
Deciphering the Role of Heme Oxygenase-1 (HO-1) Expressing Macrophages in Renal Ischemia-Reperfusion Injury


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tubular Cells and IRI-Induced AKI
3. Myeloid Cells and IRI-Induced AKI
4. Macrophages and IRI-Induced AKI
4.1. Origins of the Monocytes/Macrophages
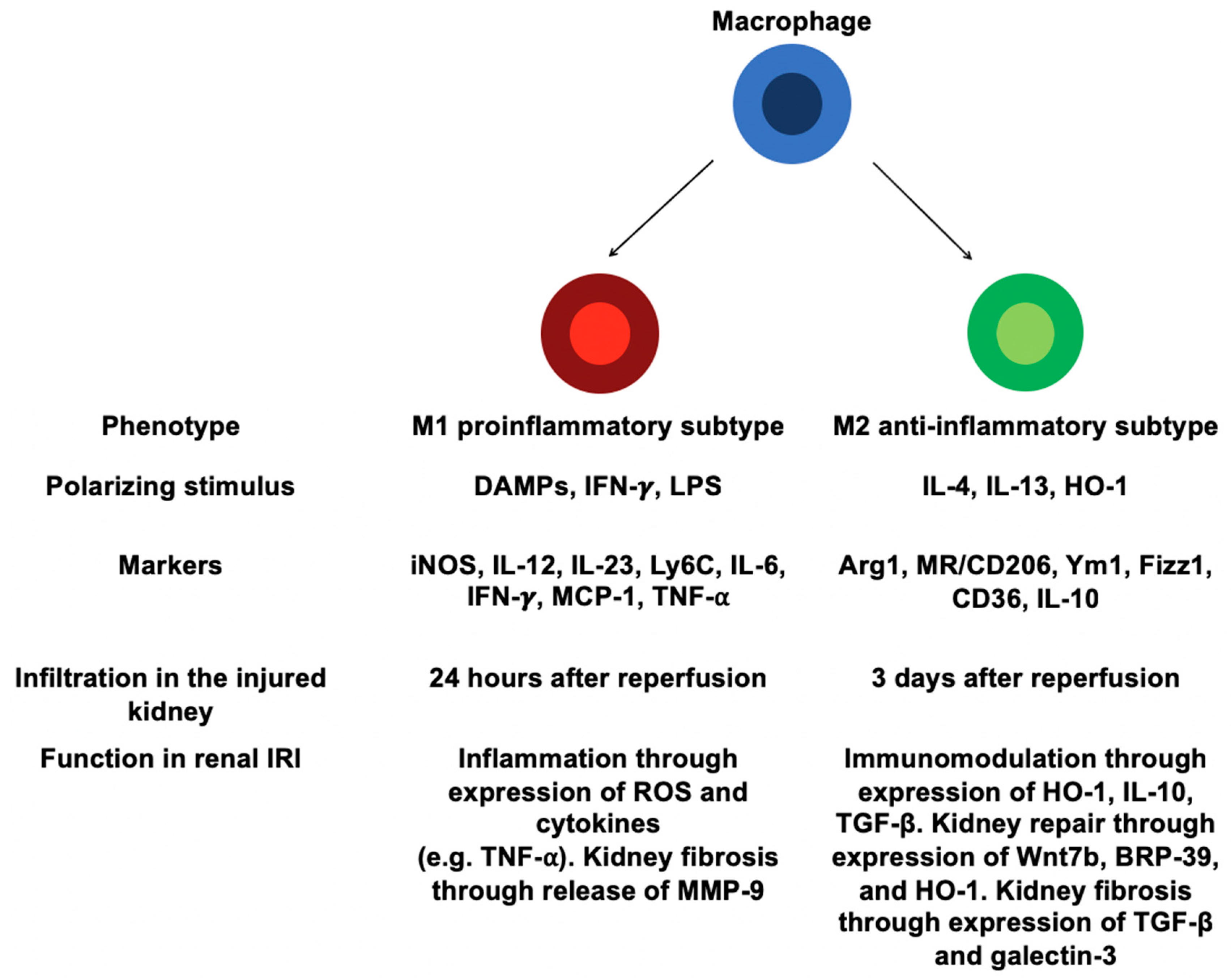
4.2. Involvement of Distinct Macrophages in Renal IRI
4.3. Macrophages and Renal Repair after IRI
4.4. Macrophages and Fibrogenesis after IRI
5. Heme Oxygenase-1 (HO-1)
5.1. Overview
5.2. Regulation of HO-1 Expression
5.3. Cytoprotective Effects of HO-1
5.4. Anti-Inflammatory Effect of HO-1
6. HO-1 Expressing Macrophages and Renal IRI
6.1. Macrophages Are Critical for HO-1 Cytoprotective Effects
6.2. HO-1 and Macrophage Polarization
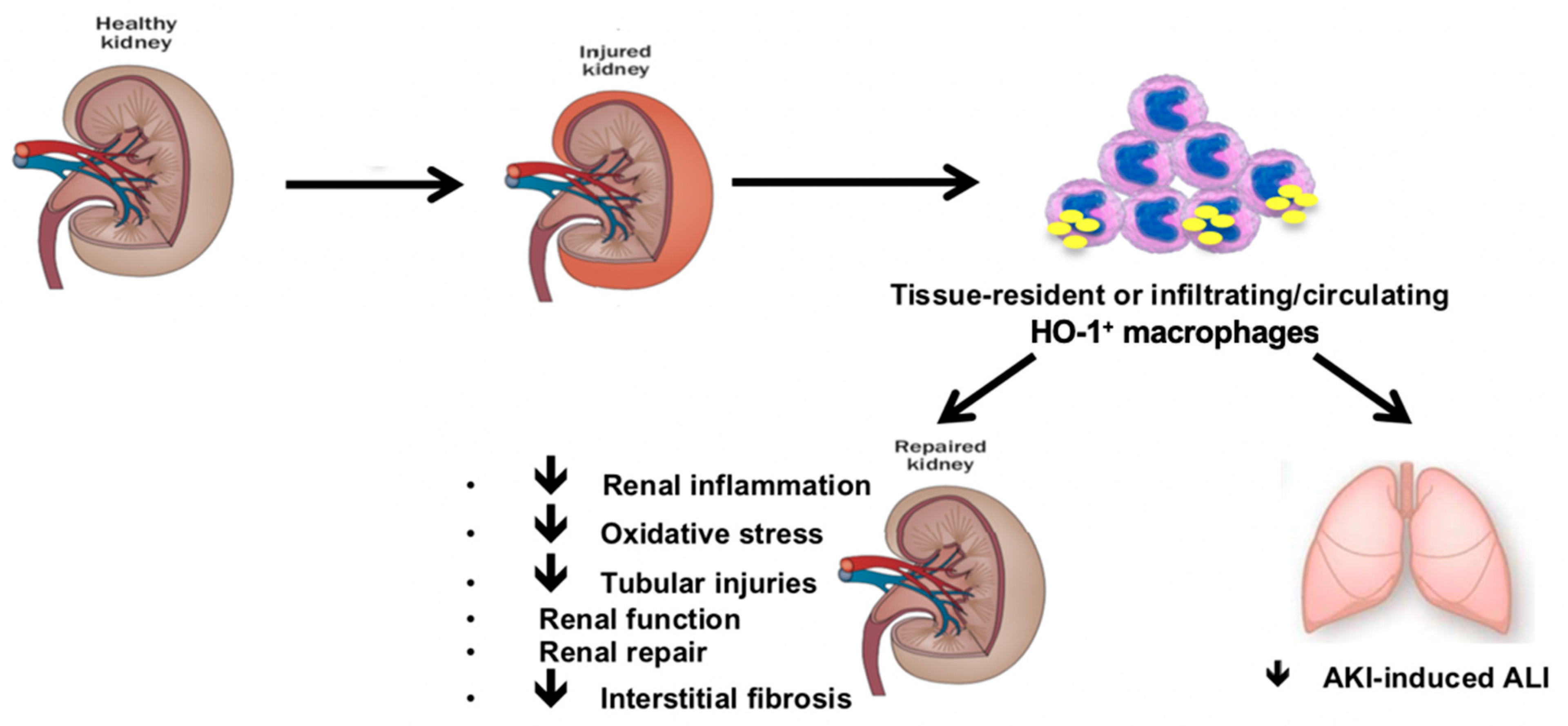
6.3. HO-1 Expressing Macrophages Mitigates Distant Organ Injuries upon Renal IRI
6.4. HO-1 Expressing Macrophages Modulates Adaptive Renal Repair after AKI
6.5. The Origin of HO-1 Expressing Macrophages
7. Macrophages and IRI-Induced AKI in Humans
8. HO-1 Expressing Macrophages: A Novel Nephron Sparing Strategy?
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lameire, N.H.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.; Kellum, J.A.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W.; et al. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef]
- Waikar, S.S.; Liu, K.D.; Chertow, G.M. Diagnosis, Epidemiology and Outcomes of Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2008, 3, 844–861. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, B.; Krick, S.; Dhillon, N.; Lerner, S.M.; Ames, S.; Bromberg, J.S.; Lin, M.; Walsh, L.; Vella, J.; Fischereder, M.; et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 3390–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Delbauve, S.; Wespes, E.; Roumeguère, T.; Leo, O.; Flamand, V.; Le Moine, A.; Hougardy, J.-M. Dual effect of hemin on renal ischemia-reperfusion injury. Biochem. Biophys. Res. Commun. 2018, 503, 2820–2825. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Delbauve, S.; Roumeguère, T.; Wespes, E.; Leo, O.; Flamand, V.; Le Moine, A.; Hougardy, J.-M. HO-1 mitigates acute kidney injury and subsequent kidney-lung cross-talk. Free Radic. Res. 2019, 53, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Harris, D.C.H.; Wang, Y. Macrophages in Kidney Injury, Inflammation, and Fibrosis. Physiology 2015, 30, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.R.; Rabb, H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 2014, 11, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 2015, 4, 20–27. [Google Scholar] [CrossRef]
- Qian, J.; You, H.; Zhu, Q.; Ma, S.; Zhou, Y.; Zheng, Y.; Liu, J.; Kuang, D.; Gu, Y.; Hao, C.; et al. Nitrotyrosine Level Was Associated with Mortality in Patients with Acute Kidney Injury. PLoS ONE 2013, 8, e79962. [Google Scholar] [CrossRef] [Green Version]
- Wahl, P.; Schoop, R.; Bilic, G.; Neuweiler, J.; Le Hir, M.; Yoshinaga, S.K.; Wüthrich, R.P. Renal tubular epithelial expression of the costimulatory molecule B7RP-1 (inducible costimulator ligand). J. Am. Soc. Nephrol. 2002, 13, 1517–1526. [Google Scholar] [CrossRef]
- Niemann-Masanek, U.; Mueller, A.; Yard, B.A.; Waldherr, R.; Van Der Woude, F.J. B7-1 (CD80) and B7-2 (CD 86) Expression in Human Tubular Epithelial Cells in vivo and in vitro1. Nephron 2002, 92, 542–556. [Google Scholar] [CrossRef]
- Kawamoto, H.; Minato, N. Myeloid cells. Int. J. Biochem. Cell Biol. 2004, 36, 1374–1379. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Hedblom, A.; Li, M.; Gallo, D.; Csizmadia, E.; Harris, C.C.; Nemeth, Z.H.; Zuckerbraun, B.S.; Soares, M.J.; Persson, J.; et al. Heme oxygenase-1 derived carbon monoxide permits maturation of myeloid cells. Cell Death Dis. 2014, 5, e1139. [Google Scholar] [CrossRef] [Green Version]
- Hull, T.D.; Kamal, A.I.; Boddu, R.; Bolisetty, S.; Guo, L.; Tisher, C.C.; Rangarajan, S.; Chen, B.; Curtis, L.M.; George, J.F.; et al. Heme Oxygenase-1 Regulates Myeloid Cell Trafficking in AKI. J. Am. Soc. Nephrol. 2015, 26, 2139–2151. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; Rouse, M.; Huang, L.; Vergis, A.L.; Reutershan, J.; Cathro, H.P.; Linden, J.; Okusa, M.D. Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury. Kidney Int. 2009, 75, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, K.J.; Williams, W.W.; Colvin, R.B.; Meehan, S.M.; Springer, T.A.; Gutierrez-Ramos, J.C.; Bonventre, J.V. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J. Clin. Investig. 1996, 97, 1056–1063. [Google Scholar] [CrossRef]
- Li, L.; Huang, L.; Vergis, A.L.; Ye, H.; Bajwa, A.; Narayan, V.; Strieter, R.M.; Rosin, D.L.; Okusa, M.D. IL-17 produced by neutrophils regulates IFN-γ–mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J. Clin. Investig. 2010, 120, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouschop, K.M.A.; Roelofs, J.J.T.H.; Claessen, N.; Martins, P.D.C.; Zwaginga, J.-J.; Pals, S.T.; Weening, J.J.; Florquin, S. Protection against Renal Ischemia Reperfusion Injury by CD44 Disruption. J. Am. Soc. Nephrol. 2005, 16, 2034–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.J.; Rees, A.J.; Griffin, M.D.; Hughes, J.; Kurts, C.; Duffield, J. The Renal Mononuclear Phagocytic System. J. Am. Soc. Nephrol. 2011, 23, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Kruger, T.; Benke, D.; Eitner, F.; Lang, A.; Wirtz, M.; Hamilton-Williams, E.E.; Engel, D.; Giese, B.; Muller-Newen, G.; Floege, J.; et al. Identification and functional characterization of dendritic cells in the healthy murine kidney and in ex-perimental glomerulonephritis. J. Am. Soc. Nephrol. 2004, 15, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Liu, K.; Helft, J.; Bogunovic, M.; Greter, M.; Hashimoto, D.; Price, J.; Yin, N.; Bromberg, J.; Lira, S.A.; et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 2009, 206, 3115–3130. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Swaminathan, S.; Bachman, L.-A.; Croatt, A.-J.; Nath, K.-A.; Griffin, M.-D. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia–reperfusion injury. Kidney Int. 2007, 71, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.Y.; Choi, H.M.; Lee, S.Y.; Kim, M.G.; Kim, H.-K.; Jo, S.-K. The role of Tregs and CD11c+ macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int. 2010, 78, 981–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-G.; Boo, C.S.; Ko, Y.S.; Lee, H.Y.; Cho, W.Y.; Kim, H.K.; Jo, S.-K. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol. Dial. Transplant. 2010, 25, 2908–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisheit, C.K.; Engel, D.R.; Kurts, C. Dendritic Cells and Macrophages: Sentinels in the Kidney. Clin. J. Am. Soc. Nephrol. 2015, 10, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunology 2003, 19, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Mildner, A.; Yona, S. Developmental and Functional Heterogeneity of Monocytes. Immunology 2018, 49, 595–613. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.; Tacke, R.; Hedrick, C.C.; Hanna, R.N. Nonclassical Patrolling Monocyte Function in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Huang, L.; Sung, S.-S.J.; Vergis, A.L.; Rosin, D.L.; Rose, C.E.; Lobo, P.I.; Okusa, M.D. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia–reperfusion injury. Kidney Int. 2008, 74, 1526–1537. [Google Scholar] [CrossRef] [Green Version]
- Tittel, A.P.; Heuser, C.; Ohliger, C.; Knolle, P.A.; Engel, D.R.; Kurts, C. Kidney Dendritic Cells Induce Innate Immunity against Bacterial Pyelonephritis. J. Am. Soc. Nephrol. 2011, 22, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Huen, S.; Nishio, H.; Nishio, S.; Lee, H.K.; Choi, B.-S.; Ruhrberg, C.; Cantley, L.G. Distinct Macrophage Phenotypes Contribute to Kidney Injury and Repair. J. Am. Soc. Nephrol. 2011, 22, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Kurts, C.; Panzer, U.; Anders, H.-J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.R.; Rabb, H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 2009, 130, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Muraille, E.; Leo, O.; Moser, M. Th1/Th2 Paradigm Extended: Macrophage Polarization as an Unappreciated Pathogen-Driven Escape Mechanism? Front. Immunol. 2014, 5, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, H.-J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S. Macrophages and Immunologic Inflammation of the Kidney. Semin. Nephrol. 2010, 30, 234–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Clements, M.; Gershenovich, M.; Chaber, C.; Campos-Rivera, J.; Du, P.; Zhang, M.; Ledbetter, S.; Zuk, A. Differential Ly6C Expression after Renal Ischemia-Reperfusion Identifies Unique Macrophage Populations. J. Am. Soc. Nephrol. 2015, 27, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Alikhan, M.A.; Jones, C.V.; Williams, T.M.; Beckhouse, A.G.; Fletcher, A.L.; Kett, M.M.; Sakkal, S.; Samuel, C.S.; Ramsay, R.G.; Deane, J.A.; et al. Colony-Stimulating Factor-1 Promotes Kidney Growth and Repair via Alteration of Macrophage Responses. Am. J. Pathol. 2011, 179, 1243–1256. [Google Scholar] [CrossRef]
- Sola, A.; Weigert, A.; Jung, M.; Vinuesa, E.; Brecht, K.; Weis, N.; Brüne, B.; Borregaard, N.; Hotter, G. Sphingosine-1-phosphate signalling induces the production of Lcn-2 by macrophages to promote kidney regeneration. J. Pathol. 2011, 225, 597–608. [Google Scholar] [CrossRef]
- Canaud, G.; Bonventre, J.V. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial. Transplant. 2014, 30, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic Epithelial Cells Repair the Kidney after Injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar] [CrossRef] [Green Version]
- Savill, J.; Smith, J.; Sarraf, C.; Ren, Y.; Abbott, F.; Rees, A. Glomerular mesangial cells and inflammatory macrophages ingest neutrophils undergoing apoptosis. Kidney Int. 1992, 42, 924–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Faubel, S.; He, Z.; Hernando, A.A.; Jani, A.; Kedl, R.; Edelstein, C.L. Depletion of Macrophages and Dendritic Cells in Ischemic Acute Kidney Injury. Am. J. Nephrol. 2012, 35, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-L.; Li, B.; Rao, S.; Yeo, E.-J.; Hudson, T.E.; Nowlin, B.T.; Pei, H.; Chen, L.; Zheng, J.J.; Carroll, T.J.; et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, I.M.; Hall, I.E.; Kale, S.; Lee, S.; He, C.-H.; Lee, Y.; Chupp, G.L.; Moeckel, G.W.; Lee, C.G.; Elias, J.A.; et al. Chitinase-Like Protein Brp-39/YKL-40 Modulates the Renal Response to Ischemic Injury and Predicts Delayed Allograft Function. J. Am. Soc. Nephrol. 2013, 24, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Sung, S.A.; Jo, S.K.; Cho, W.Y.; Won, N.H.; Kim, H.K. Reduction of Renal Fibrosis as a Result of Liposome Encapsulated Clodronate Induced Macrophage Depletion after Unilateral Ureteral Obstruction in Rats. Nephron 2006, 105, e1–e9. [Google Scholar] [CrossRef]
- Machida, Y.; Kitamoto, K.; Izumi, Y.; Shiota, M.; Uchida, J.; Kira, Y.; Nakatani, T.; Miura, K. Renal fibrosis in murine obstructive nephropathy is attenuated by depletion of monocyte lineage, not dendritic cells. J. Pharmacol. Sci. 2010, 114, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Snelgrove, S.L.; Kausman, J.Y.; Lo, C.; Lo, C.; Ooi, J.D.; Coates, P.T.; Hickey, M.J.; Holdsworth, S.R.; Kurts, C.; Engel, D.R.; et al. Renal Dendritic Cells Adopt a Pro-Inflammatory Phenotype in Obstructive Uropathy to Activate T Cells but Do Not Directly Contribute to Fibrosis. Am. J. Pathol. 2012, 180, 91–103. [Google Scholar] [CrossRef]
- Tan, T.K.; Zheng, G.; Hsu, T.-T.; Wang, Y.; Lee, V.W.; Tian, X.; Cao, Q.; Wang, Y.; Harris, D.C. Macrophage Matrix Metalloproteinase-9 Mediates Epithelial-Mesenchymal Transition in Vitro in Murine Renal Tubular Cells. Am. J. Pathol. 2010, 176, 1256–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Dong, Y.; Tian, X.; Tan, T.K.; Liu, Z.; Zhao, Y.; Zhang, Y.; Harris, D.C.; Zheng, G. Matrix metalloproteinases contribute to kidney fibrosis in chronic kidney diseases. World J. Nephrol. 2013, 2, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-β-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, N.C.; MacKinnon, A.C.; Farnworth, S.L.; Kipari, T.; Haslett, C.; Iredale, J.P.; Liu, F.-T.; Hughes, J.; Sethi, T. Galectin-3 Expression and Secretion Links Macrophages to the Promotion of Renal Fibrosis. Am. J. Pathol. 2008, 172, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lin, S.-C.; Chen, G.; He, L.; Hu, Z.; Chan, L.; Trial, J.; Entman, M.L.; Wang, Y. Adiponectin Promotes Monocyte-to-Fibroblast Transition in Renal Fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1644–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Meng, X.-M.; Ng, Y.-Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.-R.; Xiao, J.; Wang, Y.-Y.; et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2015, 7, 8809–8822. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-M.; Wang, S.; Huang, X.-R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.-F.; Nikolic-Paterson, D.J.; Lan, H.-Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Jiang, H.; Pan, J.; Huang, X.-R.; Wang, Y.-C.; Huang, H.-F.; To, K.-F.; Nikolic-Paterson, D.J.; Lan, H.-Y.; Chen, J.-H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef] [Green Version]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic catabolism of hemoglobin: Stimulation of microsomal heme oxy-genase by hemin. J. Lab. Clin. Med. 1970, 75, 410–421. [Google Scholar]
- Ryter, S.W.; Choi, A.M.K. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maines, M.D. The Heme Oxygenase System:A Regulator of Second Messenger Gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Mccoubrey, W.K.J.; Huang, T.J.; Maines, M.D. Isolation and Characterization of a cDNA from the Rat Brain that Encodes Hemoprotein Heme Oxygenase-3. J. Biol. Inorg. Chem. 1997, 247, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.R.; Shaefi, S.; Otterbein, L.E. HO-1 and CD39: It Takes Two to Protect the Realm. Front. Immunol. 2019, 10, 1765. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Hanto, D.W.; Otterbein, L.E. The social network of carbon monoxide in medicine. Trends Mol. Med. 2013, 19, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, K.; Sun, J.; Taketani, S.; Nakajima, O.; Nishitani, C.; Sassa, S.; Hayashi, N.; Yamamoto, M.; Shibahara, S.; Fujita, H.; et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001, 20, 2835–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme Induces Ubiquitination and Degradation of the Transcription Factor Bach1. Mol. Cell. Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Yamamoto, M. Molecular Mechanisms Activating the Nrf2-Keap1 Pathway of Antioxidant Gene Regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M.; Yamamoto, T.; Zhang, J.; Itoh, K.; Kyo, M.; Kamiya, T.; Aburatani, H.; Katsuoka, F.; Kurokawa, H.; Tanaka, T.; et al. Molecular Basis Distinguishing the DNA Binding Profile of Nrf2-Maf Heterodimer from That of Maf Homodimer. J. Biol. Chem. 2007, 282, 33681–33690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vile, G.F.; Basu-Modak, S.; Waltner, C.; Tyrrell, R.M. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc. Natl. Acad. Sci. USA 1994, 91, 2607–2610. [Google Scholar] [CrossRef] [Green Version]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of Cell Protection by Heme Oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Abraham, N.G.; Lavrovsky, Y.; Schwartzman, M.L.; Stoltz, R.A.; Levere, R.D.; Gerritsen, M.E.; Shibahara, S.; Kappas, A. Transfection of the human heme oxygenase gene into rabbit coronary microvessel endothelial cells: Protective effect against heme and hemoglobin toxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 6798–6802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, A.; Rodrigues, C.D.; Gregoire, I.P.; Cunha-Rodrigues, M.; et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 2007, 13, 703–710. [Google Scholar] [CrossRef]
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Seixas, E.; Gozzelino, R.; Chora, A.; Ferreira, A.; Silva, G.; Larsen, R.; Rebelo, S.; Penido, C.; Smith, N.R.; Coutinho, A.; et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. USA 2009, 106, 15837–15842. [Google Scholar] [CrossRef] [Green Version]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Soares, M.P.; Lin, Y.; Anrather, J.; Csizmadia, E.; Takigami, K.; Sato, K.; Grey, S.T.; Colvin, R.B.; Choi, A.M.; Poss, K.D.; et al. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat. Med. 1998, 4, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Ke, B.; Buelow, R.; Shen, X.-D.; Melinek, J.; Amersi, F.; Gao, F.; Ritter, T.; Volk, H.-D.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Heme Oxygenase 1 Gene Transfer Prevents CD95/Fas Ligand-Mediated Apoptosis and Improves Liver Allograft Survival via Carbon Monoxide Signaling Pathway. Hum. Gene Ther. 2002, 13, 1189–1199. [Google Scholar] [CrossRef]
- Soares, M.P.; Usheva, A.; Brouard, S.; Berberat, P.O.; Gunther, L.; Tobiasch, E.; Bach, F.H. Modulation of Endothelial Cell Apoptosis by Heme Oxygenase-1-Derived Carbon Monoxide. Antioxid. Redox Signal. 2002, 4, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon Monoxide Generated by Heme Oxygenase 1 Suppresses Endothelial Cell Apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Porras, A.; Zuluaga, S.; Black, E.; Valladares, A.; Álvarez, A.M.; Ambrosino, C.; Benito, M.; Nebreda, A.R. p38α Mitogen-activated Protein Kinase Sensitizes Cells to Apoptosis Induced by Different Stimuli. Mol. Biol. Cell 2004, 15, 922–933. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.; Cunha, A.; Grégoire, I.P.; Seldon, M.P.; Soares, M.P. The Antiapoptotic Effect of Heme Oxygenase-1 in Endothelial Cells Involves the Degradation of p38α MAPK Isoform. J. Immunol. 2006, 177, 1894–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruda, M.A.; Rossi, A.G.; De Freitas, M.S.; Barja-Fidalgo, C.; Graça-Souza, A.V. Heme Inhibits Human Neutrophil Apoptosis: Involvement of Phosphoinositide 3-Kinase, MAPK, and NF-κB. J. Immunol. 2004, 173, 2023–2030. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Alam, J.; Fu, X.-Y.; Lee, P.J. Carbon Monoxide Differentially Modulates STAT1 and STAT3 and Inhibits Apoptosis via a Phosphatidylinositol 3-Kinase/Akt and p38 Kinase-dependent STAT3 Pathway during Anoxia-Reoxygenation Injury. J. Biol. Chem. 2005, 280, 8714–8721. [Google Scholar] [CrossRef] [Green Version]
- Willis, D.; Moore, A.; Frederick, R.; Willoughby, D. Heme oxygenase: A novel target for the modulation of inflammatory response. Nat. Med. 1996, 2, 87–90. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-S.; Chau, L.-Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 2002, 8, 240–246. [Google Scholar] [CrossRef]
- Chen, S.; Kapturczak, M.H.; Wasserfall, C.; Glushakova, O.Y.; Campbell-Thompson, M.; Deshane, J.S.; Joseph, R.; Cruz, P.E.; Hauswirth, W.W.; Madsen, K.M.; et al. Interleukin 10 attenuates neointimal proliferation and inflammation in aortic allografts by a heme oxygenase-dependent pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 7251–7256. [Google Scholar] [CrossRef] [Green Version]
- Ricchetti, G.A.; Williams, L.M.; Foxwell, B.M.J. Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide. J. Leukoc. Biol. 2004, 76, 719–726. [Google Scholar] [CrossRef]
- Drechsler, Y.; Dolganiuc, A.; Norkina, O.; Romics, L.; Li, W.; Kodys, K.; Bach, F.H.; Mandrekar, P.; Szabo, G. Heme Oxygenase-1 Mediates the Anti-Inflammatory Effects of Acute Alcohol on IL-10 Induction Involving p38 MAPK Activation in Monocytes. J. Immunol. 2006, 177, 2592–2600. [Google Scholar] [CrossRef]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.; Juncos, J.; Croatt, A.; Ackerman, A.; Grande, J.; Knutson, K.; Kane, G.; Terzic, A.; Griffin, M.; Nath, K. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int. 2007, 72, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Thierry, A.; Delbauve, S.; Preyat, N.; Soares, M.P.M.; Roumeguère, T.; Leo, O.; Flamand, V.; Le Moine, A.; Hougardy, J.-M. Specific expression of heme oxygenase-1 by myeloid cells modulates renal ischemia-reperfusion injury. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Kovtunovych, G.; Ghosh, M.C.; Ollivierre, W.; Weitzel, R.P.; Eckhaus, M.A.; Tisdale, J.F.; Yachie, A.; Rouault, T.A. Wild-type macrophages reverse disease in heme oxygenase 1-deficient mice. Blood 2014, 124, 1522–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme Oxygenase-1 Modulates Early Inflammatory Responses: Evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Ramdas, V.; Spencer, N.; Marson, L.; Anegon, I.; Hughes, J.; Kluth, D.C. Macrophages Expressing Heme Oxygenase-1 Improve Renal Function in Ischemia/Reperfusion Injury. Mol. Ther. 2010, 18, 1706–1713. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Nkejabega, N.C.; McKay, J.; Choudhary, A.K.; Vernon, M.A.; Beesley, M.F.; Clay, S.; Conway, B.C.; Marson, L.P.; Kluth, D.C.; et al. The induction of macrophage hemeoxygenase-1 is protective during acute kidney injury in aging mice. Kidney Int. 2011, 79, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Sierra-Filardi, E.; Vega, M.A.; Sánchez-Mateos, P.; Corbí, A.L.; Puig-Kröger, A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 2010, 215, 788–795. [Google Scholar] [CrossRef]
- Weis, N.; Weigert, A.; Von Knethen, A.; Brüne, B. Heme Oxygenase-1 Contributes to an Alternative Macrophage Activation Profile Induced by Apoptotic Cell Supernatants. Mol. Biol. Cell 2009, 20, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Harusato, A.; Naito, Y.; Takagi, T.; Uchiyama, K.; Mizushima, K.; Hirai, Y.; Higashimura, Y.; Katada, K.; Handa, O.; Ishikawa, T.; et al. BTB and CNC Homolog 1 (Bach1) Deficiency Ameliorates TNBS Colitis in Mice. Inflamm. Bowel Dis. 2013, 19, 740–753. [Google Scholar] [CrossRef]
- Zhang, M.; Nakamura, K.; Kageyama, S.; Lawal, A.O.; Gong, K.W.; Bhetraratana, M.; Fujii, T.; Sulaiman, D.; Hirao, H.; Bolisetty, S.; et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Devey, L.; Ferenbach, D.; Mohr, E.; Sangster, K.; Bellamy, C.O.; Hughes, J.; Wigmore, S.J. Tissue-resident Macrophages Protect the Liver From Ischemia Reperfusion Injury via a Heme Oxygenase-1-Dependent Mechanism. Mol. Ther. 2009, 17, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Cozzi, M.; Bush, E.L.; Rabb, H. Distant Organ Dysfunction in Acute Kidney Injury: A Review. Am. J. Kidney Dis. 2018, 72, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Grams, M.E.; Rabb, H. The distant organ effects of acute kidney injury. Kidney Int. 2012, 81, 942–948. [Google Scholar] [CrossRef] [Green Version]
- Vieira, J.M.; Castro, I.; Curvello-Neto, A.; DeMarzo, S.; Caruso, P.; Pastore, L.; Imanishe, M.H.; Abdulkader, R.C.R.M.; Deheinzelin, D. Effect of acute kidney injury on weaning from mechanical ventilation in critically ill patients. Crit. Care Med. 2007, 35, 184–191. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Li, S.; Jiang, J. Artesunate Inhibits Renal Ischemia Reperfusion-Stimulated Lung Inflammation in Rats by Activating HO-1 Pathway. Inflammation 2017, 41, 114–121. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Ying, Y.; Kim, J.; Westphal, S.N.; Long, K.E.; Padanilam, B.J. Targeted Deletion of p53 in the Proximal Tubule Prevents Ischemic Renal Injury. J. Am. Soc. Nephrol. 2014, 25, 2707–2716. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Liu, Y.; Wei, Q.; Huo, Y.; Liu, K.; Liu, F.; Dong, Z. Tubular p53 Regulates Multiple Genes to Mediate AKI. J. Am. Soc. Nephrol. 2014, 25, 2278–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, T.A.; Hato, T.; Mai, E.; Yoshimoto, M.; Kuehl, S.; Anderson, M.; Mang, H.; Plotkin, Z.; Chan, R.J.; Dagher, P.C. p53 Is Renoprotective after Ischemic Kidney Injury by Reducing Inflammation. J. Am. Soc. Nephrol. 2012, 24, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megyesi, J.; Andrade, L.; Vieira, J.M.J.; Safirstein, R.L.; Price, P.M. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001, 60, 2164–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, S.; Nakano, D.; Kitada, K.; Sofue, T.; Ohsaki, H.; Moriwaki, K.; Hara, T.; Ohmori, K.; Kohno, M.; Nishiyama, A. The cyclin-dependent kinase inhibitor p21 is essential for the beneficial effects of renal ischemic preconditioning on renal ischemia/reperfusion injury in mice. Kidney Int. 2014, 85, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Megyesi, J.; Tarcsafalvi, A.; Li, S.; Hodeify, R.; Seng, N.S.H.L.; Portilla, D.; Price, P.M. Increased expression of p21WAF1/CIP1 in kidney proximal tubules mediates fibrosis. Am. J. Physiol. Ren. Physiol. 2015, 308, F122–F130. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, J.H.; Schildhorn, C.; Jacobi, C.; Hömme, M.; Hartner, A.; Braun, H.; Kryzer, C.; Wang, C.; Von Zglinicki, T.; Kränzlin, B.; et al. Telomere Shortening Reduces Regenerative Capacity after Acute Kidney Injury. J. Am. Soc. Nephrol. 2009, 21, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Flores, B.; Gillings, N.; Bian, A.; Cho, H.J.; Yan, S.; Liu, Y.; Levine, B.; Moe, O.W.; Hu, M.C. αKlotho Mitigates Progression of AKI to CKD through Activation of Autophagy. J. Am. Soc. Nephrol. 2015, 27, 2331–2345. [Google Scholar] [CrossRef] [Green Version]
- Gigliotti, J.C.; Huang, L.; Ye, H.; Bajwa, A.; Chattrabhuti, K.; Lee, S.; Klibanov, A.L.; Kalantari, K.; Rosin, D.L.; Okusa, M.D. Ultrasound Prevents Renal Ischemia-Reperfusion Injury by Stimulating the Splenic Cholinergic Anti-Inflammatory Pathway. J. Am. Soc. Nephrol. 2013, 24, 1451–1460. [Google Scholar] [CrossRef] [Green Version]
- Gigliotti, J.C.; Huang, L.; Bajwa, A.; Ye, H.; Mace, E.H.; Hossack, J.A.; Kalantari, K.; Inoue, T.; Rosin, D.L.; Okusa, M.D. Ultrasound Modulates the Splenic Neuroimmune Axis in Attenuating AKI. J. Am. Soc. Nephrol. 2015, 26, 2470–2481. [Google Scholar] [CrossRef]
- Hualin, C.; Wenli, X.; Dapeng, L.; Xijing, L.; Xiuhua, P.; Qingfeng, P. The Anti-inflammatory Mechanism of Heme Oxygenase-1 Induced by Hemin in Primary Rat Alveolar Macrophages. Inflammation 2011, 35, 1087–1093. [Google Scholar] [CrossRef]
- Andrés-Hernando, A.; Altmann, C.; Ahuja, N.; Lanaspa, M.A.; Nemenoff, R.; He, Z.; Ishimoto, T.; Simpson, P.A.; Weiser-Evans, M.C.; Bacalja, J.; et al. Splenectomy exacerbates lung injury after ischemic acute kidney injury in mice. Am. J. Physiol. Physiol. 2011, 301, F907–F916. [Google Scholar] [CrossRef]
- Moeckel, G.W.; Palmer, M.B.; Cantley, L.G.; Vichot, A.A. Quantification and localization of M2 macrophages in human kidneys with acute tubular injury. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 415–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, N.M.; Ferenbach, D.A.; Isenberg, J.S.; Thomson, A.W.; Hughes, J. Dendritic cells and macrophages in the kidney: A spectrum of good and evil. Nat. Rev. Nephrol. 2014, 10, 625–643. [Google Scholar] [CrossRef]
- Huen, S.C.; Cantley, L.G. Macrophages in Renal Injury and Repair. Annu. Rev. Physiol. 2017, 79, 449–469. [Google Scholar] [CrossRef]
- Grimm, P.C.; McKenna, R.; Nickerson, P.; Russell, M.E.; Gough, J.; Gospodarek, E.; Liu, B.; Jeffery, J.; Rush, D.N. Clinical rejection is distinguished from subclinical rejection by increased infiltration by a population of activated macrophages. J. Am. Soc. Nephrol. 1999, 10, 1582–1589. [Google Scholar]
- Pilmore, H.L.; Painter, D.M.; Bishop, G.A.; McCaughan, G.W.; Eris, J.M. Early Up-Regulation of Macrophages and Myofibroblasts: A New Marker for Development of Chronic Renal Allograft Rejection. Transplantation 2000, 69, 2658–2662. [Google Scholar] [CrossRef] [PubMed]
- Bharucha, A.E.; Kulkarni, A.; Choi, K.M.; Camilleri, M.; Lempke, M.; Brunn, G.J.; Gibbons, S.J.; Zinsmeister, A.R.; Farrugia, G. First-in-Human Study Demonstrating Pharmacological Activation of Heme Oxygenase-1 in Humans. Clin. Pharmacol. Ther. 2009, 87, 187–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the Diagnosis and Treatment of the Acute Porphyrias. Ann. Intern. Med. 2005, 142, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.A.B.; Czopek, A.; Bellamy, C.O.C.; McNally, S.J.; Kluth, D.C.; Marson, L.P. Hemin Preconditioning Upregulates Heme Oxygenase-1 in Deceased Donor Renal Transplant Recipients. Transplantation 2016, 100, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, M.; Korpak, K.; Doerfler, A.; Zouaoui Boudjeltia, K. Deciphering the Role of Heme Oxygenase-1 (HO-1) Expressing Macrophages in Renal Ischemia-Reperfusion Injury. Biomedicines 2021, 9, 306. https://doi.org/10.3390/biomedicines9030306
Rossi M, Korpak K, Doerfler A, Zouaoui Boudjeltia K. Deciphering the Role of Heme Oxygenase-1 (HO-1) Expressing Macrophages in Renal Ischemia-Reperfusion Injury. Biomedicines. 2021; 9(3):306. https://doi.org/10.3390/biomedicines9030306
Chicago/Turabian StyleRossi, Maxime, Kéziah Korpak, Arnaud Doerfler, and Karim Zouaoui Boudjeltia. 2021. "Deciphering the Role of Heme Oxygenase-1 (HO-1) Expressing Macrophages in Renal Ischemia-Reperfusion Injury" Biomedicines 9, no. 3: 306. https://doi.org/10.3390/biomedicines9030306
APA StyleRossi, M., Korpak, K., Doerfler, A., & Zouaoui Boudjeltia, K. (2021). Deciphering the Role of Heme Oxygenase-1 (HO-1) Expressing Macrophages in Renal Ischemia-Reperfusion Injury. Biomedicines, 9(3), 306. https://doi.org/10.3390/biomedicines9030306