
C3d Elicits Neutrophil Degranulation and Decreases Endothelial Cell Migration, with Implications for Patients with Alpha-1 Antitrypsin Deficiency

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Study PoPulation
2.3. Isolation of Plasma and Neutrophils from Whole Blood
2.4. Sodium Dodecyl Sulphate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot Analysis
2.5. C3d Analysis
2.6. Degranulation Assays
2.7. CXCL8 Secretion Analysis
2.8. In Vitro Endothelial Cell Scratch Wound Assays
2.9. Data Analysis
3. Results
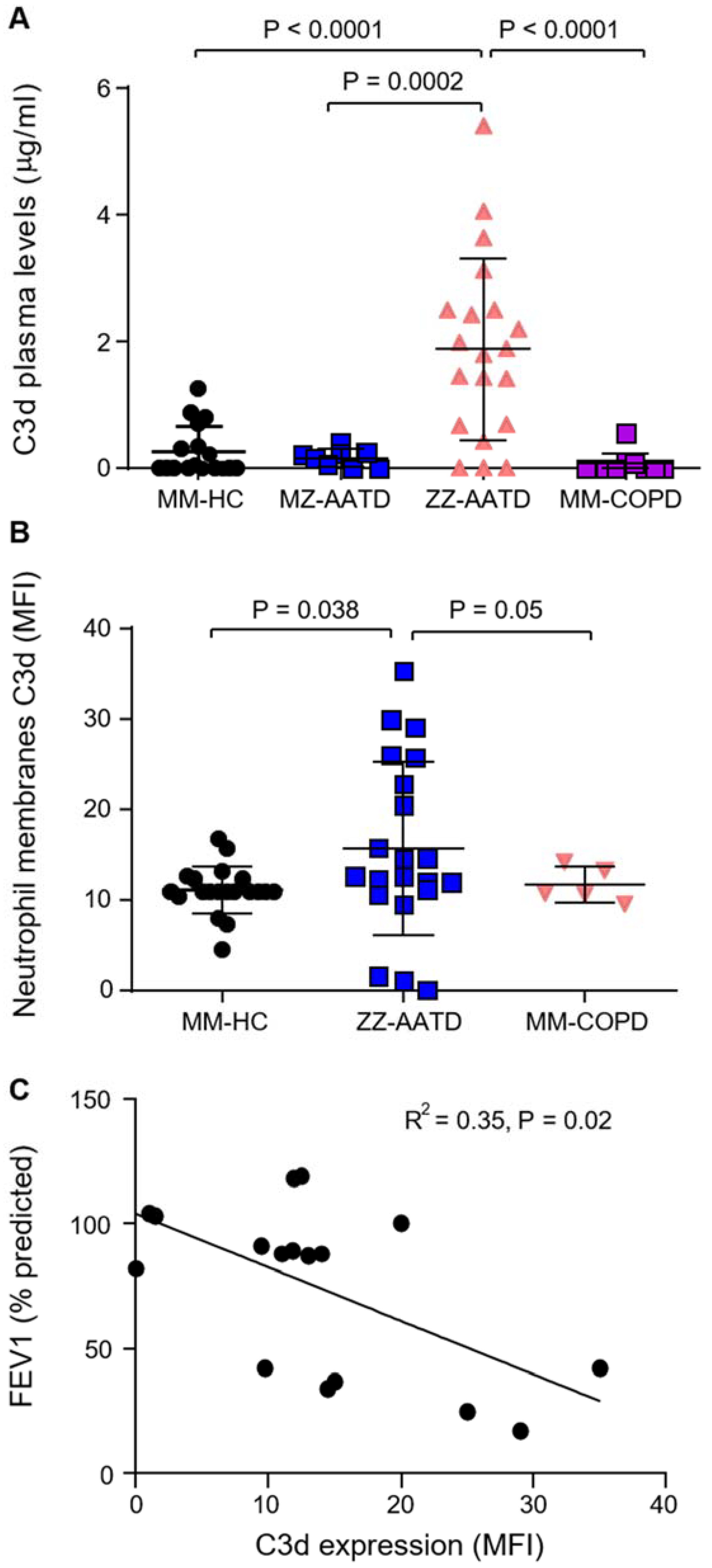
3.1. Increased C3d in Plasma and on Circulating Neutrophils in AATD
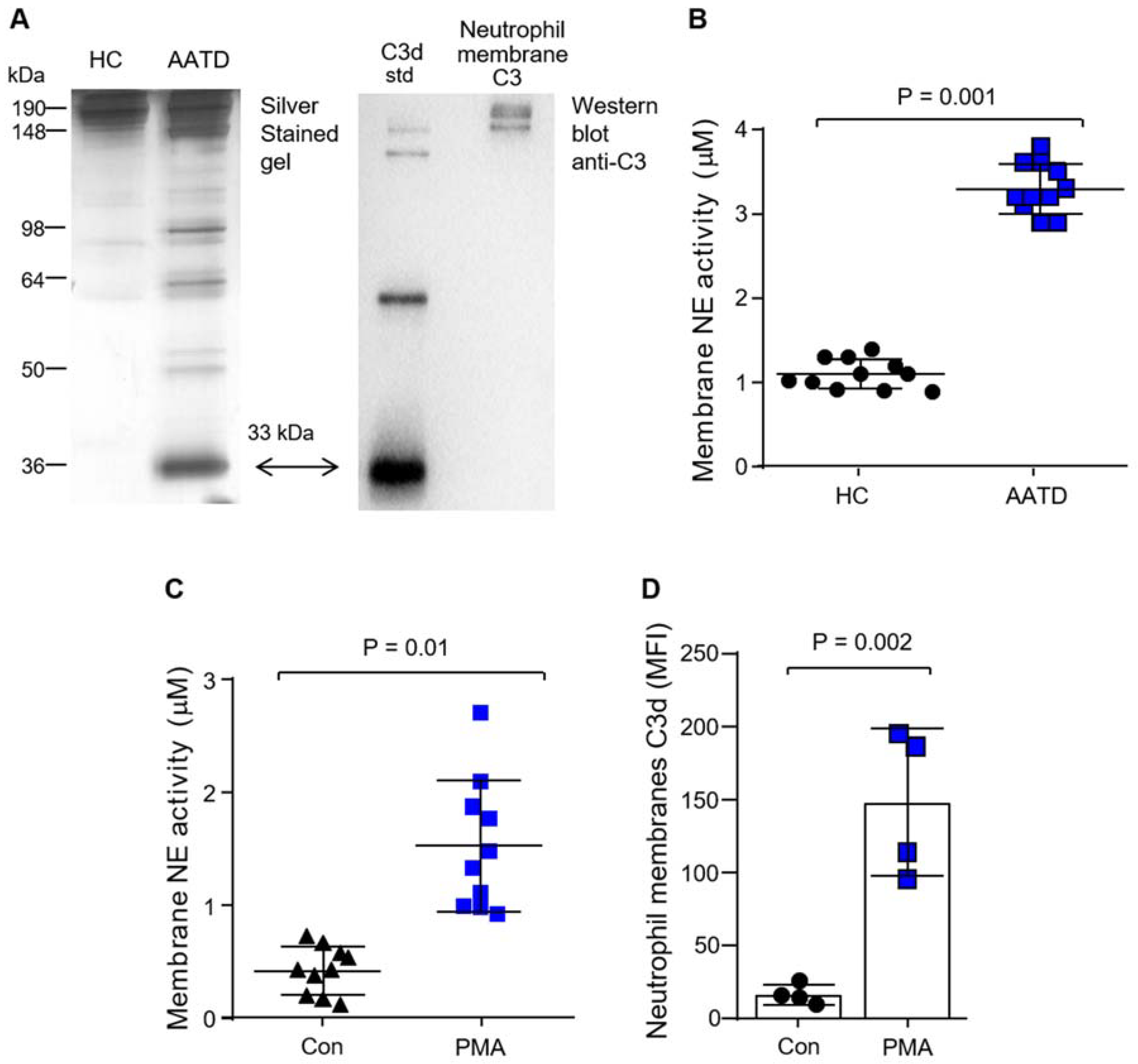
3.2. Production of C3d by Neutrophil Membrane-Bound Neutrophil Elastase
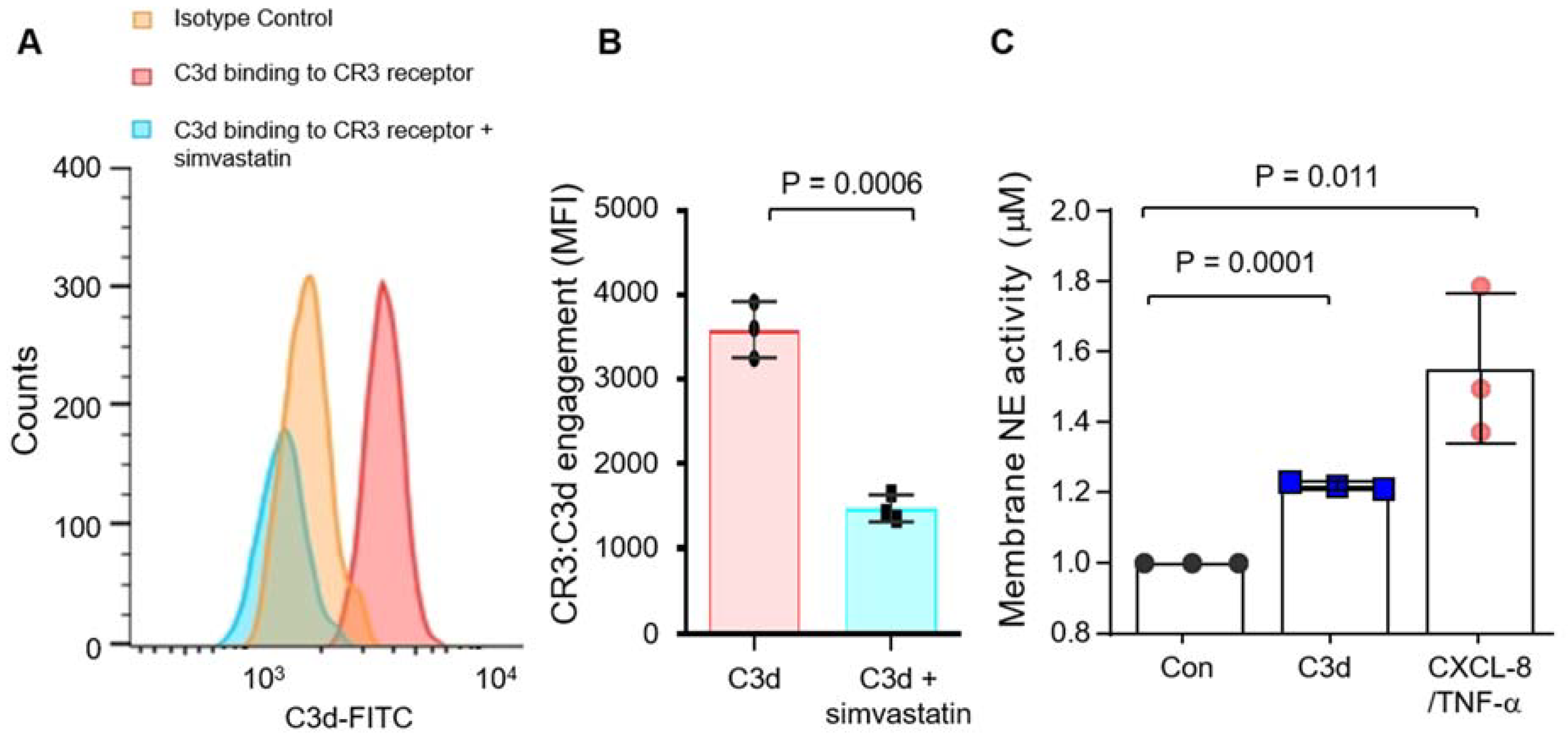
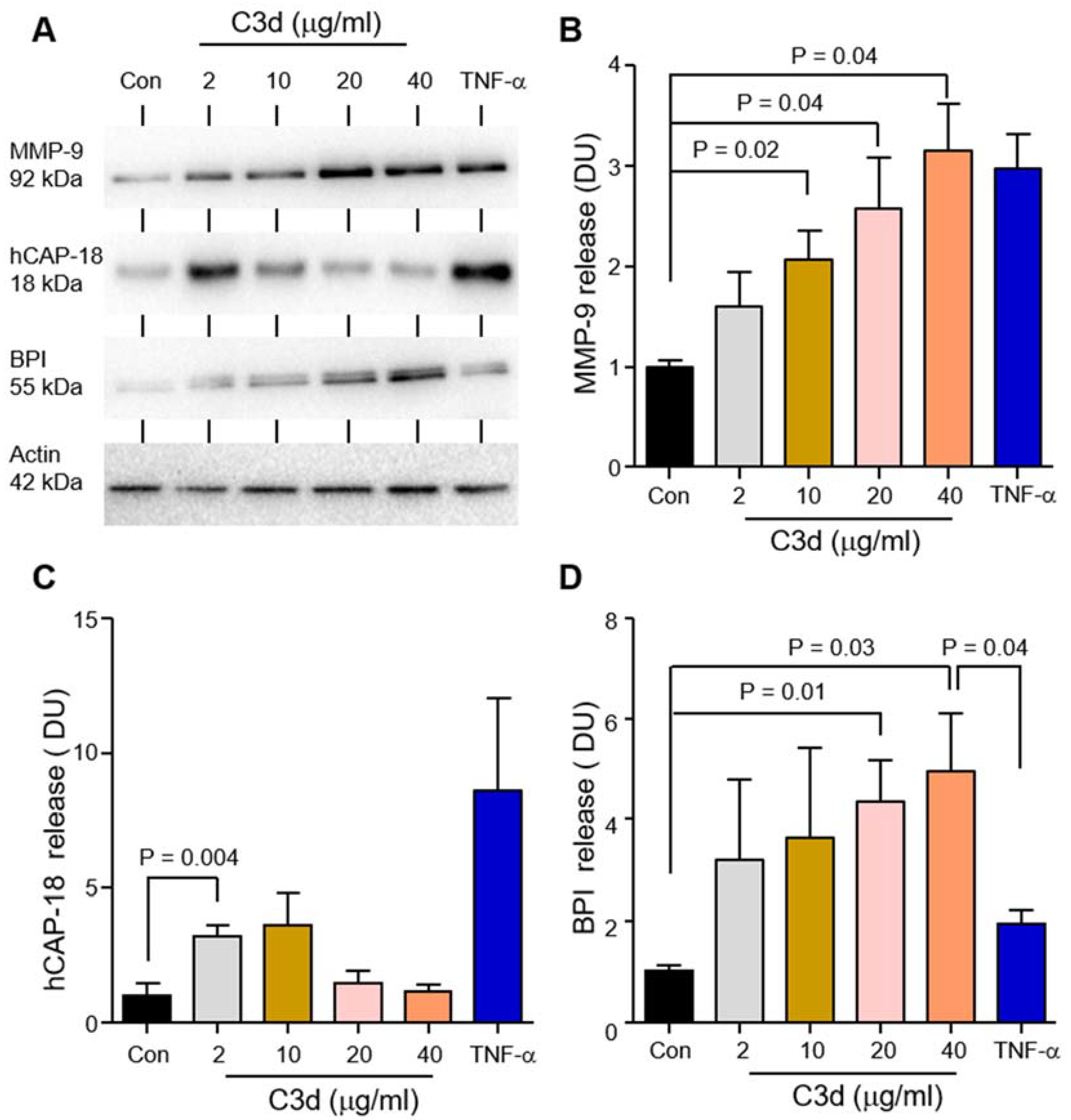
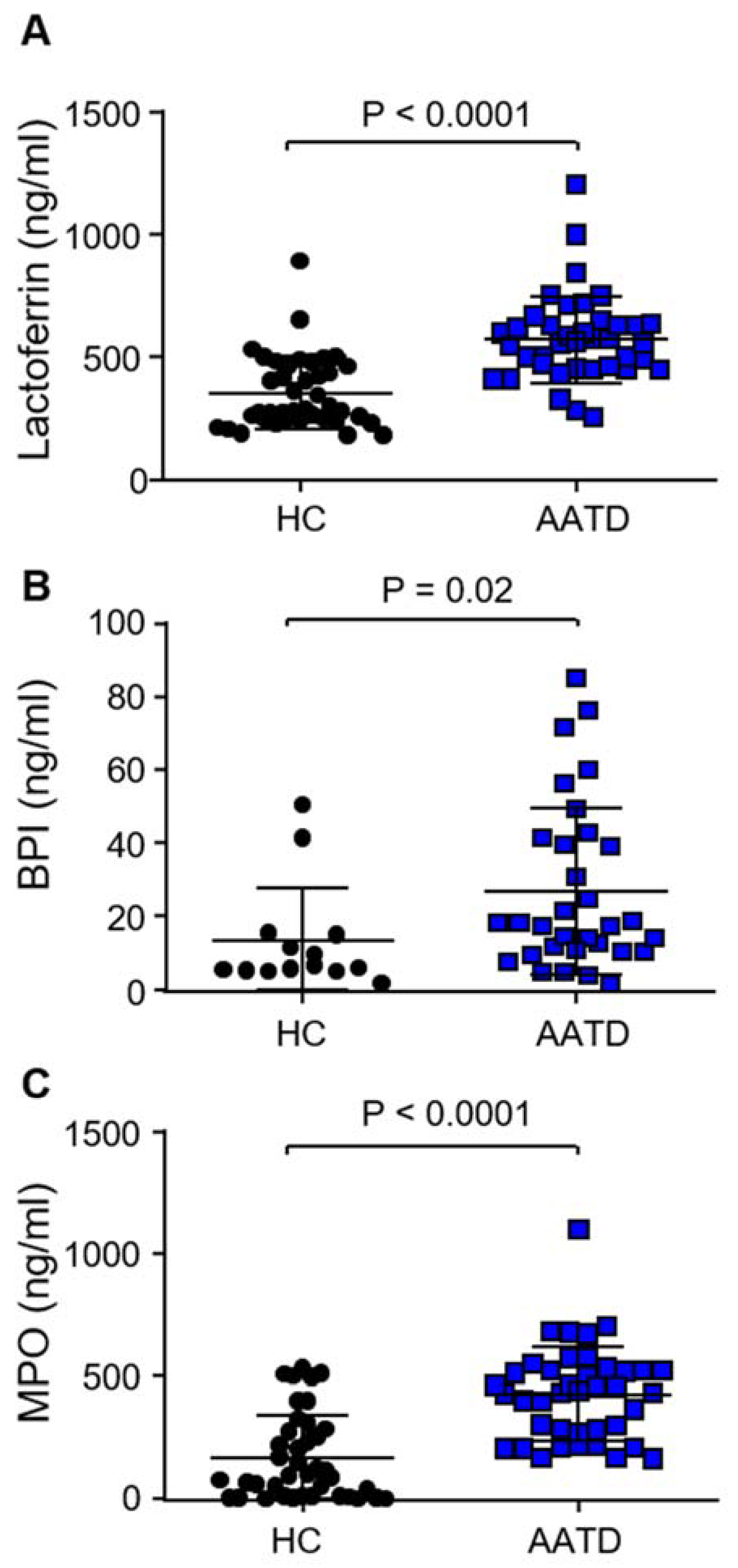
3.3. Degranulation of Neutrophil Granule Subtypes in Respone to C3d
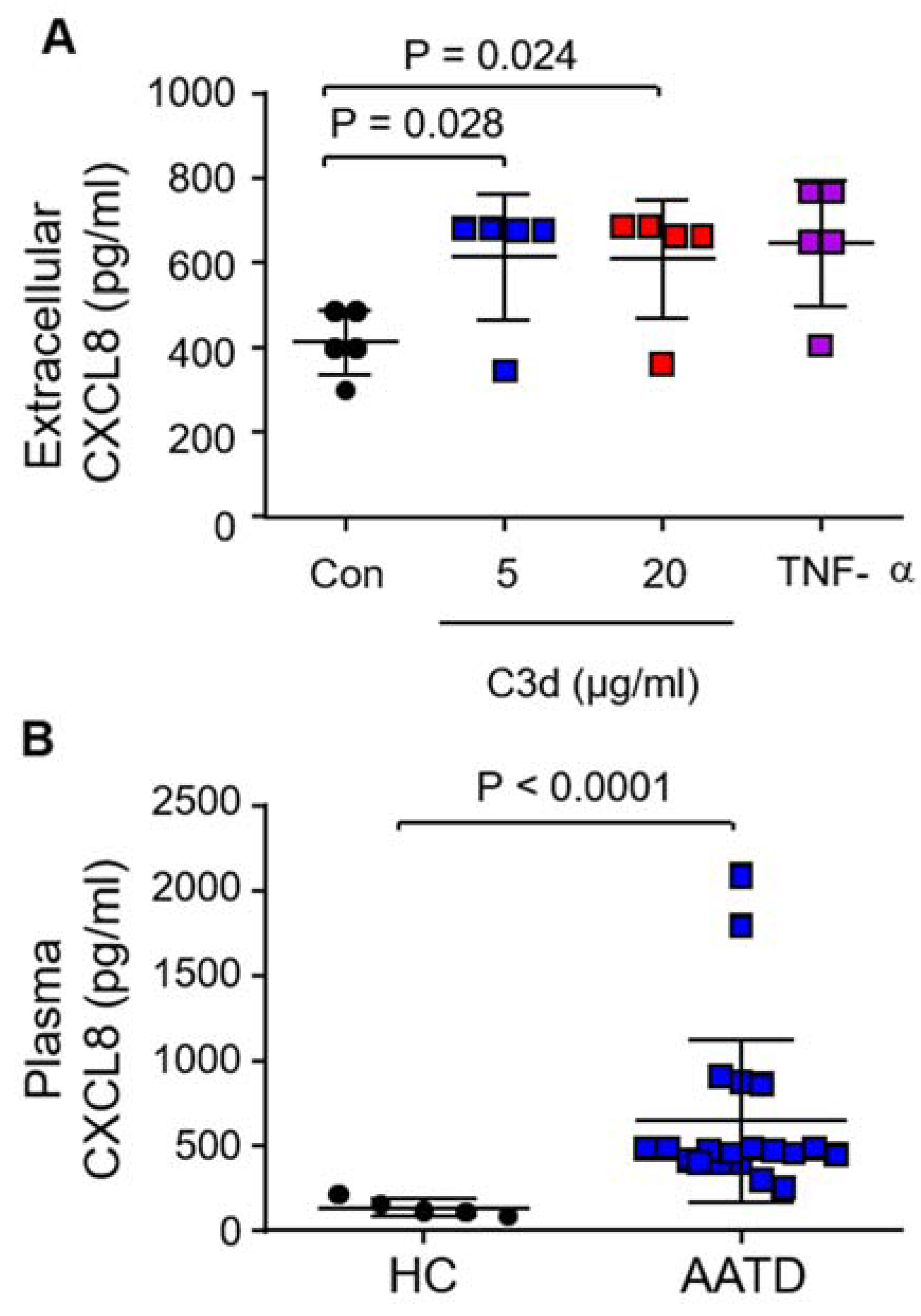
3.4. Secretion of Pro-Inflammatory CXCL8 in Response to C3d
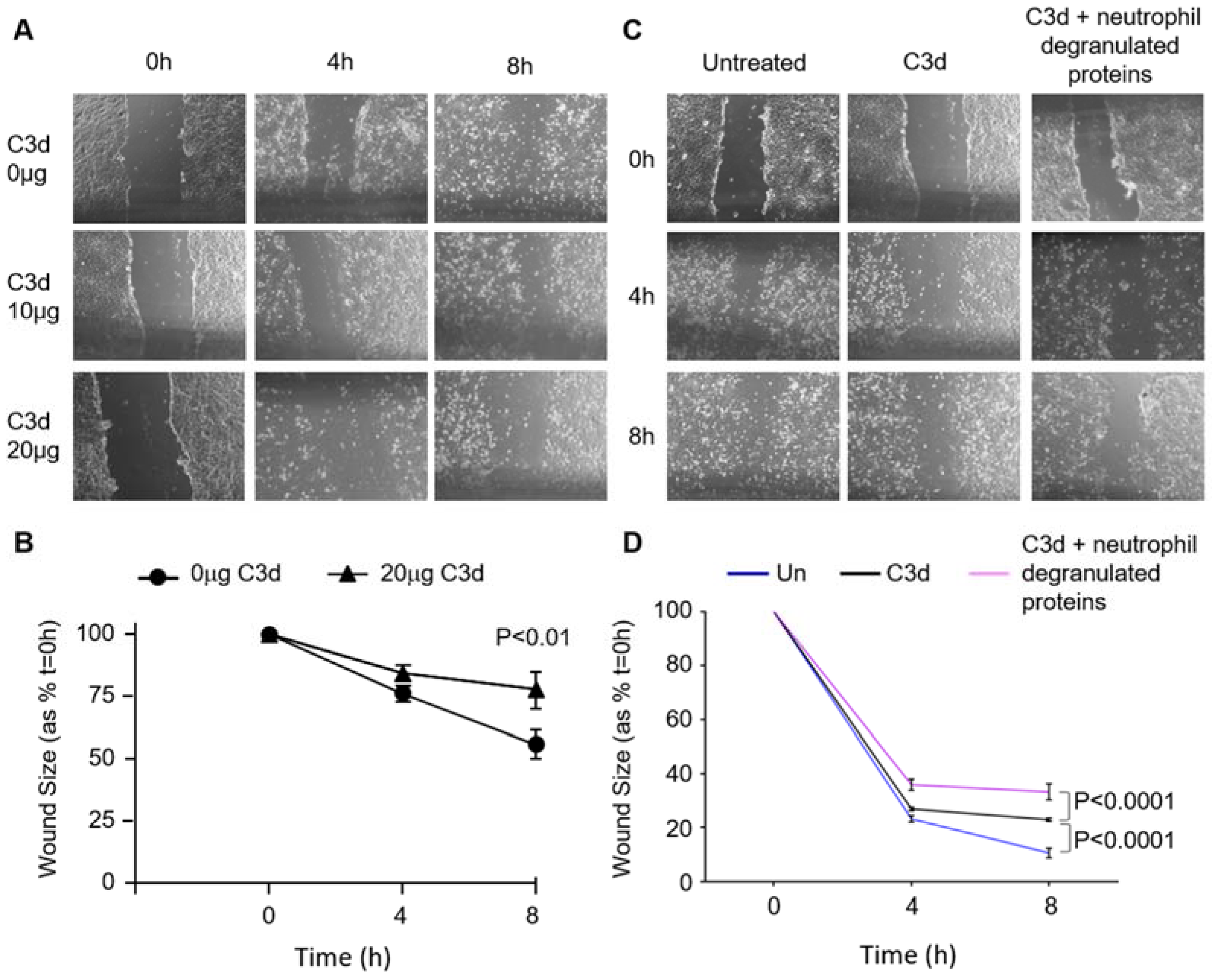
3.5. The Impact of C3d on Wound Healing of Vascular Endothelial Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolarich, D.; Turecek, P.L.; Weber, A.; Mitterer, A.; Graninger, M.; Matthiessen, P.; Nicolaes, G.A.; Altmann, F.; Schwarz, H.P. Biochemical, molecular characterization, and glycoproteomic analyses of ?1-proteinase inhibitor products used for replacement therapy. Transfusion 2006, 46, 1959–1977. [Google Scholar] [CrossRef]
- Kolarich, D.; Weber, A.; Turecek, P.L.; Schwarz, H.-P.; Altmann, F. Comprehensive glyco-proteomic analysis of human α1-antitrypsin and its charge isoforms. Proteome 2006, 6, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J. Clin. Investig. 1990, 85, 1343–1352. [Google Scholar] [CrossRef]
- Curiel, D.T.; Chytil, A.; Courtney, M.; Crystal, R.G. Serum α1-Antitrypsin Deficiency Associated with the Common S-type (Glu264 → Val) Mutation Results from Intracellular Degradation of α1- Antitrypsin Prior to Secretion. J. Biol. Chem. 1989, 264, 10477–10486. [Google Scholar] [CrossRef]
- Lomas, D.A.; Li-Evans, D.; Finch, J.T.; Carrell, R.W. The mechanism of Z α1-antitrypsin accumulation in the liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- DeMeo, D.L. 1-Antitrypsin deficiency {middle dot} 2: Genetic aspects of 1-antitrypsin deficiency: Phenotypes and genetic modifiers of emphysema risk. Thorax 2004, 59, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantly, M.; Nukiwa, T.; Crystal, R.G. Molecular basis of alpha-1-antitrypsin deficiency. Am. J. Med. 1988, 84, 13–31. [Google Scholar] [CrossRef]
- Abboud, R.; Nelson, T.; Jung, B.; Mattman, A. Alpha1-antitrypsin deficiency: A clinical-genetic overview. Appl. Clin. Genet. 2011, 4, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotondo, J.C.; Oton-Gonzalez, L.; Selvatici, R.; Rizzo, P.; Pavasini, R.; Campo, G.C.; Lanzillotti, C.; Mazziotta, C.; De Mattei, M.; Tognon, M.; et al. SERPINA1 Gene Promoter Is Differentially Methylated in Peripheral Blood Mononuclear Cells of Pregnant Women. Front. Cell Dev. Biol. 2020, 8, 550543. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.T.; Campbell, E.J.; Bayley, D.L.; Hill, S.L.; Stockley, R.A. Evidence for Excessive Bronchial Inflammation during an Acute Exacerbation of Chronic Obstructive Pulmonary Disease in Patients with α1-Antitrypsin Deficiency (PiZ). Am. J. Respir. Crit. Care Med. 1999, 160, 1968–1975. [Google Scholar] [CrossRef]
- Needham, M. Exacerbations in 1-antitrypsin deficiency. Eur. Respir. J. 2005, 25, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranton, J.; Bieth, J.G. Inhibition of Proteinase 3 by ?1-AntitrypsinIn VitroPredicts Very Fast InhibitionIn Vivo. Am. J. Respir. Cell Mol. Biol. 2003, 29, 57–61. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, C.A.; O’Brien, M.E.; Wormald, M.R.; White, M.; Banville, N.; Hurley, K.; McCarthy, C.; McElvaney, N.G.; Reeves, E.P. The BLT1 Inhibitory Function of α-1 Antitrypsin Augmentation Therapy Disrupts Leukotriene B4Neutrophil Signaling. J. Immunol. 2015, 195, 3628–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergin, D.A.; Reeves, E.P.; Hurley, K.; Wolfe, R.; Jameel, R.; Fitzgerald, S.; McElvaney, N.G. The Circulating Proteinase Inhibitor α-1 Antitrypsin Regulates Neutrophil Degranulation and Autoimmunity. Sci. Transl. Med. 2014, 6, 217ra1. [Google Scholar] [CrossRef] [PubMed]
- Bergin, D.A.; Reeves, E.P.; Meleady, P.; Henry, M.; McElvaney, O.J.; Carroll, T.P.; Condron, C.; Chotirmall, S.H.; Clynes, M.; O’Neill, S.J.; et al. α-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J. Clin. Investig. 2010, 120, 4236–4250. [Google Scholar] [CrossRef] [Green Version]
- Griese, M.; Latzin, P.; Kappler, M.; Weckerle, K.; Heinzlmaier, T.; Bernhardt, T.; Hartl, D. 1-Antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur. Respir. J. 2006, 29, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Kalis, M.; Kumar, R.; Janciauskiene, S.; Salehi, A.; Cilio, C.M. α 1-antitrypsin enhances insulin secretion and prevents cytokine-mediated apoptosis in pancreatic β-cells. Islets 2010, 2, 185–189. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, C.; Dunlea, D.M.; Saldova, R.; Henry, M.; Meleady, P.; McElvaney, O.J.; Marsh, B.; Rudd, P.M.; Reeves, E.P.; McElvaney, N.G. Glycosylation Repurposes Alpha-1 Antitrypsin for Resolution of Community-acquired Pneumonia. Am. J. Respir. Crit. Care Med. 2018, 197, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- E O’Brien, M.; Fee, L.; Browne, N.; Carroll, T.P.; Meleady, P.; Henry, M.; McQuillan, K.; Murphy, M.P.; Logan, M.; McCarthy, C.; et al. Activation of complement component 3 is associated with airways disease and pulmonary emphysema in alpha-1 antitrypsin deficiency. Thorax 2020, 75, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westall, G.P.; Snell, G.I.; McLean, C.; Kotsimbos, T.; Williams, T.; Magro, C. C3d and C4d Deposition Early After Lung Transplantation. J. Hear. Lung Transplant. 2008, 27, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.; Richards, N.; Hornby, J.; Powell, R. Relation between synovial fluid C3 degradation products and local joint inflammation in rheumatoid arthritis, osteoarthritis, and crystal associated arthropathy. Ann. Rheum. Dis. 1988, 47, 190–197. [Google Scholar] [CrossRef]
- Lyons, P.A.; Rayner, T.F.; Trivedi, S.; Holle, J.U.; Watts, R.A.; Jayne, D.R.; Baslund, B.; Brenchley, P.; Bruchfeld, A.; Chaudhry, A.N.; et al. Genetically distinct subsets within ANCA-associated vasculitis. N. Engl. J. Med. 2012, 367, 214–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segelmark, M.; Elzouki, A.-N.; Wieslander, J.; Eriksson, S. The PiZ gene of α1-antitrypsin as a determinant of outcome in PR3-ANCA-positive vasculitis. Kidney Int. 1995, 48, 844–850. [Google Scholar] [CrossRef] [Green Version]
- Cathomas, M.; Schüller, A.; Candinas, D.; Inglin, R. Severe postoperative wound healing disturbance in a patient with alpha-1-antitrypsin deficiency: The impact of augmentation therapy. Int. Wound J. 2015, 12, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Hernández Pérez, J.M.; Fumero García, S.; Alvarez Pío, A. Successful α1-antitrypsin replacement therapy in a patient with α1-antitrypsin deficiency and granulomatosis with polyangiitis. Rheumatology 2013, 52, 755–757. [Google Scholar] [CrossRef]
- Franciosi, A.N.; Ralph, J.; O’Farrell, N.J.; Buckley, C.; Gulmann, C.; O’Kane, M.; Carroll, T.P.; McElvaney, N.G. Alpha-1 antitrypsin deficiency–associated panniculitis. J. Am. Acad. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Vorup-Jensen, T.; Jensen, R.K. Structural Immunology of Complement Receptors 3 and 4. Front. Immunol. 2018, 9, 2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.M.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nat. Cell Biol. 2002, 416, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Saeki, K.; Saeki, K.; Nakahara, M.; Matsuyama, S.; Nakamura, N.; Yogiashi, Y.; Yoneda, A.; Koyanagi, M.; Kondo, Y.; Yuo, A. A Feeder-Free and Efficient Production of Functional Neutrophils from Human Embryonic Stem Cells. STEM CELLS 2009, 27, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.R.; Bajic, G.; Zhang, X.; Laustsen, A.K.; Koldsø, H.; Skeby, K.K.; Schiøtt, B.; Andersen, G.R.; Vorup-Jensen, T. Structural Basis for Simvastatin Competitive Antagonism of Complement Receptor 3. J. Biol. Chem. 2016, 291, 16963–16976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.; Sya, J.; Hup, N.; Murphy, M.; McElvaney, N.; Reeves, E. Alpha-1 Antitrypsin Augmentation Inhibits Proteolysis of Neutrophil Membrane Voltage-Gated Proton Channel-1 in Alpha-1 Deficient Individuals. Medicina 2021, 57, 814. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlo, J.R.; Spitznagel, J.K.; Studer, E.J.; Conrad, D.H.; Ruddy, S. Cleavage of membrane bound C3bi, an intermediate of the third component of complement, to C3c and C3d-like fragments by crude leucocyte lysosomal lysates and purified leucocyte elastase. Immunology 1981, 44, 381–391. [Google Scholar]
- Taylor, J.C.; Crawford, I.P.; Hugli, T.E. Limited degradation of the third component (C3) of human complement by human leukocyte elastase (HLE): Partial characterization of C3 fragments. Biochemistry 1977, 16, 3390–3396. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Kasahara, K.; Allen, R.M.; Standiford, T.J.; Rolfe, M.W.; Becker, F.S.; Chensue, S.W.; Kunkel, S.L. Cytokine-induced neutrophil-derived interleukin-8. Am. J. Pathol. 1992, 141, 397–407. [Google Scholar] [PubMed]
- Etecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-Derived Cytokines: Facts Beyond Expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef] [Green Version]
- Morris, H.; Morgan, M.D.; Wood, A.M.; Smith, S.W.; Ekeowa, U.I.; Herrmann, K.; Holle, J.U.; Guillevin, L.; Lomas, D.A.; Perez, J.; et al. ANCA-associated vasculitis is linked to carriage of the Z allele of α1 antitrypsin and its polymers. Ann. Rheum. Dis. 2011, 70, 1851–1856. [Google Scholar] [CrossRef]
- Malerba, M.; Ricciardolo, F.; Radaeli, A.; Torregiani, C.; Ceriani, L.; Mori, E.; Bontempelli, M.; Tantucci, C.; Grassi, V. Neutrophilic inflammation and IL-8 levels in induced sputum of alpha-1-antitrypsin PiMZ subjects. Thorax 2006, 61, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Rouhani, F.; Paone, G.; Smith, N.K.; Krein, P.; Barnes, P.; Brantly, M.L. Lung Neutrophil Burden Correlates With Increased Pro-Inflammatory Cytokines and Decreased Lung Function in Individuals With α1-Antitrypsin Deficiency. Chest 2000, 117, 250S–251S. [Google Scholar] [CrossRef]
- Hubbard, R.C.; Fells, G.; Gadek, J.; Pacholok, S.; Humes, J.; Crystal, R.G. Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J. Clin. Investig. 1991, 88, 891–897. [Google Scholar] [CrossRef]
- Nilsson, B.; Ekdahl, K.N. Complement Diagnostics: Concepts, Indications, and Practical Guidelines. Clin. Dev. Immunol. 2012, 2012, 962702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genschmer, K.R.; Russell, D.W.; Lal, C.; Szul, T.; Bratcher, P.E.; Noerager, B.D.; Roda, M.A.; Xu, X.; Rezonzew, G.; Viera, L.; et al. Activated PMN Exosomes: Pathogenic Entities Causing Matrix Destruction and Disease in the Lung. Cell 2019, 176, 113–126.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.; McEnery, T.; McQuillan, K.; McElvaney, O.F.; McElvaney, O.J.; Landers, S.; Coleman, O.; Bussayajirapong, A.; Hawkins, P.; Henry, M.; et al. α1 Antitrypsin therapy modulates the neutrophil membrane proteome and secretome. Eur. Respir. J. 2020, 55, 1901678. [Google Scholar] [CrossRef] [PubMed]
- Van Doren, S.R. Matrix metalloproteinase interactions with collagen and elastin. Matrix Biol. 2015, 44, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Finlay, G.A.; O’Driscoll, L.; Russell, K.J.; D’Arcy, E.M.; Masterson, J.B.; Fitzgerald, M.X.; O’Connor, C.M. Matrix Metalloproteinase Expression and Production by Alveolar Macrophages in Emphysema. Am. J. Respir. Crit. Care Med. 1997, 156, 240–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.; Sorensen, R.U.; Tosi, M.F.; Dearborn, D.G.; Döring, G. Complement receptor expression on neutrophils at an inflammatory site, the Pseudomonas-infected lung in cystic fibrosis. J. Clin. Investig. 1989, 84, 1302–1313. [Google Scholar] [CrossRef] [Green Version]
- Ying, Q.-L.; Simon, S.R. Elastolysis by Proteinase 3 and its Inhibition by α1-Proteinase Inhibitor. Am. J. Respir. Cell Mol. Biol. 2002, 26, 356–361. [Google Scholar] [CrossRef]
- Audrain, M.A.P.; Sesboüé, R.; Baranger, T.A.R.; Elliott, J.; Testa, A.; Martin, J.; Lockwood, C.M.; Esnault, V.L.M. Analysis of anti-neutrophil cytoplasmic antibodies (ANCA): Frequency and specificity in a sample of 191 homozygous (PiZZ) alpha1-antitrypsin-deficient subjects. Nephrol. Dial. Transplant. 2001, 16, 39–44. [Google Scholar] [CrossRef]
- Franciosiz, A.N.; McCarthy, C.; Carroll, T.; McElvaney, N.G. Unusual Acute Sequelae of α 1 -Antitrypsin Deficiency. Chest 2015, 148, e136–e138. [Google Scholar] [CrossRef]
- McCarthy, C.; Orr, C.; Fee, L.T.; Carroll, T.; Dunlea, D.M.; Hunt, D.; Dunne, E.; O’Connell, P.; McCarthy, G.; Kenny, D.; et al. Brief Report: Genetic Variation of the α1 -Antitrypsin Gene Is Associated With Increased Autoantibody Production in Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, K.; Gargoum, F.; Murphy, M.P.; McElvaney, O.J.; McElvaney, N.G.; Reeves, E.P. Targeting IgG Autoantibodies for Improved Cytotoxicity of Bactericidal Permeability Increasing Protein in Cystic Fibrosis. Front. Pharmacol. 2020, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Schultz, H.; Csernok, E.; Schuster, A.; Schmitz, T.S.; Ernst, M.; Gross, W.L. Anti-neutrophil cytoplasmic antibodies directed against the bactericidal/permeability-increasing protein (BPI) in pediatric cystic fibrosis patients do not recognize N-terminal regions important for the anti-microbial and lipopolysaccharide-binding activi. Pediatr. Allergy Immunol. 2000, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Mahadeva, R.; Dunn, A.C.; Westerbeek, R.C.; Sharples, L.; Whitehouse, D.B.; Carroll, N.R.; Russell, R.-I.; Webb, A.K.; Bilton, D.; Lomas, D.; et al. Anti-neutrophil cytoplasmic antibodies (ANCA) against bactericidal/permeability-increasing protein (BPI) and cystic fibrosis lung disease. Clin. Exp. Immunol. 1999, 117, 561–567. [Google Scholar] [CrossRef]
- Gou, S.-J.; Yuan, J.; Chen, M.; Yu, F.; Zhao, M.-H. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody–associated vasculitis. Kidney Int. 2013, 83, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Osman, M.; Tervaert, J.W.C.; Pagnoux, C. Avacopan for the treatment of ANCA-associated vasculitis. Expert Rev. Clin. Immunol. 2021, 17, 717–726. [Google Scholar] [CrossRef]
- Chapman, K.R.; Burdon, J.G.W.; Piitulainen, E.; Sandhaus, R.A.; Seersholm, N.; Stocks, J.M.; Stoel, B.C.; Huang, L.; Yao, Z.; Edelman, J.M.; et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 386, 360–368. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZZ-AATD | MZ-AATD | MM-COPD | HC | |
|---|---|---|---|---|
| Number of subjects | 80 | 8 | 10 | 40 |
| Age, years (mean ± SD) | 49.62 ± 13.92 | 55.86 ± 15.08 | 70.86 ± 6.71 | 30.38 ± 5.01 |
| FEV1 (% predicted) | 75.54 ± 33.79 | 70.86 ± 24.48 | 57.3 ± 23.32 | 99.56 ± 7.9 |
| FEV1/FVC (% predicted) | 59.15 ± 20.72 | 61.86 ± 17.28 | 50 ± 16.94 | 80.15 ± 6.52 |
| DLCO (% predicted) | 64.58 ± 24.98 | 81.00 ±21.43 | 36 ± 10.86 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fee, L.T.; Gogoi, D.; O’Brien, M.E.; McHugh, E.; Casey, M.; Gough, C.; Murphy, M.; Hopkins, A.M.; Carroll, T.P.; McElvaney, N.G.; et al. C3d Elicits Neutrophil Degranulation and Decreases Endothelial Cell Migration, with Implications for Patients with Alpha-1 Antitrypsin Deficiency. Biomedicines 2021, 9, 1925. https://doi.org/10.3390/biomedicines9121925
Fee LT, Gogoi D, O’Brien ME, McHugh E, Casey M, Gough C, Murphy M, Hopkins AM, Carroll TP, McElvaney NG, et al. C3d Elicits Neutrophil Degranulation and Decreases Endothelial Cell Migration, with Implications for Patients with Alpha-1 Antitrypsin Deficiency. Biomedicines. 2021; 9(12):1925. https://doi.org/10.3390/biomedicines9121925
Chicago/Turabian StyleFee, Laura T., Debananda Gogoi, Michael E. O’Brien, Emer McHugh, Michelle Casey, Ciara Gough, Mark Murphy, Ann M. Hopkins, Tomás P. Carroll, Noel G. McElvaney, and et al. 2021. "C3d Elicits Neutrophil Degranulation and Decreases Endothelial Cell Migration, with Implications for Patients with Alpha-1 Antitrypsin Deficiency" Biomedicines 9, no. 12: 1925. https://doi.org/10.3390/biomedicines9121925
APA StyleFee, L. T., Gogoi, D., O’Brien, M. E., McHugh, E., Casey, M., Gough, C., Murphy, M., Hopkins, A. M., Carroll, T. P., McElvaney, N. G., & Reeves, E. P. (2021). C3d Elicits Neutrophil Degranulation and Decreases Endothelial Cell Migration, with Implications for Patients with Alpha-1 Antitrypsin Deficiency. Biomedicines, 9(12), 1925. https://doi.org/10.3390/biomedicines9121925