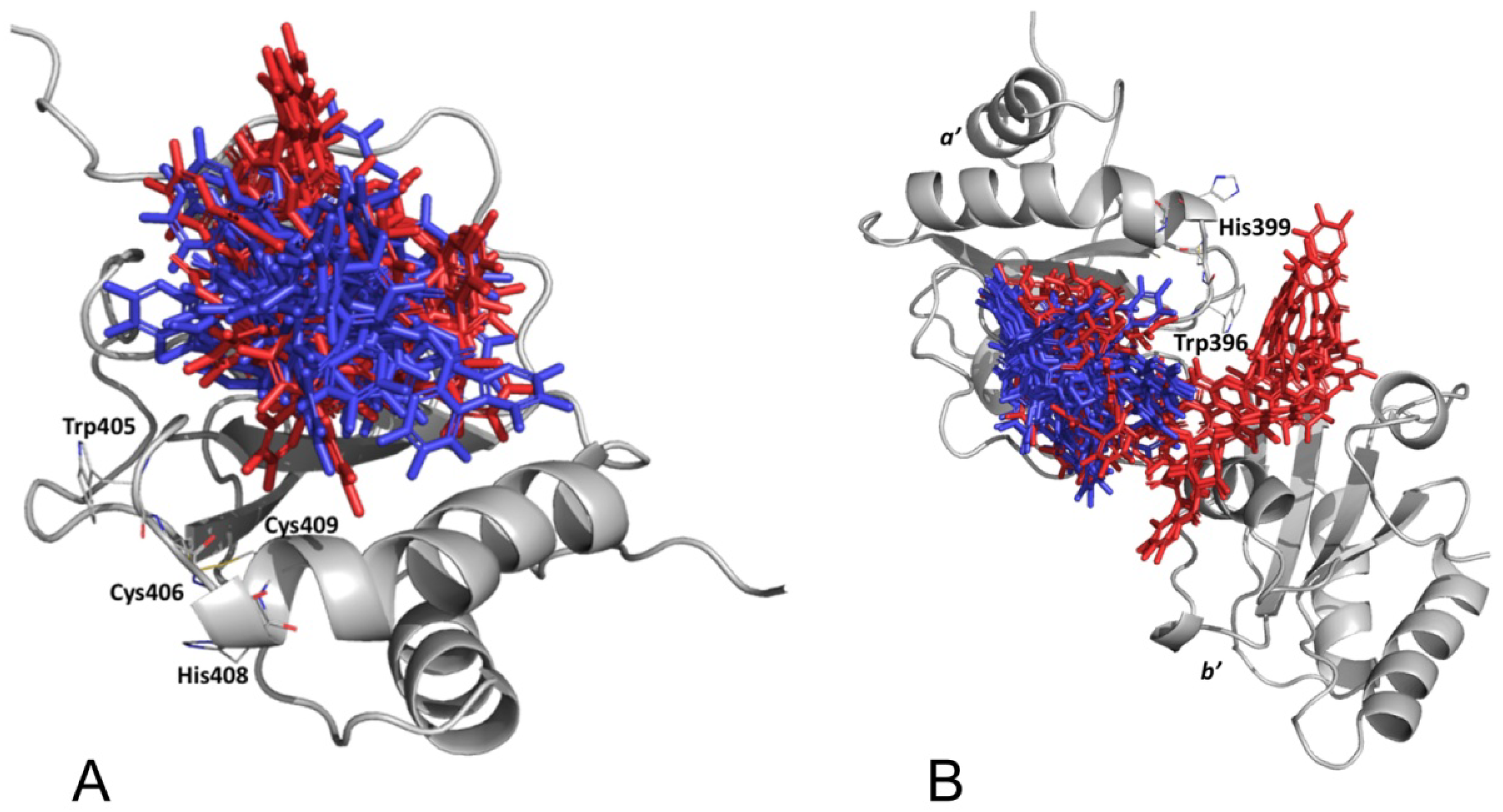
3.2.5. PDIA3/PDIA1 β-Punicalagin Binding Mode Comparison
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PDI | protein disulfide isomerase |
| IAV | influenza A virus |
| ITC | isothermal titration calorimetry |
| DSC | differential scanning calorimetry |
| MD | molecular dynamics |
| RMSD | root mean square deviation |
| KDE | kernel density estimation |
| MDCOM | mean distance between poses’ center of mass |
| SASA | solvent accessible surface area |
| RMSF | root mean square fluctuation |
References
- Hettinghouse, A.; Liu, R.; Liu, C. Multifunctional Molecule ERp57: From Cancer to Neurodegenerative Diseases. Pharmacol. Ther. 2018, 181, 34–48. [Google Scholar] [CrossRef]
- Frickel, E.M.; Frei, P.; Bouvier, M.; Stafford, W.F.; Helenius, A.; Glockshuber, R.; Ellgaard, L. ERp57 Is a Multifunctional Thiol-Disulfide Oxidoreductase. J. Biol. Chem. 2004, 279, 18277–18287. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Wearsch, P.A.; Peaper, D.R.; Cresswell, P.; Reinisch, K.M. Insights into MHC Class I Peptide Loading from the Structure of the Tapasin-ERp57 Thiol Oxidoreductase Heterodimer. Immunity 2009, 30, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Silvennoinen, L.; Myllyharju, J.; Ruoppolo, M.; Orrù, S.; Caterino, M.; Kivirikko, K.I.; Koivunen, P. Identification and Characterization of Structural Domains of Human ERp57: Association with Calreticulin Requires Several Domains. J. Biol. Chem. 2004, 279, 13607–13615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turano, C.; Gaucci, E.; Grillo, C.; Chichiarelli, S. ERp57/GRP58: A Protein with Multiple Functions. Cell. Mol. Biol. Lett. 2011, 16, 539–563. [Google Scholar] [CrossRef] [PubMed]
- Jessop, C.E.; Chakravarthi, S.; Garbi, N.; Hämmerling, G.J.; Lovell, S.; Bulleid, N.J. ERp57 Is Essential for Efficient Folding of Glycoproteins Sharing Common Structural Domains. EMBO J. 2007, 26, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.G.; Campbell, E.C.; Lynch, S.; Wong, V.; Antoniou, A.N.; Powis, S.J. Major Histocompatibility Complex Class I-ERp57-Tapasin Interactions within the Peptide-Loading Complex. J. Biol. Chem. 2007, 282, 17587–17593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemere, I.; Garbi, N.; Hammerling, G.; Hintze, K.J. Role of the 1,25D 3-MARRS Receptor in the 1,25(OH) 2D 3-Stimulated Uptake of Calcium and Phosphate in Intestinal Cells. Steroids 2012, 77, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Chichiarelli, S.; Gaucci, E.; Ferraro, A.; Grillo, C.; Altieri, F.; Cocchiola, R.; Arcangeli, V.; Turano, C.; Eufemi, M. Role of ERp57 in the Signaling and Transcriptional Activity of STAT3 in a Melanoma Cell Line. Arch. Biochem. Biophys. 2010, 494, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, N.; Korwin-Mihavics, B.R.; Nakada, E.M.; Bruno, S.R.; Heppner, D.E.; Chapman, D.G.; Hoffman, S.M.; van der Vliet, A.; Suratt, B.T.; Dienz, O.; et al. Lung Epithelial Protein Disulfide Isomerase A3 (PDIA3) Plays an Important Role in Influenza Infection, Inflammation, and Airway Mechanics. Redox Biol. 2019, 101129. [Google Scholar] [CrossRef]
- Kondo, R.; Ishino, K.; Wada, R.; Takata, H.; Peng, W.X.; Kudo, M.; Kure, S.; Kaneya, Y.; Taniai, N.; Yoshida, H.; et al. Downregulation of Protein Disulfide-Isomerase A3 Expression Inhibits Cell Proliferation and Induces Apoptosis through STAT3 Signaling in Hepatocellular Carcinoma. Int. J. Oncol. 2019, 54, 1409–1421. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhao, X.; Chang, S.; Li, Y.; Guo, M.; Guan, Y. ERp57-Small Interfering RNA Silencing Can Enhance the Sensitivity of Drug-Resistant Human Ovarian Cancer Cells to Paclitaxel. Int. J. Oncol. 2019, 54, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Fu, P.; Dou, J.; Wang, N. Downregulation of PDIA3 Inhibits Proliferation and Invasion of Human Acute Myeloid Leukemia Cells. OncoTargets Ther. 2018, 11, 2925–2935. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Wen, C.; Peng, Z.; Shao, Y.Y.; Hu, L.; Li, S.; Li, C.; Zhou, H.H. P4HB and PDIA3 Are Associated with Tumor Progression and Therapeutic Outcome of Diffuse Gliomas. Oncol. Rep. 2018, 39, 501–510. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, J.; Shi, Q.; Chao, Y.; Di, Y.; Sun, J.; Zhang, J.; Huang, L.; Guo, H.; He, C. Expression of Protein Disulfide Isomerase A3 Precursor in Colorectal Cancer. OncoTargets Ther. 2018, 11, 4159–4166. [Google Scholar] [CrossRef] [Green Version]
- Giamogante, F.; Marrocco, I.; Cervoni, L.; Eufemi, M.; Chichiarelli, S.; Altieri, F. Punicalagin, an Active Pomegranate Component, Is a New Inhibitor of PDIA3 Reductase Activity. Biochimie 2018, 147, 122–129. [Google Scholar] [CrossRef]
- Khatib, M.; Innocenti, M.; Giuliani, C.; Al-Tamimi, A.; Romani, A.; Mulinacci, N. Mesocarp and Exocarp of Laffan and Wonderful Pomegranate Varieties: By-Products as a Source of Ellagitannins. Int. J. Food Nutr. Sci. 2017, 4, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.S.; Li, J.H.; Liu, T.Y.; Lin, T.C. Folk Medicine Terminalia Catappa and Its Major Tannin Component, Punicalagin, Are Effective against Bleomycin-Induced Genotoxicity in Chinese Hamster Ovary Cells. Cancer Lett. 2000, 152, 115–122. [Google Scholar] [CrossRef]
- Marzouk, M.S.A.; El-Toumy, S.A.A.; Moharram, F.A.; Shalaby, N.M.M.; Ahmed, A.A.E. Pharmacologically Active Ellagitannins from Terminalia Myriocarpa. Planta Med. 2002, 68, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Asres, K.; Bucar, F.; Knauder, E.; Yardley, V.; Kendrick, H.; Croft, S.L. In Vitro Antiprotozoal Activity of Extract and Compounds from the Stem Bark of Combretum Molle. Phyther. Res. 2001, 15, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.; Sestili, P.; Akhtar, S. Pomegranate Peel and Fruit Extracts: A Review of Potential Anti-Inflammatory and Anti-Infective Effects. J. Ethnopharmacol. 2012, 143, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Turrini, E.; Ferruzzi, L.; Fimognari, C. Potential Effects of Pomegranate Polyphenols in Cancer Prevention and Therapy. Oxid. Med. Cell. Longev. 2015, 2015, 938475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, X.; Wang, C.C. Protein Disulfide-Isomerase, a Folding Catalyst and a Redox-Regulated Chaperone. Free Radic. Biol. Med. 2015, 83, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Määttänen, P.; Thomas, D.Y.; Gehring, K. A Structural Overview of the PDI Family of Proteins. FEBS J. 2010, 277, 3924–3936. [Google Scholar] [CrossRef] [PubMed]
- Coppari, S.; Altieri, F.; Ferraro, A.; Chichiarelli, S.; Eufemi, M.; Turano, C. Nuclear Localization and DNA Interaction of Protein Disulfide Isomerase ERp57 in Mammalian Cells. J. Cell. Biochem. 2002, 85, 325–333. [Google Scholar] [CrossRef]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.H.; Wang, C.C.; Ruddock, L.W. Two Endoplasmic Reticulum PDI Peroxidases Increase the Efficiency of the Use of Peroxide during Disulfide Bond Formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef]
- Giamogante, F.; Marrocco, I.; Romaniello, D.; Eufemi, M.; Chichiarelli, S.; Altieri, F. Comparative Analysis of the Interaction between Different Flavonoids and PDIA3. Oxid. Med. Cell. Longev. 2016, 2016, 4518281. [Google Scholar] [CrossRef]
- Raturi, A.; Mutus, B. Characterization of Redox State and Reductase Activity of Protein Disulfide Isomerase under Different Redox Environments Using a Sensitive Fluorescent Assay. Free Radic. Biol. Med. 2007, 43, 62–70. [Google Scholar] [CrossRef]
- Trnková, L.; Ricci, D.; Grillo, C.; Colotti, G.; Altieri, F. Green Tea Catechins Can Bind and Modify ERp57/PDIA3 Activity. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 2671–2682. [Google Scholar] [CrossRef]
- Ghisaidoobe, A.B.T.; Chung, S.J. Intrinsic Tryptophan Fluorescence in the Detection and Analysis of Proteins: A Focus on Förster Resonance Energy Transfer Techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Weber, G. Quenching of Fluorescence by Oxygen. Probe for Structural Fluctuations in Macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Quenching of Fluorescence; Springer: New York, NY, USA, 2006; pp. 277–330. [Google Scholar] [CrossRef]
- Ware, W.R. Oxygen Quenching of Fluorescence in Solution: An Experimental Study of the Diffusion Process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Bi, S.; Song, D.; Tian, Y.; Zhou, X.; Liu, Z.; Zhang, H. Molecular Spectroscopic Study on the Interaction of Tetracyclines with Serum Albumins. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2005, 61, 629–636. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Sun, Y.; Zhang, Y.; Liu, H.; Cheng, J.; Bi, S.; Zhang, H. Study of Interaction between Protein and Main Active Components in Citrus aurantium L. by Optical Spectroscopy. J. Lumin. 2010, 130, 270–279. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Ren, J.; Fang, J.; Ke, H.; Gong, W.; Feng, W.; Wang, C.C. Structural Insights into the Redox-Regulated Dynamic Conformations of Human Protein Disulfide Isomerase. Antioxid. Redox Signal. 2013, 19, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, H.A.; Mutus, B. Protein Disulfide Isomerase a Multifunctional Protein with Multiple Physiological Roles. Front. Chem. 2014, 2, 70. [Google Scholar] [CrossRef] [Green Version]
- Ragno, R.; Frasca, S.; Manetti, F.; Brizzi, A.; Massa, S. HIV-Reverse Transcriptase Inhibition: Inclusion of Ligand-Induced Fit by Cross-Docking Studies. J. Med. Chem. 2005, 48, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Bastos, T.M.; Botelho, M.; Soares, P.; Franco, C.H.; Alc, L.; Antonini, L.; Sabatino, M.; Mautone, N.; Freitas-junior, L.H.; Moraes, C.B.; et al. Identification of Inhibitors to Trypanosoma Cruzi Sirtuins Based on Compounds Developed to Human Enzymes. Int J. Mol. Sci. 2020, 21, 3659. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.B.; Desmond, J.L.; Byrne, L.J.; Heal, J.W.; Howard, M.J.; Sanghera, N.; Walker, K.L.; Wallis, A.K.; Wells, S.A.; Williamson, R.A.; et al. ‘Something in the Way She Moves’: The Functional Significance of Flexibility in the Multiple Roles of Protein Disulfide Isomerase (PDI). Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Römer, R.A.; Wells, S.A.; Emilio Jimenez-Roldan, J.; Bhattacharyya, M.; Vishweshwara, S.; Freedman, R.B. The Flexibility and Dynamics of Protein Disulfide Isomerase. Proteins Struct. Funct. Bioinform. 2016, 84, 1776–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Chen, S.; Wang, X.; Wang, L.; Wallis, A.K.; Freedman, R.B.; Wang, C.C. Plasticity of Human Protein Disulfide Isomerase: Evidence for Mobility around the x-Linker Region and Its Functional Significance. J. Biol. Chem. 2010, 285, 26788–26797. [Google Scholar] [CrossRef] [Green Version]
- Guyette, J.; Evangelista, B.; Tatulian, S.A.; Teter, K. Stability and Conformational Resilience of Protein Disulfide Isomerase. Biochemistry 2019, 58, 3572–3584. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA. Available online: https://pymol.org/2/ (accessed on 30 September 2021).
- Olsson, M.H.M.; SØndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical p K a Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Darby, N.J.; Creighton, T.E. Functional Properties of the Individual Thioredoxin-like Domains of Protein Disulfide Isomerase. Biochemistry 1995, 34, 11725–11735. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol. 2017, 13, 1–17. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Simmerling, C.; et al. Amber 2019; University of California: San Francisco, CA, USA, 2019. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 18, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.H.; Ferguson, D.M. Isothermal-Isobaric Molecular Dynamics Simulations with Monte Carlo Volume Sampling. Comput. Phys. Commun. 1995, 91, 283–289. [Google Scholar] [CrossRef]
- Lzaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin Stabilization of Molecular Dynamics. J. Chem. Phys. 2001, 144, 2090. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.R.; Millman, K.J.; vad der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.P. Least Squares Quantization in PCM. IEEE Trans. Inf. Theory 1982, 18, 129–137. [Google Scholar] [CrossRef]
- Scott, D.W. Multivariate Density Estimation: Theory, Practice, and Visualization, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J.P. RM1: A Reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KSV (M−1 × 103) | Kd (M) | |
|---|---|---|
| Reduced form | ||
| PDIA1 | 97.9 ± 2.1 | 11.9 × 10−6 |
| PDIA3 | 157.1 ± 1.9 | 10.0 × 10−6 |
| Not reduced form | ||
| PDIA1 | 183.2 ± 2.7 | 4.9 × 10−6 |
| PDIA3 | 240.4 ± 3.6 | 3.9 × 10−6 |
| Kd (10−6 M) | ΔH (kcal/mol) | TΔS (kcal/mol) | |
|---|---|---|---|
| PDIA1 | 1.0 ± 0.2 | −2.7 ± 0.4 | 5.6 ± 0.5 |
| PDIA3 | 1.2 ± 0.3 | −1.1 ± 0.2 | 6.8 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paglia, G.; Antonini, L.; Cervoni, L.; Ragno, R.; Sabatino, M.; Minacori, M.; Rubini, E.; Altieri, F. A Comparative Analysis of Punicalagin Interaction with PDIA1 and PDIA3 by Biochemical and Computational Approaches. Biomedicines 2021, 9, 1533. https://doi.org/10.3390/biomedicines9111533
Paglia G, Antonini L, Cervoni L, Ragno R, Sabatino M, Minacori M, Rubini E, Altieri F. A Comparative Analysis of Punicalagin Interaction with PDIA1 and PDIA3 by Biochemical and Computational Approaches. Biomedicines. 2021; 9(11):1533. https://doi.org/10.3390/biomedicines9111533
Chicago/Turabian StylePaglia, Giuliano, Lorenzo Antonini, Laura Cervoni, Rino Ragno, Manuela Sabatino, Marco Minacori, Elisabetta Rubini, and Fabio Altieri. 2021. "A Comparative Analysis of Punicalagin Interaction with PDIA1 and PDIA3 by Biochemical and Computational Approaches" Biomedicines 9, no. 11: 1533. https://doi.org/10.3390/biomedicines9111533
APA StylePaglia, G., Antonini, L., Cervoni, L., Ragno, R., Sabatino, M., Minacori, M., Rubini, E., & Altieri, F. (2021). A Comparative Analysis of Punicalagin Interaction with PDIA1 and PDIA3 by Biochemical and Computational Approaches. Biomedicines, 9(11), 1533. https://doi.org/10.3390/biomedicines9111533