
Neutrophil Extracellular Traps in Skin Diseases


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neutrophil Extracellular Traps
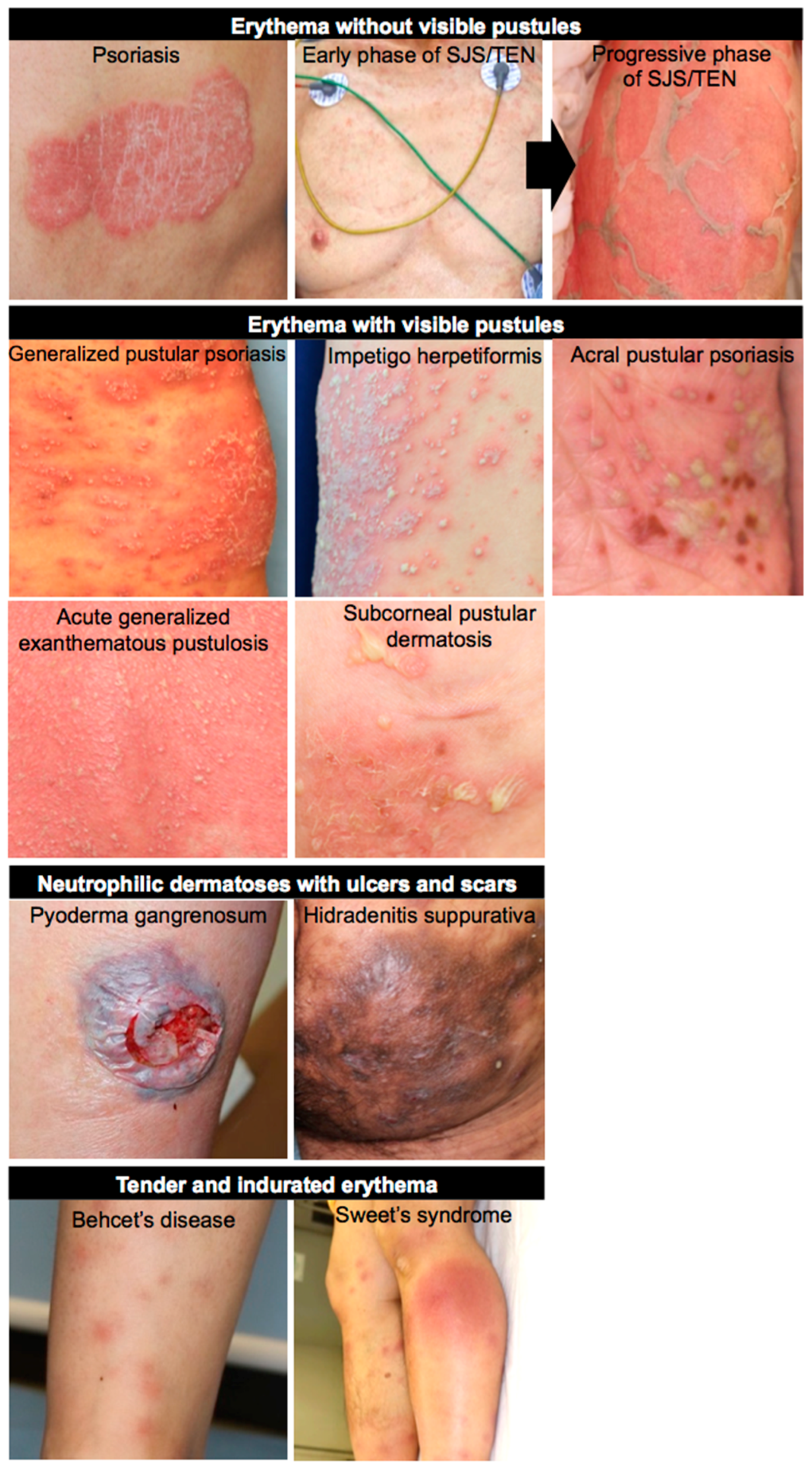
3. NETs in Skin Diseases
3.1. Psoriasis
3.2. Pustular Dermatosis
3.3. Behcet’s Disease
3.4. Pyoderma Gangrenosum
3.5. Hidradenitis Suppurativa
3.6. Other Neutrophil-Related Skin Disorders
3.7. Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Vorobjeva, N.V. Neutrophil extracellular traps: New aspects. Mosc. Univ. Biol. Sci. Bull. 2020, 75, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Wigerblad, G.; Kaplan, M.J. NETs spread ever wider in rheumatic diseases. Nat. Rev. Rheumatol. 2020, 16, 73–74. [Google Scholar] [CrossRef]
- Wang, W.M.; Jin, H.Z. Role of neutrophils in psoriasis. J. Immunol. Res. 2020, 2020, 3709749. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.H.; Enk, A.H. Neutrophil extracellular traps in dermatology: Caught in the NET. J. Dermatol. Sci. 2016, 84, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12951. [Google Scholar] [CrossRef] [Green Version]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzilotti, C.; Pratesi, F.; Tommasi, C.; Migliorini, P. Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun. Rev. 2010, 9, 158–160. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinegin, B.; Vorobjeva, N.; Pinegin, V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun. Rev. 2015, 14, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Skrzeczynska-Moncznik, J.; Zabieglo, K.; Osiecka, O.; Morytko, A.; Brzoza, P.; Drozdz, L.; Kapinska-Mrowiecka, M.; Korkmaz, B.; Pastuszczak, M.; Kosalka-Wegiel, J.; et al. Differences in staining for neutrophil elastase and its controlling inhibitor SLPI reveal heterogeneity among neutrophils in psoriasis. J. Investig. Dermatol. 2020, 140, 1371–1378.e1373. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Adrover, J.M.; Hidalgo, A. Neutrophils in homeostasis, immunity, and cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef]
- Nakabo, S.; Romo-Tena, J.; Kaplan, M.J. Neutrophils as drivers of immune dysregulation in autoimmune diseases with skin manifestations. J. Investig. Dermatol. 2021, in press. [Google Scholar] [CrossRef]
- Wojcik, P.; Garley, M.; Wronski, A.; Jablonska, E.; Skrzydlewska, E. Cannabidiol modifies the formation of NETs in neutrophils of psoriatic patients. Int. J. Mol. Sci. 2020, 21, 6795. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Fang, H.; Dang, E.; Xue, K.; Zhang, J.; Li, B.; Qiao, H.; Cao, T.; Zhuang, Y.; Shen, S.; et al. Neutrophil extracellular traps promote inflammatory responses in psoriasis via activating epidermal TLR4/IL-36R crosstalk. Front. Immunol. 2019, 10, 746. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.C.; Yu, H.S.; Yen, F.L.; Lin, C.L.; Chen, G.S.; Lan, C.C. Neutrophil extracellular trap formation is increased in psoriasis and induces human beta-defensin-2 production in epidermal keratinocytes. Sci. Rep. 2016, 6, 31119. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 2011, 187, 490–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teague, H.L.; Varghese, N.J.; Tsoi, L.C.; Dey, A.K.; Garshick, M.S.; Silverman, J.I.; Baumer, Y.; Harrington, C.L.; Stempinski, E.; Elnabawi, Y.A.; et al. Neutrophil subsets, platelets, and vascular disease in psoriasis. JACC Basic Transl. Sci. 2019, 4, 1–14. [Google Scholar] [CrossRef]
- Jiang, M.; Fang, H.; Shao, S.; Dang, E.; Zhang, J.; Qiao, P.; Yang, A.; Wang, G. Keratinocyte exosomes activate neutrophils and enhance skin inflammation in psoriasis. FASEB J. 2019, 33, 13241–13253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keijsers, R.; Hendriks, A.G.M.; van Erp, P.E.J.; van Cranenbroek, B.; van de Kerkhof, P.C.M.; Koenen, H.; Joosten, I. In vivo induction of cutaneous inflammation results in the accumulation of extracellular trap-forming neutrophils expressing RORgammat and IL-17. J. Investig. Dermatol. 2014, 134, 1276–1284. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Huang, L.; Vergis, A.L.; Ye, H.; Bajwa, A.; Narayan, V.; Strieter, R.M.; Rosin, D.L.; Okusa, M.D. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J. Clin. Investig. 2010, 120, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Tillack, K.; Breiden, P.; Martin, R.; Sospedra, M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 2012, 188, 3150–3159. [Google Scholar] [CrossRef]
- Lambert, S.; Hambro, C.A.; Johnston, A.; Stuart, P.E.; Tsoi, L.C.; Nair, R.P.; Elder, J.T. Neutrophil extracellular traps induce human Th17 cells: Effect of psoriasis-associated TRAF3IP2 genotype. J. Investig. Dermatol. 2019, 139, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Rosales, Y.A.; Langereis, J.D.; Gorris, M.A.J.; van den Reek, J.; Fasse, E.; Netea, M.G.; de Vries, I.J.M.; Gomez-Munoz, L.; van Cranenbroek, B.; Korber, A.; et al. Immunomodulatory aged neutrophils are augmented in blood and skin of psoriasis patients. J. Allergy Clin. Immunol. 2021, 148, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Skrzeczynska-Moncznik, J.; Wlodarczyk, A.; Banas, M.; Kwitniewski, M.; Zabieglo, K.; Kapinska-Mrowiecka, M.; Dubin, A.; Cichy, J. DNA structures decorated with cathepsin G/secretory leukocyte proteinase inhibitor stimulate IFNI production by plasmacytoid dendritic cells. Am. J. Clin. Exp. Immunol. 2013, 2, 186–194. [Google Scholar]
- Skrzeczynska-Moncznik, J.; Wlodarczyk, A.; Zabieglo, K.; Kapinska-Mrowiecka, M.; Marewicz, E.; Dubin, A.; Potempa, J.; Cichy, J. Secretory leukocyte proteinase inhibitor-competent DNA deposits are potent stimulators of plasmacytoid dendritic cells: Implication for psoriasis. J. Immunol. 2012, 189, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhofer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Loffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Schweckendiek, W. Treatment of psoriasis vulgaris. Med. Mon. 1959, 13, 103–104. [Google Scholar]
- Mrowietz, U.; Asadullah, K. Dimethylfumarate for psoriasis: More than a dietary curiosity. Trends Mol. Med. 2005, 11, 43–48. [Google Scholar] [CrossRef]
- Muller, S.; Behnen, M.; Bieber, K.; Moller, S.; Hellberg, L.; Witte, M.; Hansel, M.; Zillikens, D.; Solbach, W.; Laskay, T.; et al. Dimethylfumarate impairs neutrophil functions. J. Investig. Dermatol. 2016, 136, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Eng. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Simpson, M.A.; Pink, A.E.; Di Meglio, P.; Smith, C.H.; Pullabhatla, V.; Knight, J.; Spain, S.L.; Nestle, F.O.; Burden, A.D.; et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 2011, 89, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Takemoto, A.; Yamaguchi, M.; Takahashi, H.; Shoda, Y.; Mitsuma, T.; Tsuda, K.; Nishida, E.; Togawa, Y.; Nakajima, K.; et al. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J. Investig. Dermatol. 2013, 133, 2514–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, M.; Takeichi, T.; McGrath, J.A.; Sugiura, K. Autoinflammatory keratinization diseases: An emerging concept encompassing various inflammatory keratinization disorders of the skin. J. Dermatol. Sci. 2018, 90, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Iwata, Y.; Fukushima, H.; Saito, K.; Tanaka, Y.; Hasegawa, Y.; Akiyama, M.; Sugiura, K. Neutrophil extracellular traps are induced in a psoriasis model of interleukin-36 receptor antagonist-deficient mice. Sci. Rep. 2020, 10, 20149. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.; Troccaz, S.; Talabot-Ayer, D.; Rodriguez, E.; Palmer, G. The severity of imiquimod-induced mouse skin inflammation is independent of endogenous IL-38 expression. PLoS ONE 2018, 13, e0194667. [Google Scholar] [CrossRef] [PubMed]
- Tortola, L.; Rosenwald, E.; Abel, B.; Blumberg, H.; Schafer, M.; Coyle, A.J.; Renauld, J.C.; Werner, S.; Kisielow, J.; Kopf, M. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J. Clin. Investig. 2012, 122, 3965–3976. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Tu, J.; Hu, Y.; Song, G.; Yin, Z. Cathepsin G cleaves and activates IL-36gamma and promotes the inflammation of psoriasis. Drug Des. Devel. Ther. 2019, 13, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, D.M.; Henry, C.M.; Sullivan, G.P.; Martin, S.J. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017, 284, 1712–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, K.; Oiso, N.; Iinuma, S.; Matsuda, H.; Minami-Hori, M.; Ishida-Yamamoto, A.; Kawada, A.; Iizuka, H.; Akiyama, M. IL36RN mutations underlie impetigo herpetiformis. J. Investig. Dermatol. 2014, 134, 2472–2474. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, K.; Nakasuka, A.; Kono, H.; Kono, M.; Akiyama, M. Impetigo herpetiformis with IL36RN mutations in a Chinese patient: A founder haplotype of c.115+6T>C in East Asia. J. Dermatol. Sci. 2015, 79, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Ogawa, Y.; Takeichi, T.; Okamoto, T.; Osada, A.; Shimada, S.; Sugiura, K.; Akiyama, M.; Kawamura, T.; Tsukamoto, K. Impetigo herpetiformis with IL-36RN mutation successfully treated with secukinumab. Eur. J. Dermatol. 2018, 28, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Sugiura, K.; Akiyama, M.; Katoh, N. Acute generalized exanthematous pustulosis caused by dihydrocodeine phosphate in a patient with psoriasis vulgaris and a heterozygous IL36RN mutation. JAMA Dermatol. 2015, 151, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Neutrophil-mediated regulation of innate and adaptive immunity: The role of myeloperoxidase. J. Immunol. Res. 2016, 2016, 2349817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Veen, B.S.; de Winther, M.P.; Heeringa, P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal 2009, 11, 2899–2937. [Google Scholar] [CrossRef] [PubMed]
- Haskamp, S.; Bruns, H.; Hahn, M.; Hoffmann, M.; Gregor, A.; Lohr, S.; Hahn, J.; Schauer, C.; Ringer, M.; Flamann, C.; et al. Myeloperoxidase modulates inflammation in generalized pustular psoriasis and additional rare pustular skin diseases. Am. J. Hum. Genet. 2020, 107, 527–538. [Google Scholar] [CrossRef]
- Vergnano, M.; Mockenhaupt, M.; Benzian-Olsson, N.; Paulmann, M.; Grys, K.; Mahil, S.K.; Chaloner, C.; Barbosa, I.A.; August, S.; Burden, A.D.; et al. Loss-of-function myeloperoxidase mutations are associated with increased neutrophil counts and pustular skin disease. Am. J. Hum. Genet. 2020, 107, 539–543. [Google Scholar] [CrossRef]
- Frey, S.; Sticht, H.; Wilsmann-Theis, D.; Gerschutz, A.; Wolf, K.; Lohr, S.; Haskamp, S.; Frey, B.; Hahn, M.; Ekici, A.B.; et al. Rare loss-of-function mutation in SERPINA3 in generalized pustular psoriasis. J. Investig. Dermatol. 2020, 140, 1451–1455.e1413. [Google Scholar] [CrossRef]
- Twelves, S.; Mostafa, A.; Dand, N.; Burri, E.; Farkas, K.; Wilson, R.; Cooper, H.L.; Irvine, A.D.; Oon, H.H.; Kingo, K.; et al. Clinical and genetic differences between pustular psoriasis subtypes. J. Allergy Clin. Immunol. 2019, 143, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Alpsoy, E. Behcet’s disease: A comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. J. Dermatol. 2016, 43, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Safi, R.; Kallas, R.; Bardawil, T.; Mehanna, C.J.; Abbas, O.; Hamam, R.; Uthman, I.; Kibbi, A.G.; Nassar, D. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in Behcet’s disease. J. Dermatol. Sci. 2018, 92, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Perazzio, S.F.; Soeiro-Pereira, P.V.; Dos Santos, V.C.; de Brito, M.V.; Salu, B.; Oliva, M.L.V.; Stevens, A.M.; de Souza, A.W.S.; Ochs, H.D.; Torgerson, T.R.; et al. Soluble CD40L is associated with increased oxidative burst and neutrophil extracellular trap release in Behcet’s disease. Arthritis Res. Ther. 2017, 19, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yu, X.; Liu, J.; Wang, Z.; Li, C.; Shi, J.; Sun, L.; Liu, Y.; Zhang, F.; Chen, H.; et al. Neutrophil extracellular traps promote aberrant macrophages activation in Behcet’s disease. Front. Immunol. 2020, 11, 590622. [Google Scholar] [CrossRef]
- Le Joncour, A.; Martos, R.; Loyau, S.; Lelay, N.; Dossier, A.; Cazes, A.; Fouret, P.; Domont, F.; Papo, T.; Jandrot-Perrus, M.; et al. Critical role of neutrophil extracellular traps (NETs) in patients with Behcet’s disease. Ann. Rheum. Dis. 2019, 78, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Bettiol, A.; Becatti, M.; Silvestri, E.; Argento, F.R.; Fini, E.; Mannucci, A.; Galora, S.; Mattioli, I.; Urban, M.L.; Malandrino, D.; et al. Neutrophil-mediated mechanisms of damage and in-vitro protective effect of colchicine in non-vascular Behcet’s syndrome. Clin. Exp. Immunol. 2021, 206, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Vural, S.; Kerl, K.; Ertop Dogan, P.; Vollmer, S.; Puchta, U.; He, M.; Arakawa, Y.; Heper, A.O.; Karal-Oktem, A.; Hartmann, D.; et al. Lesional activation of Tc 17 cells in Behcet disease and psoriasis supports HLA class I-mediated autoimmune responses. Br. J. Dermatol. 2021. [Google Scholar] [CrossRef]
- Binus, A.M.; Qureshi, A.A.; Li, V.W.; Winterfield, L.S. Pyoderma gangrenosum: A retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br. J. Dermatol. 2011, 165, 1244–1250. [Google Scholar] [CrossRef]
- Seishima, M.; Mizutani, Y.; Shibuya, Y.; Nagasawa, C.; Aoki, T. Efficacy of granulocyte and monocyte adsorption apheresis for three cases of refractory pyoderma gangrenosum. Ther. Apher. Dial. 2007, 11, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Croia, C.; Dini, V.; Loggini, B.; Manni, E.; Romanelli, M.; Migliorini, P. Evaluation of neutrophil extracellular trap deregulated formation in pyoderma gangrenosum. Exp. Dermatol. 2021, 30, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Eid, E.; Safi, R.; El Hasbani, G.; Aftimos, V.; Abbas, O.; Kibbi, A.G.; Nassar, D. Characterizing the presence of neutrophil extracellular traps in neutrophilic dermatoses. Exp. Dermatol. 2021, 30, 988–994. [Google Scholar] [CrossRef]
- Bonnekoh, H.; Scheffel, J.; Wu, J.; Hoffmann, S.; Maurer, M.; Krause, K. Skin and systemic inflammation in Schnitzler’s syndrome are associated with neutrophil extracellular trap formation. Front. Immunol. 2019, 10, 546. [Google Scholar] [CrossRef] [Green Version]
- Disdier, P.; Harle, J.R.; Weiller-Merli, C.; Andrac, L.; Weiller, P.J. Neutrophilic dermatosis despite myeloperoxidase deficiency. J. Am. Acad. Dermatol. 1991, 24, 654–655. [Google Scholar] [CrossRef]
- Wise, C.A.; Gillum, J.D.; Seidman, C.E.; Lindor, N.M.; Veile, R.; Bashiardes, S.; Lovett, M. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum. Mol. Genet. 2002, 11, 961–969. [Google Scholar] [CrossRef]
- Mistry, P.; Carmona-Rivera, C.; Ombrello, A.K.; Hoffmann, P.; Seto, N.L.; Jones, A.; Stone, D.L.; Naz, F.; Carlucci, P.; Dell’Orso, S.; et al. Dysregulated neutrophil responses and neutrophil extracellular trap formation and degradation in PAPA syndrome. Ann. Rheum. Dis. 2018, 77, 1825–1833. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.L.; Karl, I.; Giner, T.; Poppe, H.; Schmidt, M.; Presser, D.; Goebeler, M.; Bauer, B. Keratinocytes and neutrophils are important sources of proinflammatory molecules in hidradenitis suppurativa. Br. J. Dermatol. 2016, 174, 514–521. [Google Scholar] [CrossRef]
- Schlapbach, C.; Hanni, T.; Yawalkar, N.; Hunger, R.E. Expression of the IL-23/Th17 pathway in lesions of hidradenitis suppurativa. J. Am. Acad. Dermatol. 2011, 65, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.S.; Carmona-Rivera, C.; O’Neil, L.J.; Carlucci, P.M.; Cisar, C.; Rosenberg, A.Z.; Kerns, M.L.; Caffrey, J.A.; Milner, S.M.; Sacks, J.M.; et al. Neutrophil extracellular traps, B cells, and type I interferons contribute to immune dysregulation in hidradenitis suppurativa. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Navrazhina, K.; Frew, J.W.; Gilleaudeau, P.; Sullivan-Whalen, M.; Garcet, S.; Krueger, J.G. Epithelialized tunnels are a source of inflammation in hidradenitis suppurativa. J. Allergy Clin. Immunol. 2021, 147, 2213–2224. [Google Scholar] [CrossRef]
- Viard, I.; Wehrli, P.; Bullani, R.; Schneider, P.; Holler, N.; Salomon, D.; Hunziker, T.; Saurat, J.H.; Tschopp, J.; French, L.E. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998, 282, 490–493. [Google Scholar] [CrossRef]
- Abe, R.; Shimizu, T.; Shibaki, A.; Nakamura, H.; Watanabe, H.; Shimizu, H. Toxic epidermal necrolysis and Stevens-Johnson syndrome are induced by soluble Fas ligand. Am. J. Pathol. 2003, 162, 1515–1520. [Google Scholar] [CrossRef]
- Nassif, A.; Bensussan, A.; Dorothee, G.; Mami-Chouaib, F.; Bachot, N.; Bagot, M.; Boumsell, L.; Roujeau, J.C. Drug specific cytotoxic T-cells in the skin lesions of a patient with toxic epidermal necrolysis. J. Investig. Dermatol. 2002, 118, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.H.; Hung, S.I.; Yang, J.Y.; Su, S.C.; Huang, S.P.; Wei, C.Y.; Chin, S.W.; Chiou, C.C.; Chu, S.C.; Ho, H.C.; et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat. Med. 2008, 14, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Su, S.C.; Mockenhaupt, M.; Wolkenstein, P.; Dunant, A.; Le Gouvello, S.; Chen, C.B.; Chosidow, O.; Valeyrie-Allanore, L.; Bellon, T.; Sekula, P.; et al. Interleukin-15 is associated with severity and mortality in Stevens-Johnson syndrome/toxic epidermal necrolysis. J. Investig. Dermatol. 2017, 137, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, M.; Ogawa, Y.; Hama, N.; Ujiie, I.; Hasegawa, A.; Nakajima, S.; Nomura, T.; Adachi, J.; Sato, T.; Koizumi, S.; et al. Neutrophils initiate and exacerbate Stevens-Johnson syndrome and toxic epidermal necrolysis. Sci. Transl. Med. 2021, 13, eaax2398. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Qiao, H.; Yanagi, T.; Shinkuma, S.; Nishimura, K.; Suto, A.; Fujita, Y.; Suzuki, S.; Nomura, T.; Nakamura, H.; et al. An annexin A1-FPR1 interaction contributes to necroptosis of keratinocytes in severe cutaneous adverse drug reactions. Sci. Transl. Med. 2014, 6, 245ra295. [Google Scholar] [CrossRef] [PubMed]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totani, L.; Amore, C.; Piccoli, A.; Dell’Elba, G.; Di Santo, A.; Plebani, R.; Pecce, R.; Martelli, N.; Rossi, A.; Ranucci, S.; et al. Type-4 phosphodiesterase (PDE4) blockade reduces NETosis in cystic fibrosis. Front. Pharmacol. 2021, 12, 702677. [Google Scholar] [CrossRef]
- Sharma, S.; Hofbauer, T.M.; Ondracek, A.S.; Chausheva, S.; Alimohammadi, A.; Artner, T.; Panzenboeck, A.; Rinderer, J.; Shafran, I.; Mangold, A.; et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood 2021, 137, 1104–1116. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogawa, Y.; Muto, Y.; Kinoshita, M.; Shimada, S.; Kawamura, T. Neutrophil Extracellular Traps in Skin Diseases. Biomedicines 2021, 9, 1888. https://doi.org/10.3390/biomedicines9121888
Ogawa Y, Muto Y, Kinoshita M, Shimada S, Kawamura T. Neutrophil Extracellular Traps in Skin Diseases. Biomedicines. 2021; 9(12):1888. https://doi.org/10.3390/biomedicines9121888
Chicago/Turabian StyleOgawa, Youichi, Yoshinori Muto, Manao Kinoshita, Shinji Shimada, and Tatsuyoshi Kawamura. 2021. "Neutrophil Extracellular Traps in Skin Diseases" Biomedicines 9, no. 12: 1888. https://doi.org/10.3390/biomedicines9121888
APA StyleOgawa, Y., Muto, Y., Kinoshita, M., Shimada, S., & Kawamura, T. (2021). Neutrophil Extracellular Traps in Skin Diseases. Biomedicines, 9(12), 1888. https://doi.org/10.3390/biomedicines9121888