
The Potential Role of Cytokine Storm Pathway in the Clinical Course of Viral Respiratory Pandemic

 ,
, 
 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
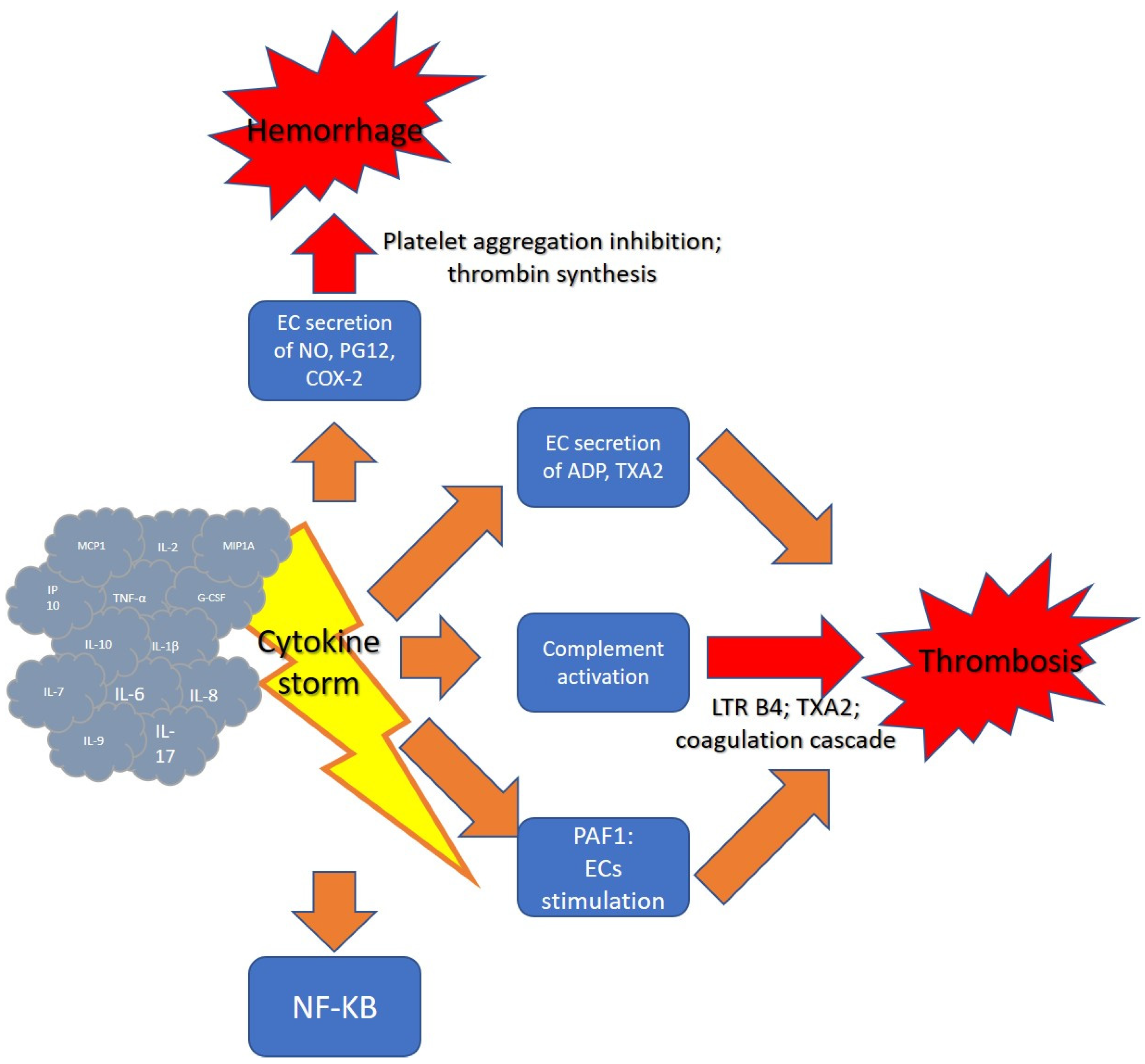
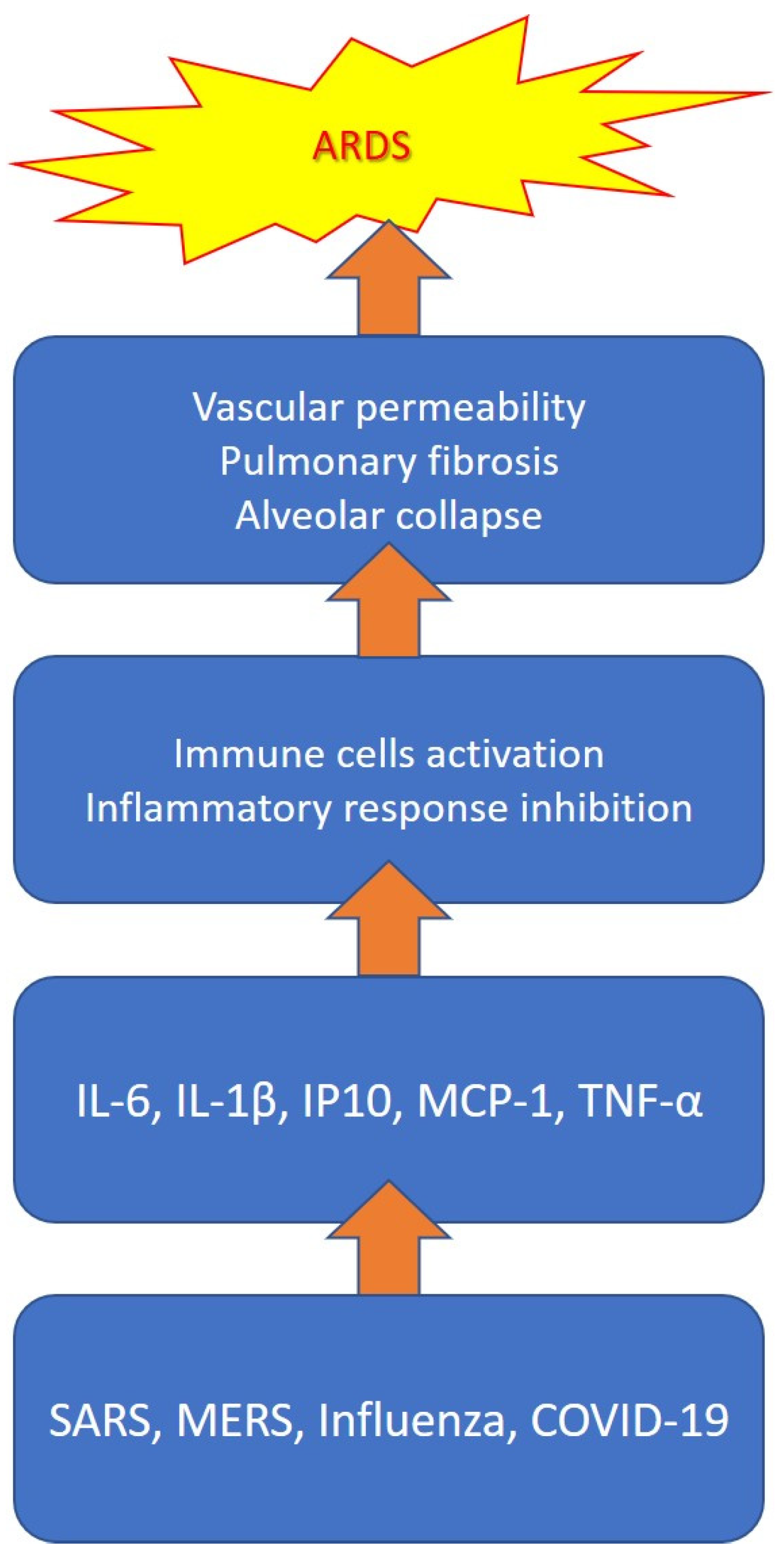
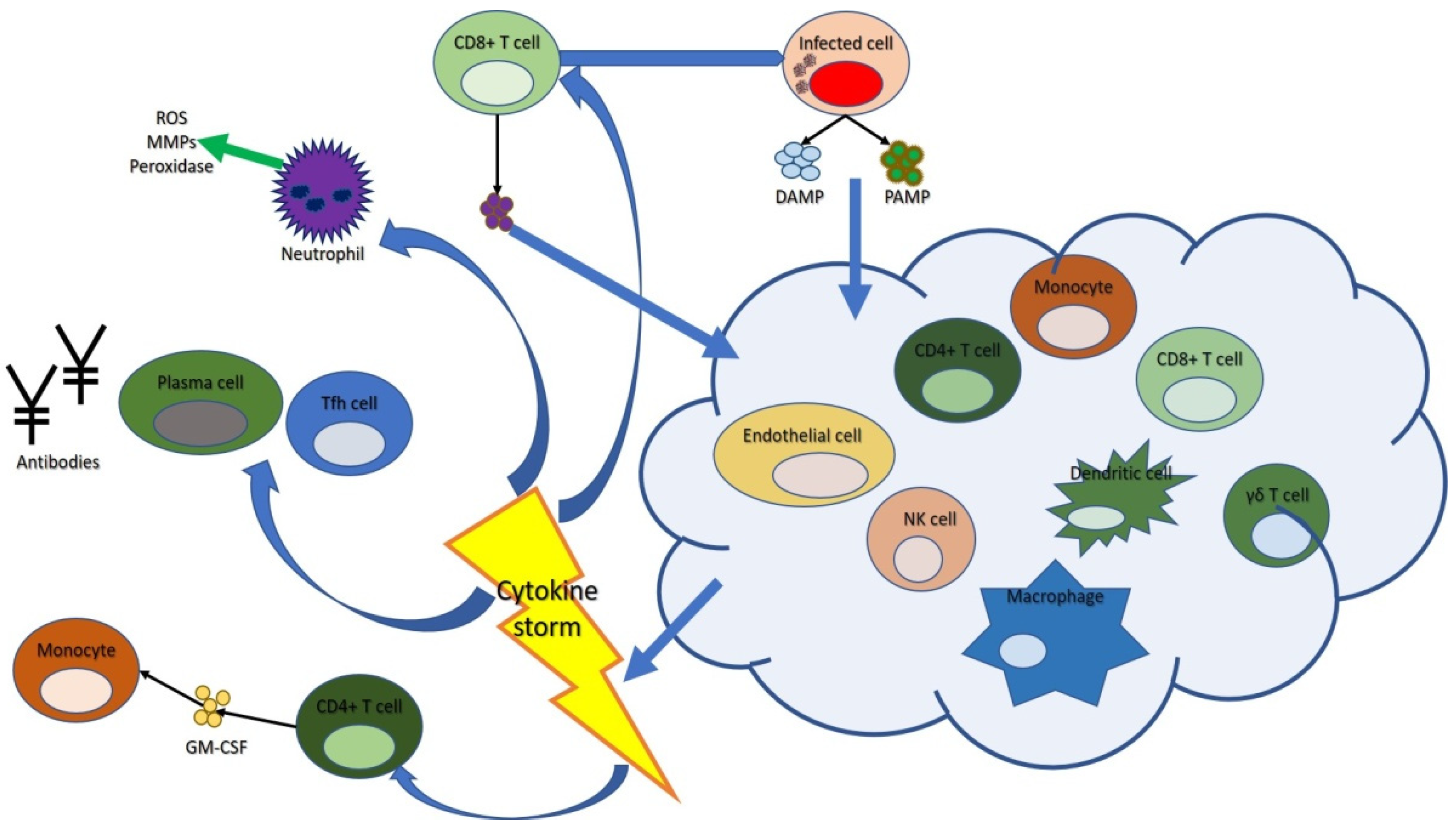
2.1. Cytokine Storm
2.2. Cytokine Storm and Hemostasis
2.3. COVID-19
2.4. SARS
2.5. MERS
2.6. H1N1 Influenza A
2.7. Spanish Flu
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Murdaca, G.; Di Gioacchino, M.; Greco, M.; Borro, M.; Paladin, F.; Petrarca, C.; Gangemi, S. Basophils and Mast Cells in COVID-19 Pathogenesis. Cells 2021, 10, 2754. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Ragab, D.; Eldin, H.S.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef] [PubMed]
- Khadke, S.; Ahmed, N.; Ahmed, N.; Ratts, R.; Raju, S.; Gallogly, M.; de Lima, M.; Sohail, M.R. Harnessing the immune system to overcome cytokine storm and reduce viral load in COVID-19: A review of the phases of illness and therapeutic agents. Virol. J. 2020, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Lukan, N. “Cytokine storm”, not only in COVID-19 patients. Mini-review. Immunol. Lett. 2020, 228, 38–44. [Google Scholar] [CrossRef]
- De La Rica, R.; Borges, M.; Gonzalez-Freire, M. COVID-19: In the Eye of the Cytokine Storm. Front. Immunol. 2020, 11, 2313. [Google Scholar] [CrossRef]
- Fara, A.; Mitrev, Z.; Rosalia, R.A.; Assas, B.M. Cytokine storm and COVID-19: A chronicle of pro-inflammatory cytokines. Open Biol. 2020, 10, 200160. [Google Scholar] [CrossRef]
- Del Turco, S.; Basta, G.; Lazzerini, G.; Evangelista, M.; Rainaldi, G.; Tanganelli, P.; Camera, M.; Tremoli, E.; De Caterina, R. Parallel decrease of tissue factor surface exposure and increase of tissue factor microparticle release by the n-3 fatty acid docosahexaenoate in endothelial cells. Thromb. Haemost. 2007, 98, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Cantan, B.; Luyt, C.-E.; Martin-Loeches, I. Influenza Infections and Emergent Viral Infections in Intensive Care Unit. Semin. Respir. Crit. Care Med. 2019, 40, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Aird, W.C. Endothelium and haemostasis. Hamostaseologie 2015, 35, 11–16. [Google Scholar] [CrossRef]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Platelet activation and prothrombotic mediators at the nexus of inflammation and atherosclerosis: Potential role of antiplatelet agents. Blood Rev. 2020, 45, 100694. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Garciá-Frade, L.J.; Lorente, J.A.; Garciá-Avello, A.; De Pablo, R.; Landín, L. Plasminogen activator inhibitor-1 levels determine the profibrinolytic response in disseminated intravascular coagulation. Am. J. Hematol. 1992, 41, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, S.M.K.; Bhat, B.V. Could stem cells be the future therapy for sepsis? Blood Rev. 2016, 30, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Elkhodary, M.S.M. Treatment of COVID-19 by Controlling the Activity of the Nuclear Factor-Kappa B. CellBio 2020, 9, 109–121. [Google Scholar] [CrossRef]
- de Martin, R.; Hoeth, M.; Hofer-Warbinek, R.; Schmid, J.A. The Transcription Factor NF-κB and the Regulation of Vascular Cell Function. Arter. Thromb. Vasc. Biol. 2000, 20, e83–e88. [Google Scholar] [CrossRef] [Green Version]
- Abu-Eid, R.; Ward, F.J. Targeting the PI3K/Akt/mTOR pathway: A therapeutic strategy in COVID-19 patients. Immunol. Lett. 2021, 240, 1–8. [Google Scholar] [CrossRef]
- Kale, S.S.; Yende, S. Effects of Aging on Inflammation and Hemostasis through the Continuum of Critical Illness. Aging Dis. 2011, 2, 501–511. [Google Scholar]
- Mari, D.; Ogliari, G.; Castaldi, D.; Vitale, G.; Bollini, E.M.; Lio, D. Hemostasis and ageing. Immun. Ageing 2008, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Morens, D.M.; Taubenberger, J.K.; Harvey, H.A.; Memoli, M.J. The 1918 influenza pandemic: Lessons for 2009 and the future. Crit. Care Med. 2010, 38 (Suppl. 4), e10. [Google Scholar] [CrossRef] [PubMed]
- Yüce, M.; Filiztekin, E.; Özkaya, K.G. COVID-19 diagnosis—A review of current methods. Biosens. Bioelectron. 2020, 172, 112752. [Google Scholar] [CrossRef]
- Salian, V.S.; Wright, J.A.; Vedell, P.T.; Nair, S.; Li, C.; Kandimalla, M.; Tang, X.; Porquera, E.M.C.; Kalari, K.R.; Kandimalla, K.K. COVID-19 Transmission, Current Treatment, and Future Therapeutic Strategies. Mol. Pharm. 2021, 18, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Alsharif, W.; Qurashi, A. Effectiveness of COVID-19 diagnosis and management tools: A review. Radiography 2020, 27, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Choi, W.J. Overview of COVID-19 inflammatory pathogenesis from the therapeutic perspective. Arch. Pharm. Res. 2021, 44, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Smith, D.M. SARS-CoV-2 Variants of Concern. Yonsei Med. J. 2021, 62, 961–968. [Google Scholar] [CrossRef]
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary trajectory of SARS-CoV-2 and emerging variants. Virol. J. 2021, 18, 166. [Google Scholar] [CrossRef]
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117. [Google Scholar] [CrossRef]
- Cantón, R.; De Lucas Ramos, P.; García-Botella, A.; García-Lledó, A.; Gómez-Pavón, J.; González Del Castillo, J.; Hernández-Sampelayo, T.; Martín-Delgado, M.C.; Martín Sánchez, F.J.; Martínez-Sellés, M.; et al. New variants of SARS-CoV-2. Rev. Esp. Quimioter. 2021, 34, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Hussman, J.P. Cellular and Molecular Pathways of COVID-19 and Potential Points of Therapeutic Intervention. Front. Pharmacol. 2020, 11, 1169. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [Green Version]
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res. 2013, 101, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R. Obstacles and advances in SARS vaccine development. Vaccine 2006, 24, 863–871. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.-J.; Jiang, S. The spike protein of SARS-CoV—A target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- de Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Tong, T.R. Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). Perspect. Med. Virol. 2006, 16, 43–95. [Google Scholar] [CrossRef] [PubMed]
- Ziebuhr, J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. Microbiol. 2004, 7, 412–419. [Google Scholar] [CrossRef]
- Openshaw, P.J. What does the peripheral blood tell you in SARS? Clin. Exp. Immunol. 2004, 136, 11–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Samkari, H.; Berliner, N. Hemophagocytic Lymphohistiocytosis. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 27–49. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Mahallawi, W.H.; Khabour, O.F.; Zhang, Q.; Makhdoum, H.M.; Suliman, B.A. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine 2018, 104, 8–13. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.K.; Wu, A.K.L.; Ip, W.K.; Lee, N.L.S.; Chan, I.H.S.; Lit, L.C.W.; Hui, D.S.C.; Chan, M.H.M.; Chung, S.S.C.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wu, K.; Wang, D.; Yue, X.; Song, D.; Zhu, Y.; Wu, J. Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-κB. Virology 2007, 365, 324–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Huang, K.-J.; Su, I.-J.; Theron, M.; Wu, Y.-C.; Lai, S.-K.; Liu, C.-C.; Lei, H.-Y. An interferon-gamma?-related cytokine storm in SARS patients. J. Med Virol. 2004, 75, 185–194. [Google Scholar] [CrossRef]
- Shah, V.K.; Firmal, P.; Alam, A.; Ganguly, D.; Chattopadhyay, S. Overview of Immune Response During SARS-CoV-2 Infection: Lessons from the Past. Front Immunol. 2020, 11, 1949. [Google Scholar] [CrossRef] [PubMed]
- Bleibtreu, A.; Bertine, M.; Bertin, C.; Houhou-Fidouh, N.; Visseaux, B. Focus on Middle East respiratory syndrome coronavirus (MERS-CoV). Med. Mal. Infect. 2019, 50, 243–251. [Google Scholar] [CrossRef]
- Chafekar, A.; Fielding, B. MERS-CoV: Understanding the Latest Human Coronavirus Threat. Viruses 2018, 10, 93. [Google Scholar] [CrossRef]
- Widagdo, W.; Na Ayudhya, S.S.; Hundie, G.B.; Haagmans, B.L. Host Determinants of MERS-CoV Transmission and Pathogenesis. Viruses 2019, 11, 280. [Google Scholar] [CrossRef] [Green Version]
- Nassar, M.; Bakhrebah, M.A.; Meo, S.A.; Alsuabeyl, M.S.; Zaher, W.A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) infection: Epidemiology, pathogenesis and clinical characteristics. Eur. Rev. Med. Pharm. Sci. 2018, 22, 4956–4961. [Google Scholar] [CrossRef]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle East respiratory syndrome. Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef] [Green Version]
- Min, C.-K.; Cheon, S.; Ha, N.-Y.; Sohn, K.M.; Kim, Y.; Aigerim, A.; Shin, H.M.; Choi, J.-Y.; Inn, K.-S.; Kim, J.H.; et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci. Rep. 2016, 6, 25359. [Google Scholar] [CrossRef]
- Sun, X.; Wang, T.; Cai, D.; Hu, Z.; Chen, J.; Liao, H.; Zhi, L.; Wei, H.; Zhang, Z.; Qiu, Y.; et al. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. 2020, 53, 38–42. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Lau, S.K.P.; To, K.K.W.; Cheng, V.C.C.; Woo, P.C.Y.; Yuen, K.-Y. Middle East Respiratory Syndrome Coronavirus: Another Zoonotic Betacoronavirus Causing SARS-Like Disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phung, T.T.B.; Sugamata, R.; Uno, K.; Aratani, Y.; Ozato, K.; Kawachi, S.; Nguyen, L.T.; Nakayama, T.; Suzuki, K. Key role of regulated upon activation normal T-cell expressed and secreted, nonstructural protein1 and myeloperoxidase in cytokine storm induced by influenza virus PR-8 (A/H1N1) infection in A549 bronchial epithelial cells. Microbiol. Immunol. 2011, 55, 874–884. [Google Scholar] [CrossRef]
- Sullivan, S.J.; Jacobson, R.M.; Dowdle, W.R.; Poland, G.A. 2009 H1N1 Influenza. Mayo Clin. Proc. 2010, 85, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.; Dennis, A.; Flutter, C.; Khan, Z. Pandemic (H1N1) 2009 influenza. Br. J. Anaesth. 2010, 104, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Khanna, M.; Gupta, N.; Gupta, A.; Vijayan, V.K. Influenza A (H1N1) 2009: A pandemic alarm. J. Biosci. 2009, 34, 481–489. [Google Scholar] [CrossRef]
- Zeng, H.; Belser, J.A.; Goldsmith, C.S.; Gustin, K.M.; Veguilla, V.; Katz, J.M.; Tumpey, T.M. A(H7N9) virus results in early induction of proinflammatory cytokine responses in both human lung epithelial and endothelial cells and shows increased human adaptation compared with avian H5N1 virus. J. Virol. 2015, 89, 4655–4667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigel, J.H.; Farrar, J.; Han, A.M.; Hayden, F.G.; Hyer, R.; de Jong, M.D.; Lochindarat, S.; Nguyen, T.K.; Nguyen, T.H.; Tran, T.H.; et al. Avian influenza A (H5N1) infection in humans. N. Engl. J. Med. 2005, 353, 1374–1385. [Google Scholar] [PubMed] [Green Version]
- Peiris, J.; Yu, W.; Leung, C.; Cheung, C.; Ng, W.; Nicholls, J.M.; Ng, T.; Chan, K.; Lai, S.; Lim, W.; et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 2004, 363, 617–619. [Google Scholar] [CrossRef]
- Monteagudo, P.L.; Muñoz-Moreno, R.; Fribourg, M.; Potla, U.; Mena, I.; Marjanovic, N.; Hartmann, B.; Sealfon, S.C.; García-Sastre, A.; Ramos, I.; et al. Differential Modulation of Innate Immune Responses in Human Primary Cells by Influenza A Viruses Carrying Human or Avian Nonstructural Protein 1. J. Virol. 2019, 94, e00999-19. [Google Scholar] [CrossRef] [Green Version]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial Cells Are Central Orchestrators of Cytokine Amplification during Influenza Virus Infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef] [Green Version]
- D’Elia, R.V.; Harrison, K.; Oyston, P.C.; Lukaszewski, R.A.; Clark, G.C. Targeting the “Cytokine Storm” for Therapeutic Benefit. Clin. Vaccine Immunol. 2013, 20, 319–327. [Google Scholar] [CrossRef]
- Jose, R.J.P.; Manuel, A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir. Med. 2020, 8, e46–e47. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2015, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Oldstone, M.B.; Teijaro, J.R.; Walsh, K.B.; Rosen, H. Dissecting influenza virus pathogenesis uncovers a novel chemical approach to combat the infection. Virology 2012, 435, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Bermejo-Martin, J.F.; De Lejarazu, R.O.; Pumarola, T.; Rello, J.; Almansa, R.; Ramírez, P.; Martin-Loeches, I.; Varillas, D.; Gallegos, M.C.; Serón, C.; et al. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit. Care 2009, 13, R201. [Google Scholar] [CrossRef] [Green Version]
- Betakova, T.; Kostrabova, A.; Lachova, V.; Turianova, L. Cytokines Induced During Influenza Virus Infection. Curr. Pharm. Des. 2017, 23, 2616–2622. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Li, Y.; Pan, R.; Zou, X. Characterizing and controlling the inflammatory network during influenza A virus infection. Sci. Rep. 2014, 4, 3799. [Google Scholar] [CrossRef] [Green Version]
- La Gruta, N.L.; Kedzierska, K.; Stambas, J.; Doherty, P.C. A question of self-preservation: Immunopathology in influenza virus infection. Immunol. Cell Biol. 2007, 85, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Coon, B.G.; Baeyens, N.; Han, J.; Budatha, M.; Ross, T.D.; Fang, J.S. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J. Cell Biol. 2015, 208, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Hagau, N.; Slavcovici, A.; Gonganau, D.N.; Oltean, S.; Dirzu, D.S.; Brezoszki, E.S.; Maxim, M.; Ciuce, C.; Mlesnite, M.; Gavrus, R.L.; et al. Clinical aspects and cytokine response in severe H1N1 influenza A virus infection. Crit. Care 2010, 14, R203. [Google Scholar] [CrossRef] [Green Version]
- Oshansky, C.M.; Gartland, A.J.; Wong, S.-S.; Jeevan, T.; Wang, D.; Roddam, P.L. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care Med. 2014, 189, 449–462. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.; Hung, I.F.; Li, I.W.; Lee, K.L.; Koo, C.K.; Yan, W.W. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2010, 50, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Tung, E.T.K.; Chan, K.; Lau, C.C.Y.; Lau, S.K.P.; Yuen, K.-Y. Cytokine Profiles Induced by the Novel Swine-Origin Influenza A/H1N1 Virus: Implications for Treatment Strategies. J. Infect. Dis. 2010, 201, 346–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.L. The Spanish Flu, Epidemics, and the Turn to Biomedical Responses. Am. J. Public Health 2018, 108, 1455–1458. [Google Scholar] [CrossRef]
- Scarpa, R.; Caso, F.; Costa, L.; Passavanti, S.; Vitale, M.G.; Trojaniello, C.; Del Puente, A.; Ascierto, P.A. May the analysis of 1918 influenza pandemic give hints to imagine the possible magnitude of Corona Virus Disease-2019 (COVID-19)? J. Transl. Med. 2020, 18, 489. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.R. Comparing the Spanish flu and COVID-19 pandemics: Lessons to carry forward. Nurs. Forum 2020, 56, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Talha, N.J.; Radia, T.J.; Abdul, H.S. H1N1 Influenza; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Nickol, M.E.; Kindrachuk, J. A year of terror and a century of reflection: Perspectives on the great influenza pandemic of 1918–1919. BMC Infect. Dis. 2019, 19, 117. [Google Scholar] [CrossRef] [Green Version]
- Taubenberger, J.K.; Morens, D.M. The 1918 Influenza Pandemic and Its Legacy. Cold Spring Harb. Perspect. Med. 2019, 10, a038695. [Google Scholar] [CrossRef] [Green Version]
- Sutton, T.C. The Pandemic Threat of Emerging H5 and H7 Avian Influenza Viruses. Viruses 2018, 10, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neto, M.; Gomes, T.D.O.; Cunha, C.S.; Souza, H.A.N.D.; Macena, M.V.M.; Fonseca, M.H.S.; Porto, F.R. Lessons from the past in the present: News from the Spanish flu pandemic to COVID-19. Rev. Bras. Enferm. 2021, 75, e20201161. [Google Scholar] [CrossRef]
- Kobasa, D.; Takada, A.; Shinya, K.; Hatta, M.; Halfmann, P.; Theriault, S.; Suzuki, H.; Nishimura, H.; Mitamura, K.; Sugaya, N.; et al. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 2004, 431, 703–707. [Google Scholar] [CrossRef]
- Kobasa, D.; Jones, S.M.; Shinya, K.; Kash, J.C.; Copps, J.; Ebihara, H.; Hatta, Y.; Kim, J.H.; Halfmann, P.; Hatta, M.; et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 2007, 445, 319–323. [Google Scholar] [CrossRef]
- Kash, J.C.; Tumpey, T.M.; Proll, S.C.; Carter, V.; Perwitasari, O.; Thomas, M.J.; Basler, C.; Palese, P.; Taubenberger, J.K.; Garcia-Sastre, A.; et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 2006, 443, 578–581. [Google Scholar] [CrossRef]
- Memoli, M.J.; Tumpey, T.M.; Jagger, B.W.; Dugan, V.G.; Sheng, Z.-M.; Qi, L.; Kash, J.C.; Taubenberger, J.K. An early ‘classical’ swine H1N1 influenza virus shows similar pathogenicity to the 1918 pandemic virus in ferrets and mice. Virology 2009, 393, 338–345. [Google Scholar] [CrossRef] [Green Version]
- de Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.D.; Chau, T.N.B.; Hoang, D.M.; Chau, N.V.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Almansa, R.; Anton, A.; Ramirez, P.; Martin-Loeches, I.; Banner, D.; Pumarola, T.; Xu, L.; Blanco, J.; Ran, L.; Lopez-Campos, G.; et al. Direct association between pharyngeal viral secretion and host cytokine response in severe pandemic influenza. BMC Infect. Dis. 2011, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Short, K.; Kedzierska, K.; Van De Sandt, C.E. Back to the Future: Lessons Learned From the 1918 Influenza Pandemic. Front. Cell. Infect. Microbiol. 2018, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; Siegers, J.Y.; Cronin, J.M.; Weatherman, S.; Brand, J.M.V.D.; Leijten, L.M.; Van Run, P.; Begeman, L.; Ham, H.-J.V.D.; Andeweg, A.C.; et al. 1918 H1N1 Influenza Virus Replicates and Induces Proinflammatory Cytokine Responses in Extrarespiratory Tissues of Ferrets. J. Infect. Dis. 2018, 217, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.B. Cases Resembling Encephalitis Lethargica occurring during the Influenza Epidemic. BMJ 1919, 1, 794–795. [Google Scholar] [CrossRef] [Green Version]
- Ravenholt, R.; Foege, W. 1918 Influenza, Encephalitis Lethargica, Parkinsonism. Lancet 1982, 320, 860–864. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Pelaia, C.; Tinello, C.; Vatrella, A.; De Sarro, G.; Pelaia, G. Lung under attack by COVID-19-induced cytokine storm: Pathogenic mechanisms and therapeutic implications. Ther. Adv. Respir. Dis. 2020, 14. [Google Scholar] [CrossRef]
- Iannaccone, G.; Scacciavillani, R.; Del Buono, M.G.; Camilli, M.; Ronco, C.; Lavie, C.J.; Abbate, A.; Crea, F.; Massetti, M.; Aspromonte, N. Weathering the Cytokine Storm in COVID-19: Therapeutic Implications. Cardiorenal Med. 2020, 10, 277–287. [Google Scholar] [CrossRef]
- Liu, B.; Li, M.; Zhou, Z.; Guan, X.; Xiang, Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J. Autoimmun. 2020, 111, 102452. [Google Scholar] [CrossRef]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm–The common denominator and the lessons to be learned. Clin. Immunol. 2020, 223, 108652. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Salinas, L.; Verdugo-Rodriguez, A.; Rodriguez, L.L.; Borca, M.V. The Role of Interleukin 6 During Viral Infections. Front. Microbiol. 2019, 10, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jariwal, R.; Raza, N.; Valdez, M.; Aboeed, A.; Garcia-Pacheco, R. Prolonged SARS-CoV2 Viral Shedding in an Elderly Patient. Cureus 2021, 13, e15128. [Google Scholar] [CrossRef]
- Gao, C.; Zhu, L.; Jin, C.C.; Tong, Y.X.; Xiao, A.T.; Zhang, S. Proinflammatory cytokines are associated with prolonged viral RNA shedding in COVID-19 patients. Clin. Immunol. 2020, 221, 108611. [Google Scholar] [CrossRef] [PubMed]
- Polidoro, R.B.; Hagan, R.S.; Santiago, R.D.S.; Schmidt, N.W. Overview: Systemic Inflammatory Response Derived From Lung Injury Caused by SARS-CoV-2 Infection Explains Severe Outcomes in COVID-19. Front. Immunol. 2020, 11, 1626. [Google Scholar] [CrossRef]
- Tleyjeh, I.M.; Kashour, Z.; Damlaj, M.; Riaz, M.; Tlayjeh, H.; Altannir, M.; Altannir, Y.; Al-Tannir, M.; Tleyjeh, R.; Hassett, L.; et al. Efficacy and safety of tocilizumab in COVID-19 patients: A living systematic review and meta-analysis. Clin. Microbiol. Infect. 2020, 27, 215–227. [Google Scholar] [CrossRef]
- Copaescu, A.; Smibert, O.; Gibson, A.; Phillips, E.J.; Trubiano, J.A. The role of IL-6 and other mediators in the cytokine storm associated with SARS-CoV-2 infection. J. Allergy Clin. Immunol. 2020, 146, 518–534.e1. [Google Scholar] [CrossRef]
- Quirch, M.; Lee, J.; Rehman, S. Hazards of the Cytokine Storm and Cytokine-Targeted Therapy in Patients With COVID-19: Review. J. Med. Internet Res. 2020, 22, e20193. [Google Scholar] [CrossRef]
- Castelli, V.; Cimini, A.; Ferri, C. Cytokine Storm in COVID-19: “When You Come Out of the Storm, You Won’t Be the Same Person Who Walked in”. Front. Immunol. 2020, 11, 2132. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics 2021, 11, 316–329. [Google Scholar] [CrossRef]
- Kooistra, E.J.; Waalders, N.J.B.; Grondman, I.; Janssen, N.A.F.; De Nooijer, A.H.; Netea, M.G.; Van De Veerdonk, F.L.; Ewalds, E.; Van Der Hoeven, J.G.; Kox, M.; et al. Anakinra treatment in critically ill COVID-19 patients: A prospective cohort study. Crit. Care 2020, 24, 688. [Google Scholar] [CrossRef]
- Ovsyannikova, I.G.; Haralambieva, I.H.; Crooke, S.; Poland, G.A.; Kennedy, R.B. The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol. Rev. 2020, 296, 205–219. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.J.; Yousuf, M.S.; Shiers, S.; Price, T.J. Neurobiology of SARS-CoV-2 interactions with the peripheral nervous system: Implications for COVID-19 and pain. PAIN Rep. 2021, 6, e885. [Google Scholar] [CrossRef] [PubMed]
- Velavan, T.P.; Pallerla, S.R.; Rüter, J.; Augustin, Y.; Kremsner, P.G.; Krishna, S.; Meyer, C.G. Host genetic factors determining COVID-19 susceptibility and severity. EBioMedicine 2021, 72, 103629. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cytokine | Source | Function | Effects in Viral Pandemics |
|---|---|---|---|
| IL-1β | Macrophages, monocytes | Proinflammation, proliferation, apoptosis, differentiation | Increase vascular permeability, pulmonar fibrosis, alveolar collapse |
| IL-6 | Macrophages, T-cells, adipocyte | Proinflammation, differentiation, cytokine production | Diffuse tissue damage, genesis of severe respiratory forms |
| CXCL10 | Leukocytes, neutrophils, eosinophils, monocytes, endhotelial cells | Chemotaxis, apoptosis, cell growth inhibition, angiostasis | Recruitment of innate immune cells into the lung with consequent tissue damage |
| MCP-1 | Macrophages, fibroblasts | Migration and infiltration of monocytes/macrophages | Responsible for infection serious clinical manifestations and poor outcomes |
| TNFα | Macrophages, NK cells, CD4+ lymphocytes, adipocyte | Proinflammation, cytokine production, cell proliferation, apoptosis | Increased perivascular infiltration of activated macrophages, neutrophils and fibroblasts, fibrin deposition and alveolar collapse, leading to acute respiratory distress syndrome (ARDS) |
| CXCL8 | Macrophages, air way smooth muscle cells, endothelial cells | Chemotactic action for neutrophils and monocytes in the respiratory tract | Involvment in the inflammation and trafficking of immune cells in the context of viral infections |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murdaca, G.; Paladin, F.; Tonacci, A.; Isola, S.; Allegra, A.; Gangemi, S. The Potential Role of Cytokine Storm Pathway in the Clinical Course of Viral Respiratory Pandemic. Biomedicines 2021, 9, 1688. https://doi.org/10.3390/biomedicines9111688
Murdaca G, Paladin F, Tonacci A, Isola S, Allegra A, Gangemi S. The Potential Role of Cytokine Storm Pathway in the Clinical Course of Viral Respiratory Pandemic. Biomedicines. 2021; 9(11):1688. https://doi.org/10.3390/biomedicines9111688
Chicago/Turabian StyleMurdaca, Giuseppe, Francesca Paladin, Alessandro Tonacci, Stefania Isola, Alessandro Allegra, and Sebastiano Gangemi. 2021. "The Potential Role of Cytokine Storm Pathway in the Clinical Course of Viral Respiratory Pandemic" Biomedicines 9, no. 11: 1688. https://doi.org/10.3390/biomedicines9111688
APA StyleMurdaca, G., Paladin, F., Tonacci, A., Isola, S., Allegra, A., & Gangemi, S. (2021). The Potential Role of Cytokine Storm Pathway in the Clinical Course of Viral Respiratory Pandemic. Biomedicines, 9(11), 1688. https://doi.org/10.3390/biomedicines9111688