
Cellular Senescence: Mechanisms and Therapeutic Potential


Abstract
:1. Introduction
1.1. DNA Damage Response
1.2. Senescence vs. Quiescence: Different Cell Cycle Arrests
2. Senescence Biomarkers
2.1. Senescence-Associated Secretory Phenotype (SASP)
2.2. Cell Cycle Arrest Factors—p16 and p53-p21 Pathways
2.3. Senescence-Associated β-Galactosidase (SA β-gal)
2.4. Morphogenesis and Chromatin Remodelling
2.5. Apoptosis Sensitivity
2.6. Metabolic Reprogramming
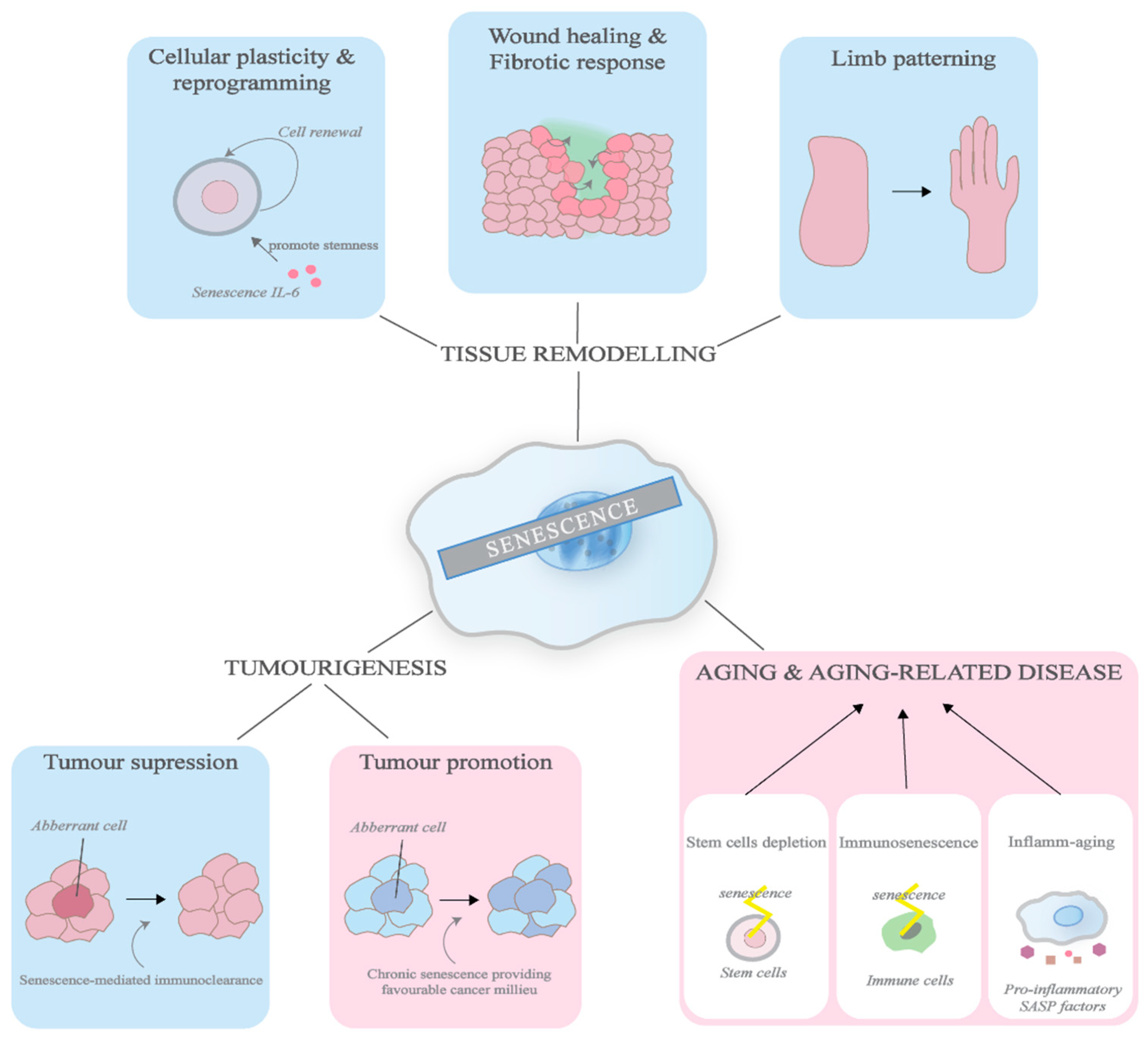
3. Physiological and Pathological Roles of Senescence
3.1. Embryogenesis
3.2. Wound Healing & Fibrotic Response
3.3. Cellular Plasticity & Reprogramming
3.4. Ageing & Ageing Related Diseases
3.5. Cancer
4. Therapeutic Significance
4.1. Senotherapy
4.1.1. Dasatinib + Quercetin
4.1.2. Navitoclax
4.1.3. Fisetin
4.1.4. HSP90 Inhibitors
4.1.5. FOXO4 Inhibitors
4.1.6. Cardiac Glycosides
4.1.7. Proteolysis Targeting Chimeras (PROTACs)
4.2. Pro-Senescence Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Z.; Chua, D.; Tan, N.S. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18, 65. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.C. Pleiotropy, natural selection, and the evolution of senescence. Evolution 1957, 11, 398–411. [Google Scholar] [CrossRef]
- di Fagagna, F.d.A. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A. Telomeres at a glance. J. Cell Sci. 2012, 125, 4173–4178. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.J.L. Loss of telomere protection: Consequences and opportunities. Front. Oncol. 2013, 3, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of survival networks in senescent cells: From mechanisms to interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef] [PubMed]
- Terzi, M.Y.; Izmirli, M.; Gogebakan, B. The cell fate: Senescence or quiescence. Mol. Biol. Rep. 2016, 43, 1213–1220. [Google Scholar] [CrossRef]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Tümpel, S.; Rudolph, K.L. Quiescence: Good and Bad of Stem Cell Aging. Trends Cell Biol. 2019, 29, 672–685. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Prieur, A.; Besnard, E.; Babled, A.; Lemaitre, J.M. p53 and p16INK4A independent induction of senescence by chromatin-dependent alteration of S-phase progression. Nat. Commun. 2011, 2, 473. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Barascu, A.; Le Chalony, C.; Pennarun, G.; Genet, D.; Imam, N.; Lopez, B.; Bertrand, P. Oxidative stress induces an ATM-independent senescence pathway through p38 MAPK-mediated lamin B1 accumulation. EMBO J. 2012, 31, 1080–1094. [Google Scholar] [CrossRef] [Green Version]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Salmonowicz, H.; Jurk, D.; Passos, J.F. Expansion and Cell-Cycle Arrest: Common Denominators of Cellular Senescence. Trends Biochem. Sci. 2019, 44, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.G.; Jackson, J.G. SASP: Tumor suppressor or promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Tan, Y.; Fan, A.; Liao, Z.; Liu, H.; Wei, P. Molecular dynamics of the recruitment of immunoreceptor signaling module DAP12 homodimer to lipid raft boundary regulated by PIP2. J. Phys. Chem. B 2019, 124, 504–510. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Sagiv, A.; Burton, D.G.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef] [Green Version]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lu, L.; Xie, Y.; Chen, X.; Tian, L.; Liang, Y.; Li, H.; Zhang, J.; Yu, X. Interleukin-6 Knockout Inhibits Senescence of Bone Mesenchymal Stem Cells in High-Fat Diet-Induced Bone Loss. Front. Endocrinol. 2021, 11, 1173. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; San Leong, H.; Lin, D.M.; Popkiss, S.; Pang, P.; Garsed, D.W.; Walkley, C.R.; Cullinane, C.; Ellul, J.; Haynes, N.M.; et al. Immune response to RB1-regulated senescence limits radiation-induced osteosarcoma formation. J. Clin. Investig. 2013, 123, 5351–5360. [Google Scholar] [CrossRef] [Green Version]
- Prata, L.G.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.M.; Zheng, L.; Wang, Q.; Hu, Y.W. The emerging role of cell senescence in atherosclerosis. Clin. Chem. Lab. Med. (CCLM) 2021, 59, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY) 2009, 1, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Ferrándiz Díaz, N.; Caraballo Otero, J.M.; García Gutiérrez, L.; Devgan, V.; Rodriguez Paredes, M.; Lafita Navarro, M.C.; Bretones Sánchez, G.; Quintanilla Cavia, A.; Muñoz Alonso, M.J.; Blanco Fernández, R.F.; et al. p21 as a transcriptional co-repressor of S-phase and mitotic control genes. PLoS ONE 2012, 7, e37759. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Paterson, M.C.; Murray, D. Single-cell analysis of p16(INK4a) and p21(WAF1) expression suggests distinct mechanisms of senescence in normal human and Li-Fraumeni Syndrome fibroblasts. J. Cell. Physiol. 2010, 223, 57–67. [Google Scholar]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors p21 and p16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagosa, C.; Simonetti, S.; Lopez-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; y Cajal, S.R. p16 Ink4a overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798. [Google Scholar] [CrossRef]
- Gary, R.K.; Kindell, S.M. Quantitative assay of senescence-asssociated beta-galactosidase activity in mammalian cell extracts. Anal. Biochem. 2005, 343, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Robbins, E.; Levine, E.; Eagle, H. Morphologic changes accompanying senescence of cultured human diploid cells. J. Exp. Med. 1970, 131, 1211–1222. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.-C.; Hu, M.-L. The limitations and validities of senescence associated-β-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp. Gerontol. 2005, 40, 813–819. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867–1884. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, A.J.; Narita, M. Old cells, new tricks: Chromatin structure in senescence. Mamm. Genome 2016, 27, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosar, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type-and insult-dependent manner and follow expression of p16ink4a. Cell Cycle 2011, 10, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta 2009, 1788, 2022–2031. [Google Scholar] [CrossRef] [Green Version]
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return Journey from Cell Death. Cancers 2021, 13, 3671. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Talbot, C.C., Jr.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000Research 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.H.; Seeber, A.; Singh, V.; Thierry, R.; Sack, R.; Amitai, A.; Kryzhanovska, M.; Eglinger, J.; Holcman, D.; Owen-Hughes, T.; et al. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat. Struct. Mol. Biol. 2017, 24, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänicke, R.U.; Sohn, D.; Schulze-Osthoff, K. The dark side of a tumor suppressor: Anti-apoptotic p53. Cell Death Differ. 2008, 15, 959–976. [Google Scholar] [CrossRef] [Green Version]
- Roninson, I.B. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): Association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002, 179, 1–14. [Google Scholar] [CrossRef]
- Al-Mohanna, M.A.; Manogaran, P.S.; Al-Mukhalafi, Z.; Al-Hussein, K.A.; Aboussekhra, A. The tumor suppressor p16INK4a gene is a regulator of apoptosis induced by ultraviolet light and cisplatin. Oncogene 2004, 23, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Barley, R.D.; Enns, L.; Paterson, M.C.; Mirzayans, R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li–Fraumeni syndrome fibroblast strains heterozygous for TP53 mutations. Oncogene 1998, 17, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Scott, A.; Andrais, B.; Pollock, S.; Murray, D. Ultraviolet light exposure triggers nuclear accumulation of p21(WAF1) and accelerated senescence in human normal and nucleotide excision repair-deficient fibroblast strains. J. Cell Physiol. 2008, 215, 55–67. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Probin, V.; Wang, Y.; Bai, A.; Zhou, D. Busulfan Selectively Induces Cellular Senescence but Not Apoptosis in WI38 Fibroblasts via a p53-Independent but Extracellular Signal-Regulated Kinase-p38 Mitogen-Activated Protein Kinase-Dependent Mechanism. J. Pharmacol. Exp. Ther. 2006, 319, 551–560. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; Guicciardi, M.E.; Splinter, N.P.; Al Suraih, M.S.; Nasser-Ghodsi, N.; Stollenwerk, D.; Gores, G.J.; LaRusso, N.F. The transcription factor ETS1 promotes apoptosis resistance of senescent cholangiocytes by epigenetically up-regulating the apoptosis suppressor BCL2L1. J. Biol. Chem. 2019, 294, 18698–18713. [Google Scholar] [CrossRef] [PubMed]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol.-Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.J.; Oh, Y.S.; Park, S.C. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 2007, 14, 1020–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, M.; Hino, S.I.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasileiou, P.V.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, K. The emerging role of senescent cells in tissue homeostasis and pathophysiology. Pathobiol. Aging Age Relat. Dis. 2015, 5, 27743. [Google Scholar] [CrossRef]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Development 2019, 146, dev151837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.I.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Jayanthi, P.; Joshua, E.; Ranganathan, K. Ageing and its implications. J. Oral Maxillofac. Pathol. 2010, 14, 48–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004, 18, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Gutarra, S.; Garcia-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardi, M.; Ballestar, E.; Gonzalez, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in age-related chronic inflammatory diseases. NPJ Aging Mech. Dis. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Prattichizzo, F.; Grillari, J.; Balistreri, C.R. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediat. Inflamm. 2018, 2018, 9076485. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.M.M.; Chua, Z.J.Y.; Tan, J.C.; Yang, Y.; Liao, Z.; Zhao, Y. From pre-diabetes to diabetes: Diagnosis, treatments and translational research. Medicina 2019, 55, 546. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yang, T.; Liu, S.; Cao, Z.; Zhao, Y.; Su, X.; Liao, Z.; Teng, X.; Hua, J. Concentrated ambient PM2.5 exposure affects mice sperm quality and testosterone biosynthesis. PeerJ 2019, 7, e8109. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment–Accomplices in tumor malignancy. Cell. Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, Y.; Liao, Z. Diabetes and cancer: Epidemiological and biological links. World J. Diabetes 2020, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xie, F.; Lin, J.; Zhao, Y.; Zhang, Q.; Liao, Z.; Wei, P. Diagnostic and Prognostic Value of Circulating CircRNAs in Cancer. Front. Med. 2021, 8, 231. [Google Scholar] [CrossRef]
- Wang, M.; Tan, Y.; Shi, Y.; Wang, X.; Liao, Z.; Wei, P. Diabetes and Sarcopenic Obesity: Pathogenesis, Diagnosis, and Treatments. Front. Endocrinol. 2020, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Wong, S.W.; Yeo, H.L.; Zhao, Y. Nanocarriers for cancer treatment: Clinical impact and safety. NanoImpact 2020, 20, 100253. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, M.; Yang, K.; Chi, T.; Liao, Z.; Wei, P. PPAR-α Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 707. [Google Scholar] [CrossRef] [PubMed]
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An aPPARent functional consequence in skeletal muscle physiology via peroxisome proliferator-activated receptors. Int. J. Mol. Sci. 2018, 19, 1425. [Google Scholar] [CrossRef] [Green Version]
- Mavrogonatou, E.; Pratsinis, H.; Kletsas, D. The role of senescence in cancer development. In Seminars in Cancer Biology; Academic Press: Salt Lake City, UT, USA, 2019. [Google Scholar]
- Liu, X.L.; Ding, J.; Meng, L.H. Oncogene-induced senescence: A double edged sword in cancer. Acta Pharm. Sin. 2018, 39, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooi, W.J.; Peeper, D.S. Oncogene-induced cell senescence—Halting on the road to cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef]
- Gonzalez-Molina, J.; Gramolelli, S.; Liao, Z.; Carlson, J.W.; Ojala, P.M.; Lehti, K. MMP14 in sarcoma: A regulator of tumor microenvironment communication in connective tissues. Cells 2019, 8, 991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, G.H.; Liu, Y.; Lu, Y.; Liu, J.; Wu, C.; Duan, H.F. IL-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS ONE 2014, 9, e113572. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Yi, J.; Yang, X.; Liu, L.; Lou, X.; Zhang, Z.; Qi, H.; Wang, Z.; Zou, J.; Zhu, W.G.; et al. MDM2-mediated degradation of WRN promotes cellular senescence in a p53-independent manner. Oncogene 2019, 38, 2501–2515. [Google Scholar] [CrossRef] [PubMed]
- Tonnessen-Murray, C.A.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; Keam, S.P.; Haupt, Y. Cannibalism in Breast Cancer: The Dangers of Overeating. Trends Cancer 2019, 5, 761–762. [Google Scholar] [CrossRef]
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S.; et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Almassi, N.; Jarrard, D.F. Drug-induced senescence bystander proliferation in prostate cancer cells in vitro and in vivo. Br. J. Cancer 2008, 98, 1244–1249. [Google Scholar] [CrossRef]
- Burton, D.G.; Giribaldi, M.G.; Munoz, A.; Halvorsen, K.; Patel, A.; Jorda, M.; Perez-Stable, C.; Rai, P. Androgen deprivation-induced senescence promotes outgrowth of androgen-refractory prostate cancer cells. PLoS ONE 2013, 8, e68003. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, R.; Lacelle, C.; Wang, E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 2004, 125, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.J.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabisz, M.; Skladanowski, A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: Implications for drug resistance and in vitro drug screening models. Cell Cycle 2009, 8, 3208–3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, T.; Tyutyunyk-Massey, L.; Murray, G.F.; Alotaibi, M.R.; Kawale, A.S.; Elsayed, Z.; Henderson, S.C.; Yakovlev, V.; Elmore, L.W.; Toor, A.; et al. Tumor cell escape from therapy-induced senescence. Biochem. Pharmacol. 2019, 162, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Chitikova, Z.V.; Gordeev, S.A.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Pospelova, T.V. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle 2014, 13, 1424–1439. [Google Scholar] [CrossRef] [Green Version]
- Was, H.; Barszcz, K.; Czarnecka, J.; Kowalczyk, A.; Bernas, T.; Uzarowska, E.; Koza, P.; Klejman, A.; Piwocka, K.; Kaminska, B.; et al. Bafilomycin A1 triggers proliferative potential of senescent cancer cells in vitro and in NOD/SCID mice. Oncotarget 2017, 8, 9303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wu, P.C.; Roberson, R.S.; Luk, B.V.; Ivanova, I.; Chu, E.; Wu, D.Y. Survivin and escaping in therapy-induced cellular senescence. Int. J. Cancer 2011, 128, 1546–1558. [Google Scholar] [CrossRef] [Green Version]
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Cancer: A matter of life cycle? Cell Biol. Int. 2007, 31, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Zhang, J.; Zhang, N.; Mercado-Uribe, I.; Tao, F.; Han, Z.; Pathak, S.; Multani, A.S.; Kuang, J.; Yao, J.; et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis 2016, 5, e281. [Google Scholar] [CrossRef] [Green Version]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.S.; Perez, G.; Ngo, L.; Gui, C.Y.; Marks, P.A. Induction of polyploidy by histone deacetylase inhibitor: A pathway for antitumor effects. Cancer Res. 2005, 65, 7832–7839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Chan, C.M.; Jing, X.; Pike, L.A.; Zhou, Q.; Lim, D.J.; Sams, S.B.; Lund, G.S.; Sharma, V.; Haugen, B.R.; Schweppe, R.E. Targeted inhibition of Src kinase with dasatinib blocks thyroid cancer growth and metastasis. Clin. Cancer Res. 2012, 18, 3580–3591. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Cellular senescence: A translational perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Saccon, T.D.; Nagpal, R.; Yadav, H.; Cavalcante, M.B.; Nunes, A.D.D.C.; Schneider, A.; Gesing, A.; Hughes, B.; Yousefzadeh, M.; Tchkonia, T.; et al. Senolytic combination of Dasatinib and Quercetin alleviates intestinal senescence and inflammation and modulates the gut microbiome in aged mice. J. Gerontol. Ser. A 2021, 76, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Gunti, S.; Robbins, Y.; Schmitt, N.C. Senolytic drug ABT-263 enhances cisplatin-and TIL-induced cell death in vitro but has limited in vivo activity in preclinical models of head and neck cancer. In Proceedings of the AACR Annual Meeting, Philadelphia, PA, USA, 27–28 April 2020. [Google Scholar]
- Yousefzadeh, M.J.; Zhu, Y.I.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Tong, X.; Huang, J.; Wu, M.; Zhang, S.; Wang, D.; Liu, S.; Fan, H. Fisetin Alleviated Bleomycin-Induced Pulmonary Fibrosis Partly by Rescuing Alveolar Epithelial Cells From Senescence. Front. Pharmacol. 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Dutta Gupta, S.; Pan, C.H. Recent update on discovery and development of Hsp90 inhibitors as senolytic agents. Int. J. Biol. Macromol. 2020, 161, 1086–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xie, Y.; Chen, H.; Lv, L.; Yao, J.; Zhang, M.; Xia, K.; Feng, X.; Li, Y.; Liang, X.; et al. FOXO4-DRI alleviates age-related testosterone secretion insufficiency by targeting senescent Leydig cells in aged mice. Aging 2020, 12, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Shi, H.; Mao, X.; Zhong, Y.; Liu, Y.; Zhao, X.; Yu, K.; Zhu, R.; Wei, Y.; Zhu, J.; Sun, H.; et al. Digoxin reduces atherosclerosis in apolipoprotein E-deficient mice. Br. J. Pharmacol. 2016, 173, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, X.; Liu, Z.; Xu, X.; Xiao, H.; Zhang, X.; Dai, H.; Wang, C. Ouabain ameliorates bleomycin induced pulmonary fibrosis by inhibiting proliferation and promoting apoptosis of lung fibroblasts. Am. J. Transl. Res. 2018, 10, 2967–2974. [Google Scholar] [PubMed]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.N.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.; et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 2020, 11, 1996. [Google Scholar] [CrossRef] [PubMed]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.I.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.C.; Seoane, S.; Ocaña, A.; Pandiella, A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: Possible combinations in solid tumors. Clin. Cancer Res. 2011, 17, 5546–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Q.; Jorgensen, C.; Pawson, T.; Hedley, D. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br. J. Cancer 2008, 99, 1074–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Rix, U.; Fang, B.; Bai, Y.; Edwards, A.; Colinge, J.; Bennett, K.L.; Gao, J.; Song, L.; Eschrich, S.; et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 2010, 6, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, A.V.A.; Arulmoli, R.; Parasuraman, S. Overviews of biological importance of quercetin: A bioactive flavonoid. Pharmacogn. Rev. 2016, 10, 84. [Google Scholar]
- Olave, N.C.; Grenett, M.H.; Cadeiras, M.; Grenett, H.E.; Higgins, P.J. Upstream stimulatory factor-2 mediates quercetin-induced suppression of PAI-1 gene expression in human endothelial cells. J. Cell. Biochem. 2010, 111, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J.; Chen, Y.; Sobel, B.E. The effect of plasminogen activator inhibitor type 1 on apoptosis. Thromb. Haemost. 2008, 100, 1037–1040. [Google Scholar] [PubMed]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri Jr, W.A.; Hogaboam, C.M. Quercetin enhances ligand-induced apoptosis in senescent idiopathic pulmonary fibrosis fibroblasts and reduces lung fibrosis in vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Prata, L.G.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib+Quercetin (D+Q) does not enhance the efficacy of senescence-inducing chemotherapy in liver cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef] [PubMed]
- Lafontaine, J.; Cardin, G.B.; Malaquin, N.; Boisvert, J.S.; Rodier, F.; Wong, P. Senolytic Targeting of Bcl-2 Anti-Apoptotic Family Increases Cell Death in Irradiated Sarcoma Cells. Cancers 2021, 13, 386. [Google Scholar] [CrossRef]
- Zhu, Y.I.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chen, C.; Chen, H.; Duan, X.; Li, J.; Zhou, Y.; Zeng, W.; Yang, L. Navitoclax (ABT263) reduces inflammation and promotes chondrogenic phenotype by clearing senescent osteoarthritic chondrocytes in osteoarthritis. Aging 2020, 12, 12750. [Google Scholar] [CrossRef]
- Sierra-Ramirez, A.; Lopez-Aceituno, J.L.; Costa-Machado, L.F.; Plaza, A.; Barradas, M.; Fernandez-Marcos, P.J. Transient metabolic improvement in obese mice treated with navitoclax or dasatinib/quercetin. Aging 2020, 12, 11337–11348. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 353–361. [Google Scholar] [CrossRef]
- Sharma, A.K.; Roberts, R.L.; Benson Jr, R.D.; Pierce, J.L.; Yu, K.; Hamrick, M.W.; McGee-Lawrence, M.E. The senolytic drug navitoclax (ABT-263) causes trabecular bone loss and impaired osteoprogenitor function in aged mice. Front. Cell Dev. Biol. 2020, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- González-Gualda, E.; Pàez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R., III; González-López, C.; Ou, H.L.; Mirón-Barroso, S.; Zhang, Z.; Lérida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cheng, Y.; Qu, W.; Sun, Y.; Wang, Z.; Wang, H.; Tian, B. Fisetin, a Dietary Flavonoid, Induces Cell Cycle Arrest and Apoptosis through Activation of p53 and Inhibition of NF-Kappa B Pathways in Bladder Cancer Cells. Basic Clin. Pharmacol. Toxicol. 2011, 108, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Pal, H.C.; Sharma, S.; Elmets, C.A.; Athar, M.; Afaq, F. Fisetin inhibits growth, induces G2/M arrest and apoptosis of human epidermoid carcinoma A431 cells: Role of mitochondrial membrane potential disruption and consequent caspases activation. Exp. Dermatol. 2013, 22, 470–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundarraj, K.; Raghunath, A.; Perumal, E. A review on the chemotherapeutic potential of fisetin: In vitro evidences. Biomed. Pharmacother. 2018, 97, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Jia, S.-S. Fisetin inhibits laryngeal carcinoma through regulation of AKT/NF-κB/mTOR and ERK1/2 signaling pathways. Biomed. Pharmacother. 2016, 83, 1164–1174. [Google Scholar] [CrossRef]
- Park, H.K.; Yoon, N.G.; Lee, J.E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Han, S.Y.; Ko, A.; Kitano, H.; Choi, C.H.; Lee, M.S.; Seo, J.; Fukuoka, J.; Kim, S.Y.; Hewitt, S.M.; Chung, J.Y.; et al. Molecular Chaperone HSP90 Is Necessary to Prevent Cellular Senescence via Lysosomal Degradation of p14ARF. Cancer Res. 2017, 77, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.H.; Kim, R.; Chen, W.; Hu, S.; Shin, K.H.; Park, N.H.; Kang, M.K. Association of hsp90 to the hTERT promoter is necessary for hTERT expression in human oral cancer cells. Carcinogenesis 2008, 29, 2425–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Keizer, P.L.; Packer, L.M.; Szypowska, A.A.; Riedl-Polderman, P.E.; van den Broek, N.J.; de Bruin, A.; Dansen, T.B.; Marais, R.; Brenkman, A.B.; Burgering, B.M. Activation of Forkhead Box O Transcription Factors by Oncogenic BRAF Promotes p21cip1-Dependent Senescence. Cancer Res. 2010, 70, 8526–8536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Ahn, D.; Park, C.-J. Biophysical investigation of the dual binding surfaces of human transcription factors FOXO4 and p53. bioRxiv 2021. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.; Putavet, D.A.; Klein, J.D.; Derks, K.W.; Bourgeois, B.R.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jiang, X.; Feng, F.; Liu, W.; Sun, H. Degradation of proteins by PROTACs and other strategies. Acta Pharm. Sin. B 2020, 10, 207–238. [Google Scholar] [CrossRef]
- Bitto, A.; Ito, T.K.; Pineda, V.V.; LeTexier, N.J.; Huang, H.Z.; Sutlief, E.; Tung, H.; Vizzini, N.; Chen, B.; Smith, K.; et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. eLife 2016, 5, e16351. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Pospelova, T.V.; Leontieva, O.V.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Blagosklonny, M.V. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle 2012, 11, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.U.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.S.; Vautier, M.; Allenbach, Y.; Zahr, N.; Benveniste, O.; Funck-Brentano, C.; Salem, J.E. Sirolimus and mTOR inhibitors: A review of side effects and specific management in solid organ transplantation. Drug Saf. 2019, 42, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M. Rapamycin and aging: When, for how long, and how much? J. Genet. Genom. Yi Chuan Xue Bao 2014, 41, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Xia, D.; Pan, Z.; Xu, D.; Zhou, Y.; Wu, Y.; Cai, N.; Tang, Q.; Wang, C.; Yan, M.; et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis. 2016, 7, e2441. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.E.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κ B activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Lim, J.S.; Kim, H.S.; Park, S.C.; Park, J.T.; Kim, H.S.; Oh, W.K.; Cho, K.A. Identification of a novel senomorphic agent, avenanthramide C, via the suppression of the senescence-associated secretory phenotype. Mech. Ageing Dev. 2020, 192, 111355. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
- Karin, O.; Agrawal, A.; Porat, Z.; Krizhanovsky, V.; Alon, U. Senescent cell turnover slows with age providing an explanation for the Gompertz law. Nat. Commun. 2019, 10, 5495. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.A.; Schwartz, G.K. Cell cycle-mediated drug resistance: An emerging concept in cancer therapy. Clin. Cancer Res. 2001, 7, 2168–2181. [Google Scholar] [PubMed]
- Finch, C.E. Longevity, Senescence, and the Genome; University of Chicago Press: Chicago, IL, USA, 1994. [Google Scholar]
- Cailliet, G.M.; Andrews, A.H.; Burton, E.J.; Watters, D.L.; Kline, D.E.; Ferry-Graham, L.A. Age determination and validation studies of marine fishes: Do deep-dwellers live longer? Exp. Gerontol. 2001, 36, 739–764. [Google Scholar] [CrossRef]
- Matise, M. Ending Aging: The Rejuvenation Breakthroughs That Could Reverse Human Aging in Our Lifetime; St. Martin’s Press: New York, NY, USA, 2018. [Google Scholar]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Dahl, E.S.; Buj, R.; Leon, K.E.; Newell, J.M.; Imamura, Y.; Bitler, B.G.; Snyder, N.W.; Aird, K.M. Targeting IDH1 as a Prosenescent Therapy in High-grade Serous Ovarian Cancer. Mol. Cancer Res. 2019, 17, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Ruscetti, M.; Morris IV, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441.e21. [Google Scholar] [CrossRef]
- Calcinotto, A.; Alimonti, A. Aging tumour cells to cure cancer: “Pro-senescence” therapy for cancer. Swiss Med. Wkl. 2017, 147, w14367. [Google Scholar]
- Toso, A.; Di Mitri, D.; Alimonti, A. Enhancing chemotherapy efficacy by reprogramming the senescence-associated secretory phenotype of prostate tumors: A way to reactivate the antitumor immunity. Oncoimmunology 2015, 4, e994380. [Google Scholar] [CrossRef] [Green Version]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| SASP Factors | Examples | Effects and Mechanisms |
|---|---|---|
| Cytokine | IL-6 | |
| Growth Factors | TGF- β1 |
|
| Proteases | MMPs |
|
| Non-Protein Factors | miRNA |
|
| Senolytic Drugs | Mechanisms | Recent Updates |
|---|---|---|
| Dasatinib + Quercetin (D+Q) | Promoting apoptosis by disrupting multiple SCAPs including ephrin-dependent suppression of apoptosis, PI3K/Akt signalling as well as by upregulating Fas-1 and Caveolin-1. |
|
| Navitoclax | Interfering with BCL-2 protein family/BH123 protein family interaction and mediates mitochondria-dependent apoptosis. |
|
| Fisetin | Promoting apoptosis via SCAPs interference: upregulate pro-apoptotic BH123 proteins, downregulation of NF-κB and anti-apoptotic BCL-2 family proteins and modulation of the PI3K/Akt/mTOR pathway. | |
| HSP90 inhibitor | Downregulating the anti-apoptotic PI3K/Akt signalling pathway. | |
| FOXO4 inhibitor | Disrupting the FOXO4/p53 axis by competing with FOXO4 for p53 binding, leading to the nuclear exclusion of p53 and its interaction with BCL-XL which subsequently causes cytochrome c release and thus causing mitochondrial-mediated apoptosis via caspase3/7 activation. |
|
| Cardiac Glycoside | Inhibiting Na+/K+ ATPase which causes the blockade of the NA+/H+ exchanger, thus preventing the export of H+ which results in intracellular acidification and ultimately, apoptosis. Additionally, may also cause apoptosis by elevating levels of pro-apoptotic BCL-2 family proteins. | |
| PROTAC | Technology which improves the pharmacological profiles of senolytic drugs. PROTACs consists of 3 components: a ligand that recognises the target, a ligand that recognises an E3 enzyme and a linker that connects the two ligands. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Z.; Yeo, H.L.; Wong, S.W.; Zhao, Y. Cellular Senescence: Mechanisms and Therapeutic Potential. Biomedicines 2021, 9, 1769. https://doi.org/10.3390/biomedicines9121769
Liao Z, Yeo HL, Wong SW, Zhao Y. Cellular Senescence: Mechanisms and Therapeutic Potential. Biomedicines. 2021; 9(12):1769. https://doi.org/10.3390/biomedicines9121769
Chicago/Turabian StyleLiao, Zehuan, Han Lin Yeo, Siaw Wen Wong, and Yan Zhao. 2021. "Cellular Senescence: Mechanisms and Therapeutic Potential" Biomedicines 9, no. 12: 1769. https://doi.org/10.3390/biomedicines9121769
APA StyleLiao, Z., Yeo, H. L., Wong, S. W., & Zhao, Y. (2021). Cellular Senescence: Mechanisms and Therapeutic Potential. Biomedicines, 9(12), 1769. https://doi.org/10.3390/biomedicines9121769