
Endothelial Cell Glucose Metabolism and Angiogenesis


Abstract
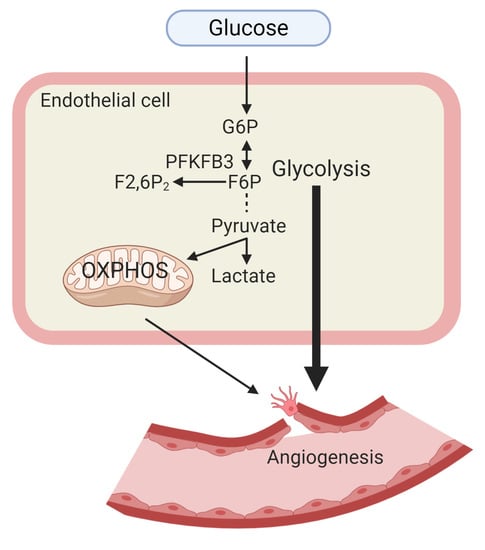
:
1. Introduction
1.1. Endothelial Cell Glucose Metabolism
1.2. Endothelial Cell Fatty Acid Oxidation
1.3. Endothelial Cell Glutamine Metabolism
2. Glucose Metabolism in Quiescent ECs
3. EC Glucose Metabolism in Pathological Angiogenesis
3.1. Ocular Angiogenesis (Diabetic Retinopathy and Retinal Angiomatous Proliferation)
3.2. Diabetic Angiogenesis
3.3. Peripheral Arterial Disease (PAD) and EC Glycolytic Flux
3.4. Tumor Angiogenesis
3.4.1. Tumor Endothelial Cells (TECs) Adapt Their Metabolism to the Tumor Hypoxic Environment
3.4.2. TECs Release More Lactate and Utilize Lactate for Proliferation
3.4.3. TECs Display High Glycolytic Flux
3.4.4. TECs Exhibit Increased Autophagy
4. EC Metabolic Regulators of Antiangiogenesis and Vessel Normalization
5. Conclusions and Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3PG | 3-phosphoglyceric acid |
| 3PO | 3-(3-pyridinyl)-1-(4-pyridynyl)-2-propen-1-one |
| AD | aldose reductase |
| ALDOA/C | aldolase A/aldolase C |
| AMPK | AMP-activated protein kinase |
| ATP | adenosine triphosphate |
| CPT1A | carnitine palmitoyltransferase 1A |
| DAG | diacylglycerol |
| DHAP | dihydroxyacetone phosphate |
| ECFC | endothelial colony-forming cell |
| ECs | endothelial cells |
| ENO1 | enolase 1 |
| F6P | fructose 6-phosphate |
| FABP | fatty acid-binding protein |
| FAO | fatty acid oxidation |
| FASN | fatty acid synthase |
| FAT | fatty acid translocase |
| FATP | fatty acid transport protein |
| G3P | glyceraldehyde 3-phosphate |
| G6P | glucose-6-phosphate |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GLS1 | glutaminase 1 |
| GLUT1/3 | glucose transporter 1/3 |
| GPI | glucose-6-phosphate isomerase |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HK1/2 | hexokinase 1/2 |
| HMGB1 | high-mobility group box 1 |
| HUVECs | human umbilical vein endothelial cells |
| IRS1/2 | insulin receptor substrate 1/2 |
| LDHA/LDHB | lactate dehydrogenase A/lactate dehydrogenase B |
| MCT1/4 | monocarboxylate transporter 1/4 |
| mTOR | mammalian target of rapamycin |
| NAD | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| OXPHOS | oxidative phosphorylation |
| PAD | peripheral artery disease |
| PDK1 | pyruvate dehydrogenase kinase 1 |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 |
| PFK1 | phosphofructokinase 1 |
| PGK | phosphoglycerate kinase |
| PHD | prolyl hydroxylase domain protein |
| PKC | protein kinase C |
| PKM2 | pyruvate kinase M2 |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| PPARα | peroxisome proliferator-activated receptor alpha |
| ROS | reactive oxygen species |
| SDH | sorbitol dehydrogenase |
| SIRT1 | sirtuin 1 |
| TECs | tumor endothelial cells |
| TFEB | transcription factor EB |
References
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar]
- Blancas, A.A.; Wong, L.E.; Glaser, D.E.; McCloskey, K.E. Specialized tip/stalk-like and phalanx-like endothelial cells from embryonic stem cells. Stem Cells Dev. 2013, 22, 1398–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Rohlenova, K.; Goveia, J.; Garcia-Caballero, M.; Subramanian, A.; Kalucka, J.; Treps, L.; Falkenberg, K.D.; de Rooij, L.; Zheng, Y.; Lin, L.; et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab. 2020, 31, 862–877.e14. [Google Scholar] [CrossRef] [PubMed]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.B.; Geck, R.C.; Toker, A. Metabolic pathway alterations in microvascular endothelial cells in response to hypoxia. PLoS ONE 2020, 15, e0232072. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yeh, Y.T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.L.; Li, Y.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [Green Version]
- Patella, F.; Schug, Z.T.; Persi, E.; Neilson, L.J.; Erami, Z.; Avanzato, D.; Maione, F.; Hernandez-Fernaud, J.R.; Mackay, G.; Zheng, L.; et al. Proteomics-based metabolic modeling reveals that fatty acid oxidation (FAO) controls endothelial cell (EC) permeability. Mol. Cell. Proteom. 2015, 14, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graupera, M.; Claret, M. Endothelial Cells: New Players in Obesity and Related Metabolic Disorders. Trends Endocrinol. Metab. 2018, 29, 781–794. [Google Scholar] [CrossRef]
- Kubota, T.; Kubota, N.; Kadowaki, T. The role of endothelial insulin signaling in the regulation of glucose metabolism. Rev. Endocr. Metab. Disord. 2013, 14, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011, 13, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, T.; Brown, J.D.; Orasanu, G.; Vogel, S.; Gonzalez, F.J.; Sartoretto, J.; Michel, T.; Plutzky, J. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J. Clin. Investig. 2009, 119, 110–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, N.H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, S.; Szabo, G. TFEB, a master regulator of lysosome biogenesis and autophagy, is a new player in alcoholic liver disease. Dig. Med. Res. 2018, 1. [Google Scholar] [CrossRef]
- Sun, J.; Lu, H.; Liang, W.; Zhao, G.; Ren, L.; Hu, D.; Chang, Z.; Liu, Y.; Garcia-Barrio, M.T.; Zhang, J.; et al. Endothelial TFEB (Transcription Factor EB) Improves Glucose Tolerance via Upregulation of IRS (Insulin Receptor Substrate) 1 and IRS2. Arter. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Krutzfeldt, A.; Spahr, R.; Mertens, S.; Siegmund, B.; Piper, H.M. Metabolism of exogenous substrates by coronary endothelial cells in culture. J. Mol. Cell. Cardiol. 1990, 22, 1393–1404. [Google Scholar] [CrossRef]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef]
- Yetkin-Arik, B.; Vogels, I.M.C.; Nowak-Sliwinska, P.; Weiss, A.; Houtkooper, R.H.; Van Noorden, C.J.F.; Klaassen, I.; Schlingemann, R.O. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci. Rep. 2019, 9, 12608. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kumar, A.; Carmeliet, P. Metabolic Pathways Fueling the Endothelial Cell Drive. Annu. Rev. Physiol. 2019, 81, 483–503. [Google Scholar] [CrossRef] [PubMed]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.W.; Wang, X.; Zecchin, A.; Thienpont, B.; Cornelissen, I.; Kalucka, J.; Garcia-Caballero, M.; Missiaen, R.; Huang, H.; Bruning, U.; et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 2017, 542, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Elmasri, H.; Ghelfi, E.; Yu, C.W.; Traphagen, S.; Cernadas, M.; Cao, H.; Shi, G.P.; Plutzky, J.; Sahin, M.; Hotamisligil, G.; et al. Endothelial cell-fatty acid binding protein 4 promotes angiogenesis: Role of stem cell factor/c-kit pathway. Angiogenesis 2012, 15, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Harjes, U.; Bridges, E.; Gharpure, K.M.; Roxanis, I.; Sheldon, H.; Miranda, F.; Mangala, L.S.; Pradeep, S.; Lopez-Berestein, G.; Ahmed, A.; et al. Antiangiogenic and tumour inhibitory effects of downregulating tumour endothelial FABP4. Oncogene 2017, 36, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.S.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef]
- Pan, M.; Wasa, M.; Ryan, U.; Souba, W. Inhibition of pulmonary microvascular endothelial glutamine transport by glucocorticoids and endotoxin. JPEN J. Parenter. Enter. Nutr. 1995, 19, 477–481. [Google Scholar] [CrossRef]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [Green Version]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [Green Version]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Bruning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid beta-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e13. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Redox-mediated signal transduction by cardiovascular Nox NADPH oxidases. J. Mol. Cell. Cardiol. 2014, 73, 70–79. [Google Scholar] [CrossRef]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Veys, K.; Fan, Z.; Ghobrial, M.; Bouche, A.; Garcia-Caballero, M.; Vriens, K.; Conchinha, N.V.; Seuwen, A.; Schlegel, F.; Gorski, T.; et al. Role of the GLUT1 Glucose Transporter in Postnatal CNS Angiogenesis and Blood-Brain Barrier Integrity. Circ. Res. 2020, 127, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, J.; Dong, L.J.; He, X.; Cheng, R.; Zhou, K.; Liu, J.; Qiu, F.; Li, X.R.; Ma, J.X. A Protective Effect of PPARalpha in Endothelial Progenitor Cells Through Regulating Metabolism. Diabetes 2019, 68, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.S.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.T.; Prosser, H.C.; Dunn, L.L.; Vanags, L.Z.; Ridiandries, A.; Tsatralis, T.; Lecce, L.; Clayton, Z.E.; Yuen, S.C.; Robertson, S.; et al. High-Density Lipoproteins Rescue Diabetes-Impaired Angiogenesis via Scavenger Receptor Class B Type I. Diabetes 2016, 65, 3091–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef]
- Rudnicki, M.; Abdifarkosh, G.; Nwadozi, E.; Ramos, S.V.; Makki, A.; Sepa-Kishi, D.M.; Ceddia, R.B.; Perry, C.G.; Roudier, E.; Haas, T.L. Endothelial-specific FoxO1 depletion prevents obesity-related disorders by increasing vascular metabolism and growth. Elife 2018, 7. [Google Scholar] [CrossRef]
- Dinh, T.; Veves, A. Microcirculation of the diabetic foot. Curr. Pharm. Des. 2005, 11, 2301–2309. [Google Scholar] [CrossRef]
- Lin, C.J.; Lan, Y.M.; Ou, M.Q.; Ji, L.Q.; Lin, S.D. Expression of miR-217 and HIF-1alpha/VEGF pathway in patients with diabetic foot ulcer and its effect on angiogenesis of diabetic foot ulcer rats. J. Endocrinol. Investig. 2019, 42, 1307–1317. [Google Scholar] [CrossRef]
- Wetterau, M.; George, F.; Weinstein, A.; Nguyen, P.D.; Tutela, J.P.; Knobel, D.; Cohen Ba, O.; Warren, S.M.; Saadeh, P.B. Topical prolyl hydroxylase domain-2 silencing improves diabetic murine wound closure. Wound Repair Regen. 2011, 19, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Sawada, N.; Jiang, A.; Takizawa, F.; Safdar, A.; Manika, A.; Tesmenitsky, Y.; Kang, K.T.; Bischoff, J.; Kalwa, H.; Sartoretto, J.L.; et al. Endothelial PGC-1alpha mediates vascular dysfunction in diabetes. Cell Metab. 2014, 19, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Tarpey, M.D.; Amorese, A.J.; Yamaguchi, D.J.; Goldberg, E.J.; Inigo, M.M.; Karnekar, R.; O’Rourke, A.; Ervasti, J.M.; et al. PFKFB3-mediated glycolysis rescues myopathic outcomes in the ischemic limb. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, H.; Liang, W.; Garcia-Barrio, M.T.; Guo, Y.; Zhang, J.; Zhu, T.; Hao, Y.; Zhang, J.; Chen, Y.E. Endothelial TFEB (Transcription Factor EB) Positively Regulates Postischemic Angiogenesis. Circ. Res. 2018, 122, 945–957. [Google Scholar] [CrossRef]
- Lu, H.; Sun, J.; Hamblin, M.H.; Chen, Y.E.; Fan, Y. Transcription factor EB regulates cardiovascular homeostasis. EBioMedicine 2021, 63, 103207. [Google Scholar] [CrossRef]
- Lu, H.; Sun, J.; Liang, W.; Chang, Z.; Rom, O.; Zhao, Y.; Zhao, G.; Xiong, W.; Wang, H.; Zhu, T.; et al. Cyclodextrin Prevents Abdominal Aortic Aneurysm via Activation of Vascular Smooth Muscle Cell Transcription Factor EB. Circulation 2020, 142, 483–498. [Google Scholar] [CrossRef]
- Long, X.; Boluyt, M.O.; Hipolito, M.L.; Lundberg, M.S.; Zheng, J.S.; O’Neill, L.; Cirielli, C.; Lakatta, E.G.; Crow, M.T. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J. Clin. Investig. 1997, 99, 2635–2643. [Google Scholar] [CrossRef]
- Annan, D.A.; Maishi, N.; Soga, T.; Dawood, R.; Li, C.; Kikuchi, H.; Hojo, T.; Morimoto, M.; Kitamura, T.; Alam, M.T.; et al. Carbonic anhydrase 2 (CAII) supports tumor blood endothelial cell survival under lactic acidosis in the tumor microenvironment. Cell Commun. Signal. 2019, 17, 169. [Google Scholar] [CrossRef] [Green Version]
- Schworer, S.; Vardhana, S.A.; Thompson, C.B. Cancer Metabolism Drives a Stromal Regenerative Response. Cell Metab. 2019, 29, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Eichten, A.; Parveen, A.; Adler, C.; Huang, Y.; Wang, W.; Ding, Y.; Adler, A.; Nevins, T.; Ni, M.; et al. Single-Cell Transcriptome Analyses Reveal Endothelial Cell Heterogeneity in Tumors and Changes following Antiangiogenic Treatment. Cancer Res. 2018, 78, 2370–2382. [Google Scholar] [CrossRef] [Green Version]
- Qian, C.N.; Tan, M.H.; Yang, J.P.; Cao, Y. Revisiting tumor angiogenesis: Vessel co-option, vessel remodeling, and cancer cell-derived vasculature formation. Chin. J. Cancer 2016, 35, 10. [Google Scholar] [CrossRef] [Green Version]
- Andonegui-Elguera, M.A.; Alfaro-Mora, Y.; Caceres-Gutierrez, R.; Caro-Sanchez, C.H.S.; Herrera, L.A.; Diaz-Chavez, J. An Overview of Vasculogenic Mimicry in Breast Cancer. Front. Oncol. 2020, 10, 220. [Google Scholar] [CrossRef] [Green Version]
- Hunt, T.K.; Aslam, R.S.; Beckert, S.; Wagner, S.; Ghani, Q.P.; Hussain, M.Z.; Roy, S.; Sen, C.K. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid. Redox Signal. 2007, 9, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Dings, R.P.; van der Linden, E.; Zwaans, B.M.; Ramaekers, F.C.; Mayo, K.H.; Griffioen, A.W. Gene expression of tumor angiogenesis dissected: Specific targeting of colon cancer angiogenic vasculature. Blood 2006, 108, 2339–2348. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, R.; Kato, M.; Okamura, N.; Nakagawa, T.; Komada, Y.; Tominaga, N.; Shimojo, M.; Fukasawa, M. Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J. Biochem. 1997, 122, 122–128. [Google Scholar] [CrossRef]
- Yeh, W.L.; Lin, C.J.; Fu, W.M. Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol. Pharmacol. 2008, 73, 170–177. [Google Scholar] [CrossRef]
- Loponte, S.; Lovisa, S.; Deem, A.K.; Carugo, A.; Viale, A. The Many Facets of Tumor Heterogeneity: Is Metabolism Lagging Behind? Cancers 2019, 11, 1574. [Google Scholar] [CrossRef] [Green Version]
- Filippi, I.; Saltarella, I.; Aldinucci, C.; Carraro, F.; Ria, R.; Vacca, A.; Naldini, A. Different Adaptive Responses to Hypoxia in Normal and Multiple Myeloma Endothelial Cells. Cell. Physiol. Biochem. 2018, 46, 203–212. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Houbaert, D.; Mece, O.; Agostinis, P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. 2019, 26, 665–679. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, H.P.; Jin, Y.; Choi, A.M.; Ryter, S.W. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011, 7, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Vizan, P.; Sanchez-Tena, S.; Alcarraz-Vizan, G.; Soler, M.; Messeguer, R.; Pujol, M.D.; Lee, W.N.; Cascante, M. Characterization of the metabolic changes underlying growth factor angiogenic activation: Identification of new potential therapeutic targets. Carcinogenesis 2009, 30, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Pilar Lopez, M.; Gomez-Lechon, M.J.; Castell, J.V. Role of glucose, insulin, and glucagon in glycogen mobilization in human hepatocytes. Diabetes 1991, 40, 263–268. [Google Scholar] [CrossRef]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Wuest, T.R.; Min, Y.; Lin, P.C. Oxygen Tension Regulates Lysosomal Activation and Receptor Tyrosine Kinase Degradation. Cancers 2019, 11, 1653. [Google Scholar] [CrossRef] [Green Version]
- Mansueto, G.; Armani, A.; Viscomi, C.; D’Orsi, L.; De Cegli, R.; Polishchuk, E.V.; Lamperti, C.; Di Meo, I.; Romanello, V.; Marchet, S.; et al. Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 2017, 25, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.C.; Vandekeere, S.; Bouche, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7, 12240. [Google Scholar] [CrossRef]
- Gomez-Escudero, J.; Clemente, C.; Garcia-Weber, D.; Acin-Perez, R.; Millan, J.; Enriquez, J.A.; Bentley, K.; Carmeliet, P.; Arroyo, A.G. PKM2 regulates endothelial cell junction dynamics and angiogenesis via ATP production. Sci. Rep. 2019, 9, 15022. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Hsiao, Y.H.; Hsieh, M.J.; Yang, S.F.; Chen, S.P.; Tsai, W.C.; Chen, P.N. Phloretin suppresses metastasis by targeting protease and inhibits cancer stemness and angiogenesis in human cervical cancer cells. Phytomedicine 2019, 62, 152964. [Google Scholar] [CrossRef]
- Yang, S.H.; Lin, J.K.; Chen, W.S.; Chiu, J.H. Anti-angiogenic effect of silymarin on colon cancer LoVo cell line. J. Surg. Res. 2003, 113, 133–138. [Google Scholar] [CrossRef]
- Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Saikawa, S.; Nakanishi, K.; Furukawa, M.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Cancer 2018, 142, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Chen, X.; Guan, S.; Yan, Y.; Lin, H.; Hua, Z.C. Curcumin inhibits angiogenesis and improves defective hematopoiesis induced by tumor-derived VEGF in tumor model through modulating VEGF-VEGFR2 signaling pathway. Oncotarget 2015, 6, 19469–19482. [Google Scholar] [CrossRef] [Green Version]
- Ocana, M.C.; Martinez-Poveda, B.; Mari-Beffa, M.; Quesada, A.R.; Medina, M.A. Fasentin diminishes endothelial cell proliferation, differentiation and invasion in a glucose metabolism-independent manner. Sci. Rep. 2020, 10, 6132. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Zhen, W.; Velayutham Anandh Babu, P.; Liu, D. Phytoestrogen genistein protects against endothelial barrier dysfunction in vascular endothelial cells through PKA-mediated suppression of RhoA signaling. Endocrinology 2013, 154, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Su, S.J.; Yeh, T.M.; Chuang, W.J.; Ho, C.L.; Chang, K.L.; Cheng, H.L.; Liu, H.S.; Cheng, H.L.; Hsu, P.Y.; Chow, N.H. The novel targets for anti-angiogenesis of genistein on human cancer cells. Biochem. Pharmacol. 2005, 69, 307–318. [Google Scholar] [CrossRef]
- Singh, S.; Pandey, S.; Chawla, A.S.; Bhatt, A.N.; Roy, B.G.; Saluja, D.; Dwarakanath, B.S. Dietary 2-deoxy-D-glucose impairs tumour growth and metastasis by inhibiting angiogenesis. Eur. J. Cancer 2019, 123, 11–24. [Google Scholar] [CrossRef]
- Huang, C.C.; Wang, S.Y.; Lin, L.L.; Wang, P.W.; Chen, T.Y.; Hsu, W.M.; Lin, T.K.; Liou, C.W.; Chuang, J.H. Glycolytic inhibitor 2-deoxyglucose simultaneously targets cancer and endothelial cells to suppress neuroblastoma growth in mice. Dis. Models Mech. 2015, 8, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- Merchan, J.R.; Kovacs, K.; Railsback, J.W.; Kurtoglu, M.; Jing, Y.; Pina, Y.; Gao, N.; Murray, T.G.; Lehrman, M.A.; Lampidis, T.J. Antiangiogenic activity of 2-deoxy-D-glucose. PLoS ONE 2010, 5, e13699. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K.; et al. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma Cells. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Del Bufalo, D.; Trisciuoglio, D.; Scarsella, M.; D’Amati, G.; Candiloro, A.; Iervolino, A.; Leonetti, C.; Zupi, G. Lonidamine causes inhibition of angiogenesis-related endothelial cell functions. Neoplasia 2004, 6, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Li, D.; Xiang, X.; Tong, L.; Qi, M.; Pu, J.; Huang, K.; Tong, Q. Methyl jasmonate abolishes the migration, invasion and angiogenesis of gastric cancer cells through down-regulation of matrix metalloproteinase 14. BMC Cancer 2013, 13, 74. [Google Scholar] [CrossRef] [Green Version]
- Schoors, S.; De Bock, K.; Cantelmo, A.R.; Georgiadou, M.; Ghesquiere, B.; Cauwenberghs, S.; Kuchnio, A.; Wong, B.W.; Quaegebeur, A.; Goveia, J.; et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014, 19, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J.; et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A., 2nd; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef] [Green Version]
- Komi, Y.; Suzuki, Y.; Shimamura, M.; Kajimoto, S.; Nakajo, S.; Masuda, M.; Shibuya, M.; Itabe, H.; Shimokado, K.; Oettgen, P.; et al. Mechanism of inhibition of tumor angiogenesis by beta-hydroxyisovalerylshikonin. Cancer Sci. 2009, 100, 269–277. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Trarbach, T.; Folprecht, G.; Shi, M.M.; Lebwohl, D.; Jalava, T.; Laurent, D.; et al. Prognostic and predictive role of lactate dehydrogenase 5 expression in colorectal cancer patients treated with PTK787/ZK 222584 (vatalanib) antiangiogenic therapy. Clin. Cancer Res. 2011, 17, 4892–4900. [Google Scholar] [CrossRef] [Green Version]
- Ulus, G.; Koparal, A.T.; Baysal, K.; Yetik Anacak, G.; Karabay Yavasoglu, N.U. The anti-angiogenic potential of (+/-) gossypol in comparison to suramin. Cytotechnology 2018, 70, 1537–1550. [Google Scholar] [CrossRef]
- Pang, X.; Wu, Y.; Wu, Y.; Lu, B.; Chen, J.; Wang, J.; Yi, Z.; Qu, W.; Liu, M. (-)-Gossypol suppresses the growth of human prostate cancer xenografts via modulating VEGF signaling-mediated angiogenesis. Mol. Cancer Ther. 2011, 10, 795–805. [Google Scholar] [CrossRef] [Green Version]
- El-Sisi, A.E.; Sokar, S.S.; Abu-Risha, S.E.; El-Mahrouk, S.R. Oxamate potentiates taxol chemotherapeutic efficacy in experimentally-induced solid ehrlich carcinoma (SEC) in mice. Biomed. Pharmacother. 2017, 95, 1565–1573. [Google Scholar] [CrossRef]
- Zhang, J.; Muri, J.; Fitzgerald, G.; Gorski, T.; Gianni-Barrera, R.; Masschelein, E.; D’Hulst, G.; Gilardoni, P.; Turiel, G.; Fan, Z.; et al. Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization. Cell Metab. 2020, 31, 1136–1153.e7. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013, 32, 1638–1650. [Google Scholar] [CrossRef] [Green Version]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M.; et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef]
- Jaidee, R.; Kongpetch, S.; Senggunprai, L.; Prawan, A.; Kukongviriyapan, U.; Kukongviriyapan, V. Phenformin inhibits proliferation, invasion, and angiogenesis of cholangiocarcinoma cells via AMPK-mTOR and HIF-1A pathways. Naunyn-Schmiedeberg Arch. Pharmacol. 2020, 393, 1681–1690. [Google Scholar] [CrossRef]
- Navarro, P.; Bueno, M.J.; Zagorac, I.; Mondejar, T.; Sanchez, J.; Mouron, S.; Munoz, J.; Gomez-Lopez, G.; Jimenez-Renard, V.; Mulero, F.; et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Rep. 2016, 15, 2705–2718. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, L.; Zhan, S.; Chen, L.; Wang, Y.; Zhang, Y.; Du, J.; Wu, Y.; Gu, L. Arsenic Trioxide Suppressed Migration and Angiogenesis by Targeting FOXO3a in Gastric Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3739. [Google Scholar] [CrossRef] [Green Version]
- Agius, L.; Meredith, E.J.; Sherratt, H.S. Stereospecificity of the inhibition by etomoxir of fatty acid and cholesterol synthesis in isolated rat hepatocytes. Biochem. Pharmacol. 1991, 42, 1717–1720. [Google Scholar] [CrossRef]
- Ashrafian, H.; Horowitz, J.D.; Frenneaux, M.P. Perhexiline. Cardiovasc. Drug Rev. 2007, 25, 76–97. [Google Scholar] [CrossRef]
- Iwamoto, H.; Abe, M.; Yang, Y.; Cui, D.; Seki, T.; Nakamura, M.; Hosaka, K.; Lim, S.; Wu, J.; He, X.; et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab. 2018, 28, 104–117.e5. [Google Scholar] [CrossRef] [Green Version]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.; Xia, S.; Liu, Y.; et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–5336. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Ruiz-Rodado, V.; Dowdy, T.; Huang, S.; Issaq, S.H.; Beck, J.; Wang, H.; Tran Hoang, C.; Lita, A.; Larion, M.; et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 2020, 8, 4. [Google Scholar] [CrossRef]
- Rohlenova, K.; Veys, K.; Miranda-Santos, I.; De Bock, K.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol. 2018, 28, 224–236. [Google Scholar] [CrossRef]
- Ribatti, D.; Annese, T.; Ruggieri, S.; Tamma, R.; Crivellato, E. Limitations of Anti-Angiogenic Treatment of Tumors. Transl. Oncol. 2019, 12, 981–986. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Compound | Tumor Type | Status |
|---|---|---|---|
| Glycolysis | |||
| Glucose transporters Glut1 | Phloretin Silybin (Silibinin) Canagliflozin Curcumin Fasentin Genistein | Cervical cancer cell Prostate cancer Liver cancer cell Multiple cancers Breast cancer Multiple cancers | Preclinical [83] Clinical phase II [84] Preclinical [85] Clinical phase I/II [86] Preclinical [87] Clinical phase I/II [88,89] |
| Hexokinases | 2-DG 1 Ketoconazole Lonidamine Methyl jasmonate | Multiple cancers Glioblastoma Melanoma, breast cancer, glioblastoma, lung cancer, prostate cancer Gastric cancer | Clinical phase I/II [90,91,92] Preclinical [93] Clinical phase III [94] Preclinical [95] |
| PFKFB3 2 | 3PO 3 PFK158 | Melanoma, lung carcinoma, pancreatic cancer Advanced solid malignancies | Preclinical [5,96,97] Clinical phase I [98] |
| Pyruvate kinase-M2 (PK-M2) | TLN-232 Shikonin | Metastatic renal cell Lung carcinoma | Clinical I/II [99] Preclinical [100] |
| Lactate dehydrogenase | PTK787/ZK 222584 (Vatalanib) Gossypol Oxamate | Colon cancer, advanced colorectal cancer Multiple cancers Breast cancer | Preclinical [101] Clinical phase I/II [102,103] Preclinical [104] |
| Lactate | Lonidamine AZD3965 | Prostate cancer Gastric cancer, prostate cancer lymphoma | Clinical phase III [94] Clinical phase I [105] |
| TCA cycle | |||
| PDK1 4 | Dichloroacetate (DCA) | Non-small-cell lung cancer, breast cancer | Preclinical [106] |
| OXPHOS | |||
| Mitochondrial complex I/III | Metformin Phenformin Arsenic trioxide | Breast cancer Cholangiocarcinoma Gastric cancer cells | Preclinical [107] Clinical phase I [108,109] Preclinical [110] |
| FAO | |||
| CPT1 5 | Etomoxir Perhexiline | Lung carcinoma, prostate cancer cell line Prostate cancer, glioma | Preclinical [23,111,112] Preclinical [112,113] |
| Glutamine metabolism | |||
| GLS1 6 | BPTES CB-839 | Osteosarcoma, pancreatic cancer Multiple cancers | Preclinical [114,115] Clinical phase I/II [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, W.; Ren, L.; Hamblin, M.H.; Fan, Y. Endothelial Cell Glucose Metabolism and Angiogenesis. Biomedicines 2021, 9, 147. https://doi.org/10.3390/biomedicines9020147
Du W, Ren L, Hamblin MH, Fan Y. Endothelial Cell Glucose Metabolism and Angiogenesis. Biomedicines. 2021; 9(2):147. https://doi.org/10.3390/biomedicines9020147
Chicago/Turabian StyleDu, Wa, Lu Ren, Milton H. Hamblin, and Yanbo Fan. 2021. "Endothelial Cell Glucose Metabolism and Angiogenesis" Biomedicines 9, no. 2: 147. https://doi.org/10.3390/biomedicines9020147
APA StyleDu, W., Ren, L., Hamblin, M. H., & Fan, Y. (2021). Endothelial Cell Glucose Metabolism and Angiogenesis. Biomedicines, 9(2), 147. https://doi.org/10.3390/biomedicines9020147