
Screening of Natural Compounds as P-Glycoprotein Inhibitors against Multidrug Resistance

 ,
, 
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Threading
2.2. Molecular Dynamics
2.3. Molecular Docking
2.4. Flavonoid Compounds
2.5. Cell Lines and Cultivation Conditions
2.6. Drug Accumulation Assay
2.7. Cell Viability Assay
2.8. Statistical Analysis
3. Results
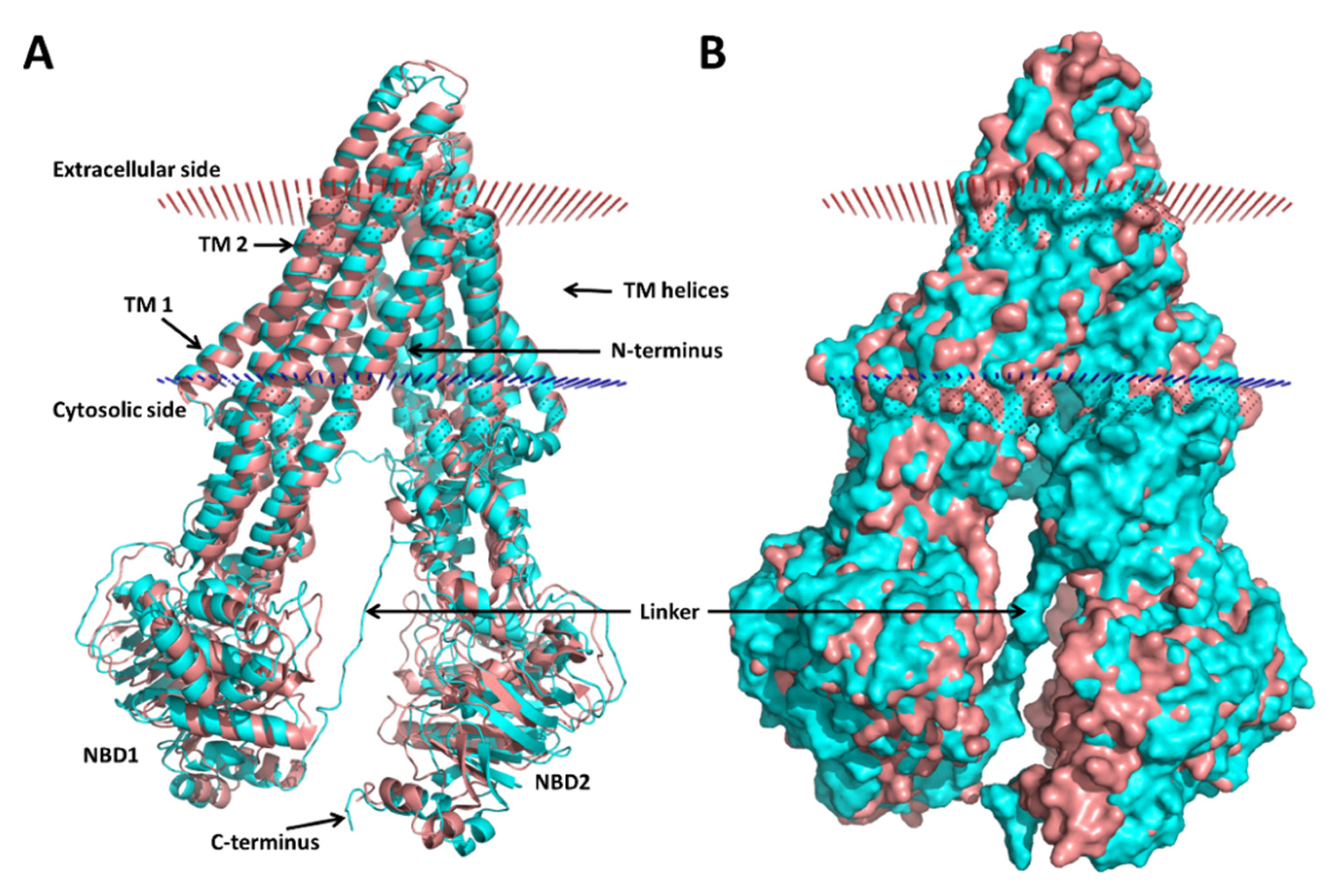
3.1. Modeling of the Human P-gp
3.2. Molecular Dynamics
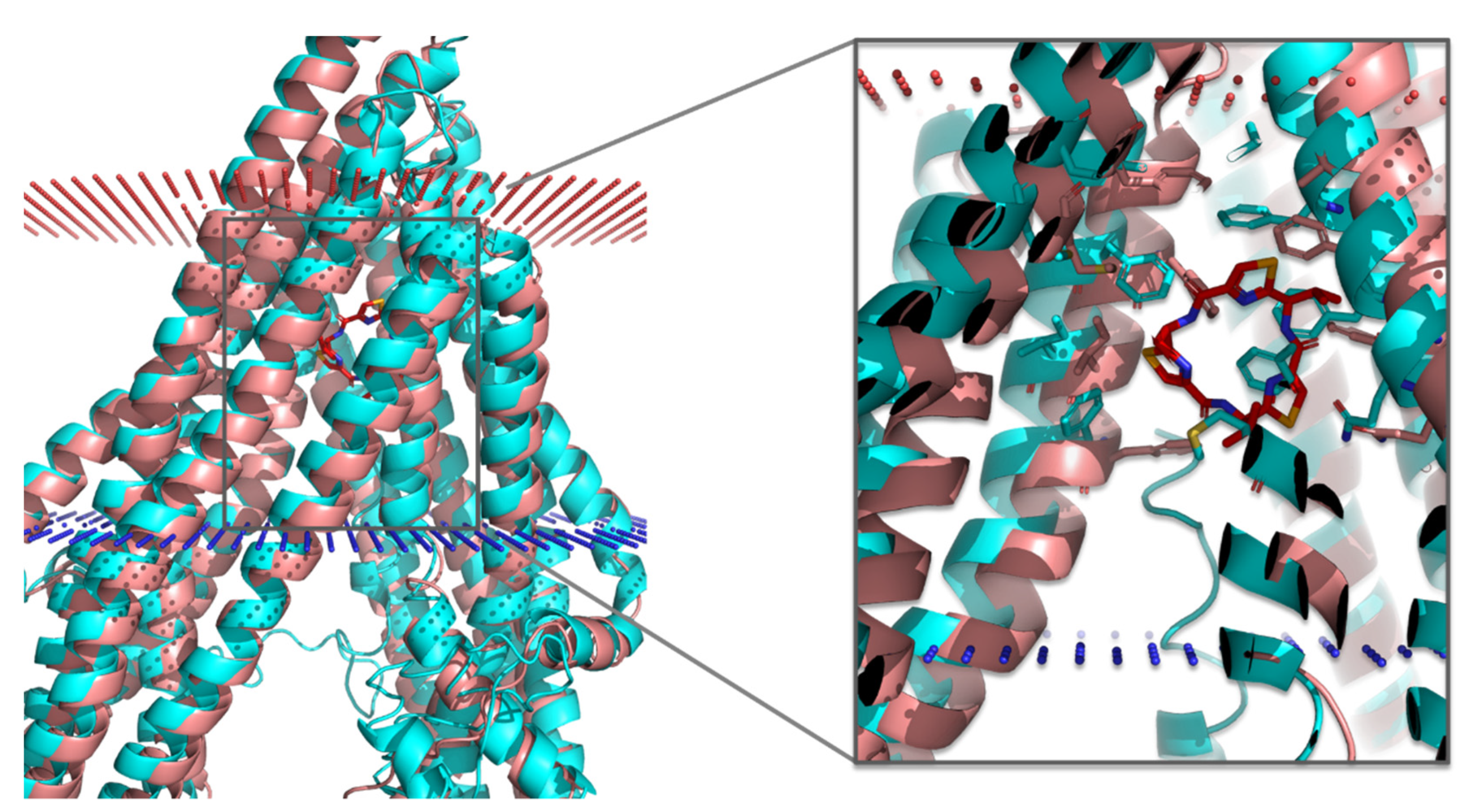
3.3. Molecular Docking of the Training Set
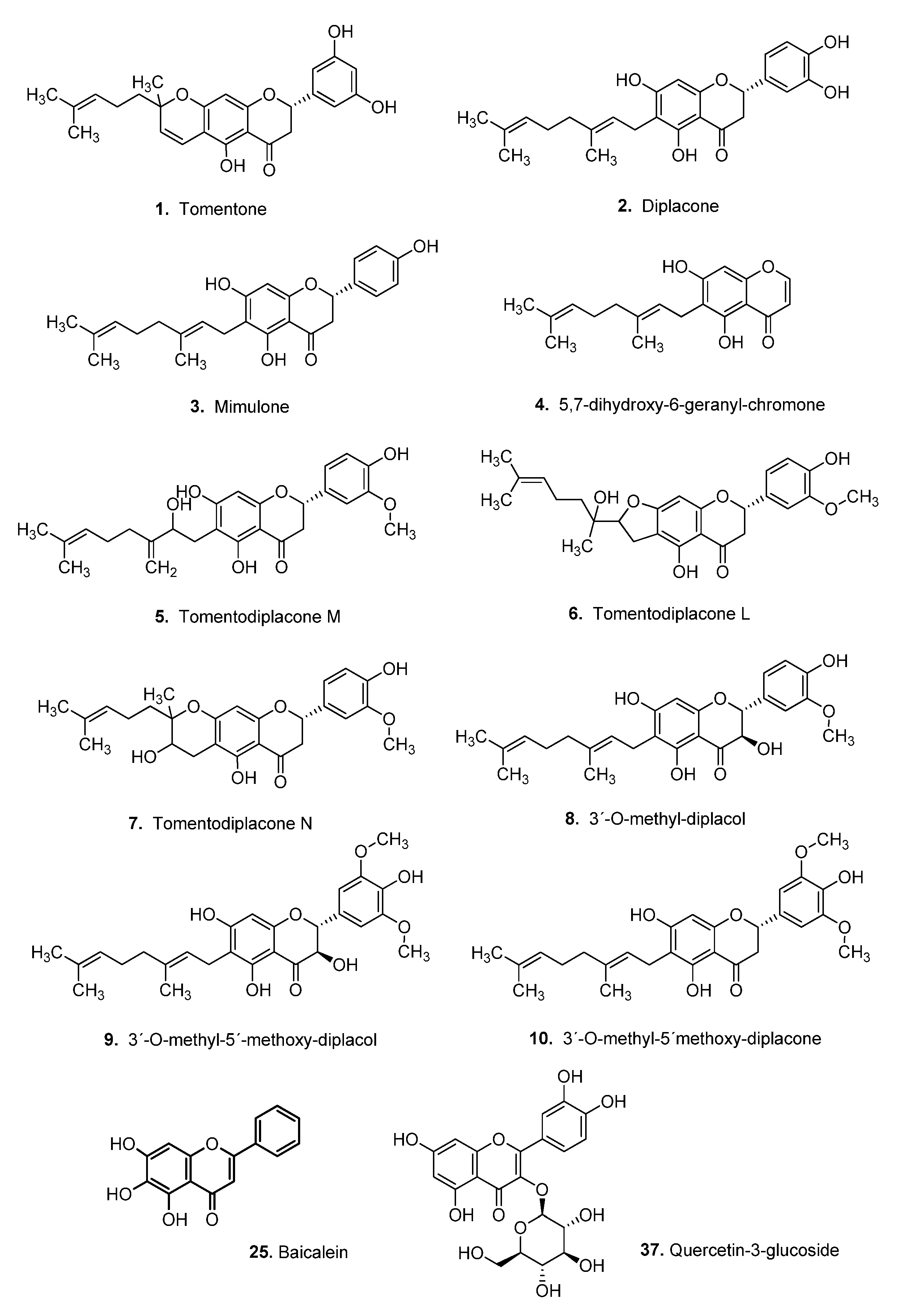
3.4. Virtual Screening of Natural Flavonoids
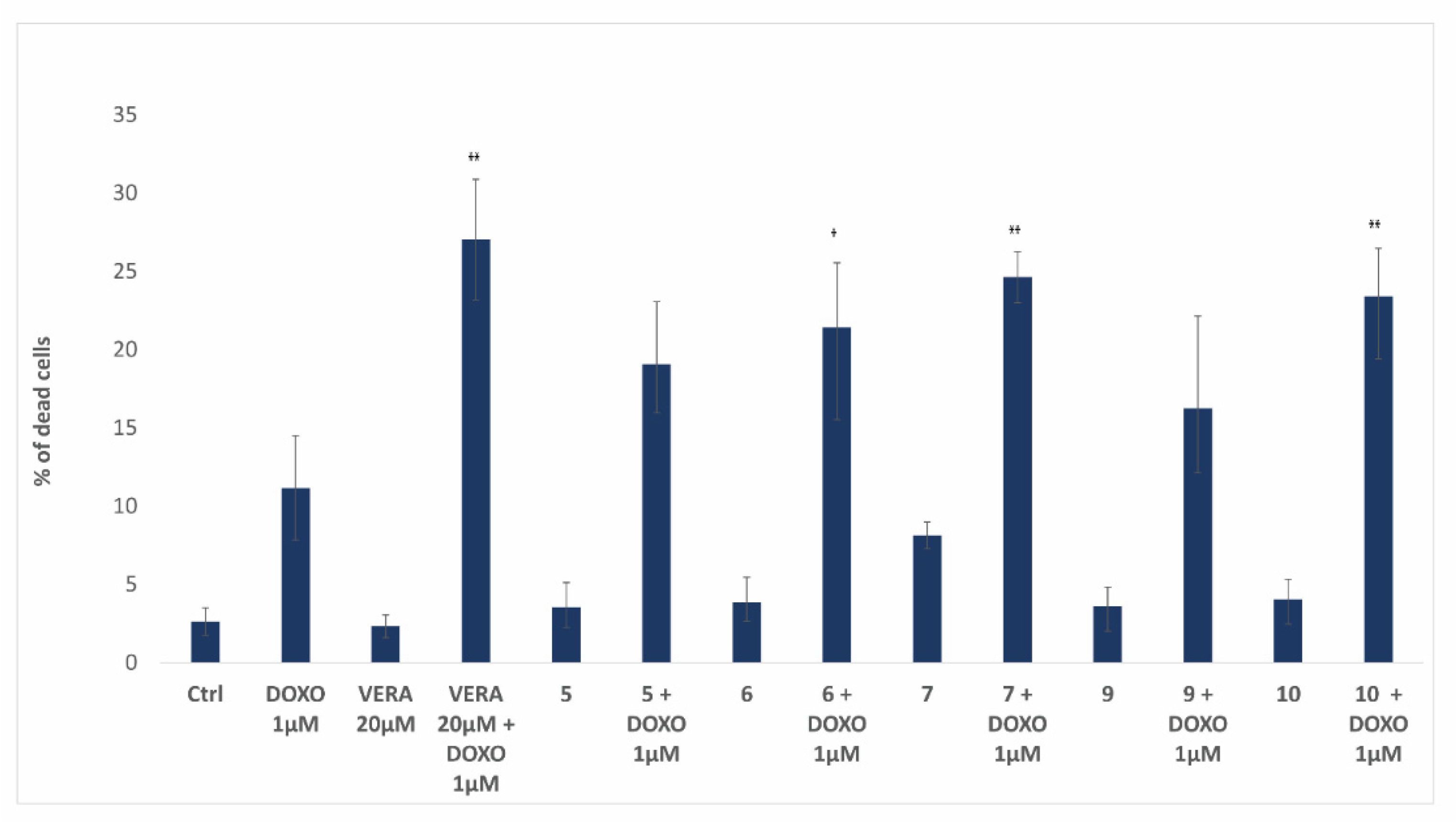
3.5. Biological Activity of Flavonoids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohnuma, S.; Ambudkar, S.V. Discovering Natural Product Modulators to Overcome Multidrug Resistance in Cancer Chemotherapy. Curr. Pharm. Biotechnol. 2011, 12, 609–620. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef] [Green Version]
- Velingkar, V.S.; Dandekar, V.D. Modulation of P-Glycoprotein Mediated Multidrug Resistance (MDR) in Cancer Using Chemosensitizers. Int. J. Pharma Sci. Res. 2010, 1, 104–111. [Google Scholar]
- Dai, Y.; Zhang, F.; Chen, N.; Wang, G.; Jia, S.; Zheng, H. Analysis of P-glycoprotein structure and binding sites. In Proceedings of the the 2nd International Conference on Information Science and Engineering; Institute of Electrical and Electronics Engineers: Wuhan, China, 2010; pp. 101–103. [Google Scholar] [CrossRef]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of Conventional Anti-cancer Drugs as New Tools against Multidrug Resistant Tumors. Drug Resist. Updat. 2020, 50, 100682. [Google Scholar] [CrossRef]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC Transporters: The Power to Change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.-H.; Chen, Z.-S. Overcoming ABC Transporter-Mediated Multidrug Resistance: Molecular Mechanisms and Novel Therapeutic Drug Strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Ling, V. Positively Cooperative Sites for Drug Transport by P-Glycoprotein with Distinct Drug Specificities. JBIC J. Biol. Inorg. Chem. 1997, 250, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.J.; Ferreira, M.-J.U.; Dos Santos, D.J.V.A. Molecular Docking Characterizes Substrate-Binding Sites and Efflux Modulation Mechanisms within P-Glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef]
- Ward, A.A.; Szewczyk, P.P.; Grimard, V.; Lee, C.-W.; Martinez, L.L.; Doshi, R.R.; Caya, A.A.; Villaluz, M.M.; Pardon, E.E.; Cregger, C.C.; et al. Structures of P-Glycoprotein Reveal Its Conformational Flexibility and an Epitope on the NucleotiDe-binding Domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Chen, J. Molecular Structure of Human P-Glycoprotein in the ATP-Bound, Outward-Facing Conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gong, W.; Wang, Y.; Liu, Y.; Li, C. Exploring Movement and Energy in Human P-Glycoprotein Conformational Rearrangement. J. Biomol. Struct. Dyn. 2018, 37, 1104–1119. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural Insight into Substrate and Inhibitor Discrimination by Human P-Glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Matsuoka, K.; Kimura, Y.; Ueda, K.; Kato, H. Inward-and Outward-Facing X-ray Crystal Structures of Homodimeric P-Glycoprotein CmABCB1. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.E.; Zhang, S. Flavonoid–Drug Interactions: Effects of Flavonoids on ABC Transporters. Life Sci. 2006, 78, 2116–2130. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Dubay, M.M.; Sausville, E.A.; Brizuela, L.; Worland, P.J. Flavopiridol Induces G1 Arrest with Inhibition of Cy-clin-Dependent Kinase (CDK) 2 and CDK4 in Human Breast Carcinoma Cells. Cancer Res. 1996, 56, 2973–2978. [Google Scholar] [PubMed]
- Raj, N.K.; Sripal, R.M.; Chaluvadi, M.R.; Krishna, D.R. Bioflavonoids Classification, Pharmacological, Biochemical Effects and Therapeutic Potential. Ind. J. Pharmacol. 2001, 33, 2. [Google Scholar]
- Di Pietro, A.; Dumontet, C.; Barron, D. Recent Advances in the Discovery of Flavonoids and Analogs with High-Affinity Binding to P-Glycoprotein Responsible for Cancer Cell Multidrug Resistance. Med. Res. Rev. 2002, 22, 512–529. [Google Scholar] [CrossRef]
- Brezani, V.; Smejkal, K.; Hosek, J.; Tomasova, V. Anti-inflammatory Natural Prenylated Phenolic Compounds—Potential Lead Substances. Curr. Med. Chem. 2018, 25, 1094–1159. [Google Scholar] [CrossRef]
- Schneiderová, K.; Šmejkal, K. Phytochemical profile of Paulownia tomentosa (Thunb). Steud. Phytochem. Rev. 2014, 14, 799–833. [Google Scholar] [CrossRef] [PubMed]
- Molčanová, L.; Janošíková, D.; Dall’acqua, S.; Šmejkal, K. C-Prenylated Flavonoids with Potential Cytotoxic Activity against Solid Tumor Cell Lines. Phytochem. Rev. 2019, 18, 1051–1100. [Google Scholar] [CrossRef]
- Šmejkal, K. Cytotoxic Potential of C-prenylated Flavonoids. Phytochem. Rev. 2014, 13, 245–275. [Google Scholar] [CrossRef]
- Slanina, J.; Páchniková, G.; Čarnecká, M.; Koubíková, L.P.; Adámková, L.; Humpa, O.; Šmejkal, K.; Slaninová, I. Identification of Key Structural Characteristics of Schisandra chinensis Lignans Involved in P-Glycoprotein Inhibition. J. Nat. Prod. 2014, 77, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Domicevica, L.; Biggin, P.C. Homology Modelling of Human P-glycoprotein. Biochem. Soc. Trans. 2015, 43, 952–958. [Google Scholar] [CrossRef]
- Bonito, C.A.; Ferreira, R.J.; Ferreira, M.-J.U.; Gillet, J.-P.; Cordeiro, M.N.D.S.; Dos Santos, D.J.V.A. Theoretical Insights on Helix Repacking as the Origin of P-Glycoprotein Promiscuity. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Condic-Jurkic, K.; Subramanian, N.; Mark, A.E.; O’Mara, M.L. The Reliability of Molecular Dynamics Simulations of the Multidrug Transporter P-glycoprotein in a Membrane Environment. PLoS ONE 2018, 13, e0191882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shityakov, S.; Förster, C. In Silico Structure-Based Screening of Versatile P-Glycoprotein Inhibitors Using Polynomial Empirical Scoring Functions. Adv. Appl. Bioinform. Chem. 2014, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Chen, J.; Shi, X.; Xu, L.; Xi, Z.; You, L.; An, R.; Wang, X. Development of in Silico Models for Predicting P-Glycoprotein Inhibitors Based on a Two-Step Approach for Feature Selection and Its Application to Chinese Herbal Medicine Screening. Mol. Pharm. 2015, 12, 3691–3713. [Google Scholar] [CrossRef]
- Ngo, T.-D.; Tran, T.-D.; Le, M.-T.; Thai, K.-M. Computational Predictive Models for P-Glycoprotein Inhibition of in-House Chalcone Derivatives and Drug-Bank Compounds. Mol. Divers. 2016, 20, 945–961. [Google Scholar] [CrossRef]
- Wongrattanakamon, P.; Lee, V.S.; Nimmanpipug, P.; Sirithunyalug, B.; Chansakaow, S.; Jiranusornkul, S. Insight into the Molecular Mechanism of P-Glycoprotein Mediated Drug Toxicity Induced by Bioflavonoids: An Integrated Computational Approach. Toxicol. Mech. Methods 2017, 5, 1–19. [Google Scholar] [CrossRef]
- Wongrattanakamon, P.; Nimmanpipug, P.; Sirithunyalug, B.; Chansakaow, S.; Jiranusornkul, S. A Significant Mechanism of Molecular Recognition between Bioflavonoids and P-Glycoprotein Leading to Herb-Drug Interactions. Toxicol. Mech. Methods 2017, 28, 1–11. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal Duplication and Homology with Bacterial Transport Proteins in the mdr1 (P-glycoprotein) Gene from Multidrug-Resistant Human Cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL Workspace: A Web-Based Environment for Protein Structure Homology Modelling. Bioinformatics 2005, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from Standard Atomic Volumes as a Quality Measure for Protein Crystal Structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of Protein Models with Three-Dimensional Profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the Estimation of the Absolute Quality of Individual Protein Structure Models. Bioinformatics 2010, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; De Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure Validation by Cα Geometry: ϕ,ψ and Cβ Deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.3.2; Schrödinger, LLC: New York, NY, USA, 2019.
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A Server for Estimating pKas and Adding Missing Hydrogens to Macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without Compromise—A Mixed Precision Model for GPU Accelerated Molecular Dynamics Simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2011, 40, D370–D376. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, I.T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Chemin. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. NNScore 2.0: A Neural-Network Receptor–Ligand Scoring Function. J. Chem. Inf. Model. 2011, 51, 2897–2903. [Google Scholar] [CrossRef]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of Machine-Learning Scoring Functions in Structure-Based Virtual Screening. Sci. Rep. 2017, 7, srep46710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanáková, Z.; Hošek, J.; Babula, P.; Dall’Acqua, S.; Václavík, J.; Šmejkal, K. C-Geranylated Flavanones from Paulownia tomentosa Fruits as Potential Anti-inflammatory Compounds Acting via Inhibition of TNF-α Production. J. Nat. Prod. 2015, 78, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Šmejkal, K.; Babula, P.; Šlapetová, T.; Brognara, E.; Dall’Acqua, S.; Žemlička, M.; Innocenti, G.; Cvačka, J. Cytotoxic Activity of C-Geranyl Compounds from Paulownia tomentosa Fruits. Planta Med. 2008, 74, 1488–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šmejkal, K.; Grycová, L.; Marek, R.; Lemière, F.; Jankovská, D.; Forejtníková, H.; Vančo, J.; Suchý, V. C-Geranyl Compounds from Paulownia tomentosa Fruits. J. Nat. Prod. 2007, 70, 1244–1248. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.-J.U.; Dos Santos, D.J.V.A. Insights on P-Glycoprotein’s Efflux Mechanism Obtained by Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 1853–1864. [Google Scholar] [CrossRef]
- Hošek, J.; Bartos, M.; Chudík, S.; Dall’Acqua, S.; Innocenti, G.; Kartal, M.; Kokoška, L.; Kollár, P.; Kutil, Z.; Landa, P.; et al. Natural Compound Cudraflavone B Shows Promising Anti-inflammatory Properties In Vitro. J. Nat. Prod. 2011, 74, 614–619. [Google Scholar] [CrossRef]
- Argyropoulou, A.; Aligiannis, N.; Trougakos, I.P.; Skaltsounis, A.-L. Natural Compounds with Anti-ageing Activity. Nat. Prod. Rep. 2013, 30, 1412–1437. [Google Scholar] [CrossRef]
- Navrátilová, A.; Nešuta, O.; Vančatová, I.; Čížek, A.; Varela-M, R.E.; López-Abán, J.; Villa-Pulgarin, J.A.; Mollinedo, F.; Muro, A.; Žemličková, H.; et al. C-Geranylated Flavonoids from Paulownia tomentosa fruits with Antimicrobial Potential and Synergistic Activity with Antibiotics. Pharm. Biol. 2016, 54, 1398–1407. [Google Scholar] [CrossRef] [Green Version]
- Hanáková, Z.; Hošek, J.; Kutil, Z.; Temml, V.; Landa, P.; Vaněk, T.; Schuster, D.; Dall’Acqua, S.; Cvačka, J.; Polanský, O.; et al. Anti-inflammatory Activity of Natural Geranylated Flavonoids: Cyclooxygenase and Lipoxygenase Inhibitory Properties and Proteomic Analysis. J. Nat. Prod. 2017, 80, 999–1006. [Google Scholar] [CrossRef]
- Ferreira, A.; Santos, A.O.; Falcão, A.; Alves, G. In Vitro Screening of Dual Flavonoid Combinations for Reversing P-glycoprotein-Mediated Multidrug Resistance: Focus on Antiepileptic Drugs. Food Chem. Toxicol. 2018, 111, 84–93. [Google Scholar] [CrossRef]
- Li, J.; Duan, B.; Guo, Y.; Zhou, R.; Sun, J.; Bie, B.; Yang, S.; Huang, C.; Yang, J.; Li, Z. Baicalein Sensitizes Hepatocellular Carcinoma Cells to 5-FU and Epirubicin by Activating Apoptosis and Ameliorating P-glycoprotein Activity. Biomed. Pharmacother. 2018, 98, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Latest Global Cancer Data: Cancer Burden Rises to 18.1 Million New Cases and 9.6 Million Cancer Deaths in 2018. Int. Agency Res. Cancer 2018, 263, 1–3.
- Dong, J.; Qin, Z.; Zhang, W.-D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R.; Chen, Z.-S.; Cheng, X.-D.; Qin, J.-J. Medicinal Chemistry Strategies to Discover P-glycoprotein Inhibitors: An Update. Drug Resist. Updat. 2020, 49, 100681. [Google Scholar] [CrossRef]
- Sato, T.; Kodan, A.; Kimura, Y.; Ueda, K.; Nakatsu, T.; Kato, H. Functional Role of the Linker Region in Purified Human P-glycoprotein. FEBS J. 2009, 276, 3504–3516. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Jin, J.; Sun, H.; Liu, G.T. Reversal of P-glycoprotein-mediated Multidrug Resistance of Cancer Cells by Five Schizandrins Isolated from the Chinese Herb Fructus schizandrae. Cancer Chemother. Pharmacol. 2008, 62, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Lu, Q.; Zhang, K.; Hu, X. Dibenzocyclooctadiene lingnans: A Class of Novel Inhibitors of P-glycoprotein. Cancer Chemother. Pharmacol. 2005, 58, 99–106. [Google Scholar] [CrossRef]
- Slaninova, I.; Březinová, L.; Koubíková, L.; Slanina, J. Dibenzocyclooctadiene lignans Overcome Drug Resistance in Lung Cancer Cells—Study of Structure–Activity Relationship. Toxicol. Vitr. 2009, 23, 1047–1054. [Google Scholar] [CrossRef]
- Šmejkal, K.; Svačinová, J.; Šlapetová, T.; Schneiderová, K.; Dall’Acqua, S.; Innocenti, G.; Závalová, V.; Kollár, P.; Chudík, S.; Marek, R.; et al. Cytotoxic Activities of Several Geranyl-Substituted Flavanones. J. Nat. Prod. 2010, 73, 568–572. [Google Scholar] [CrossRef]
- Kitagawa, S.; Nabekura, T.; Takahashi, T.; Nakamura, Y.; Sakamoto, H.; Tano, H.; Hirai, M.; Tsukahara, G. Structure-Activity Relationships of the Inhibitory Effects of Flavonoids on P-Glycoprotein-Mediated Transport in KB-C2 Cells. Biol. Pharm. Bull. 2005, 28, 2274–2278. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, R.H. Synergistic Effect of Apple Extracts and Quercetin 3-β-d-Glucoside Combination on Antiproliferative Activity in MCF-7 Human Breast Cancer Cells in Vitro. J. Agric. Food Chem. 2009, 57, 8581–8586. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yu, J.; Zhan, J.; Yang, L.; Guo, L.; Xu, Y. Pharmacokinetics, Tissue Distribution, and Metabolism Study of Icariin in Rat. BioMed Res. Int. 2017, 2017, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Yim, S.-H.; Han, F.; Kang, B.Y.; Choi, H.J.; Jung, D.-W.; Williams, D.R.; Gustafson, K.R.; Kennelly, E.J.; Lee, I.-S. Biotransformed Metabolites of the Hop Prenylflavanone Isoxanthohumol. Molecules 2019, 24, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
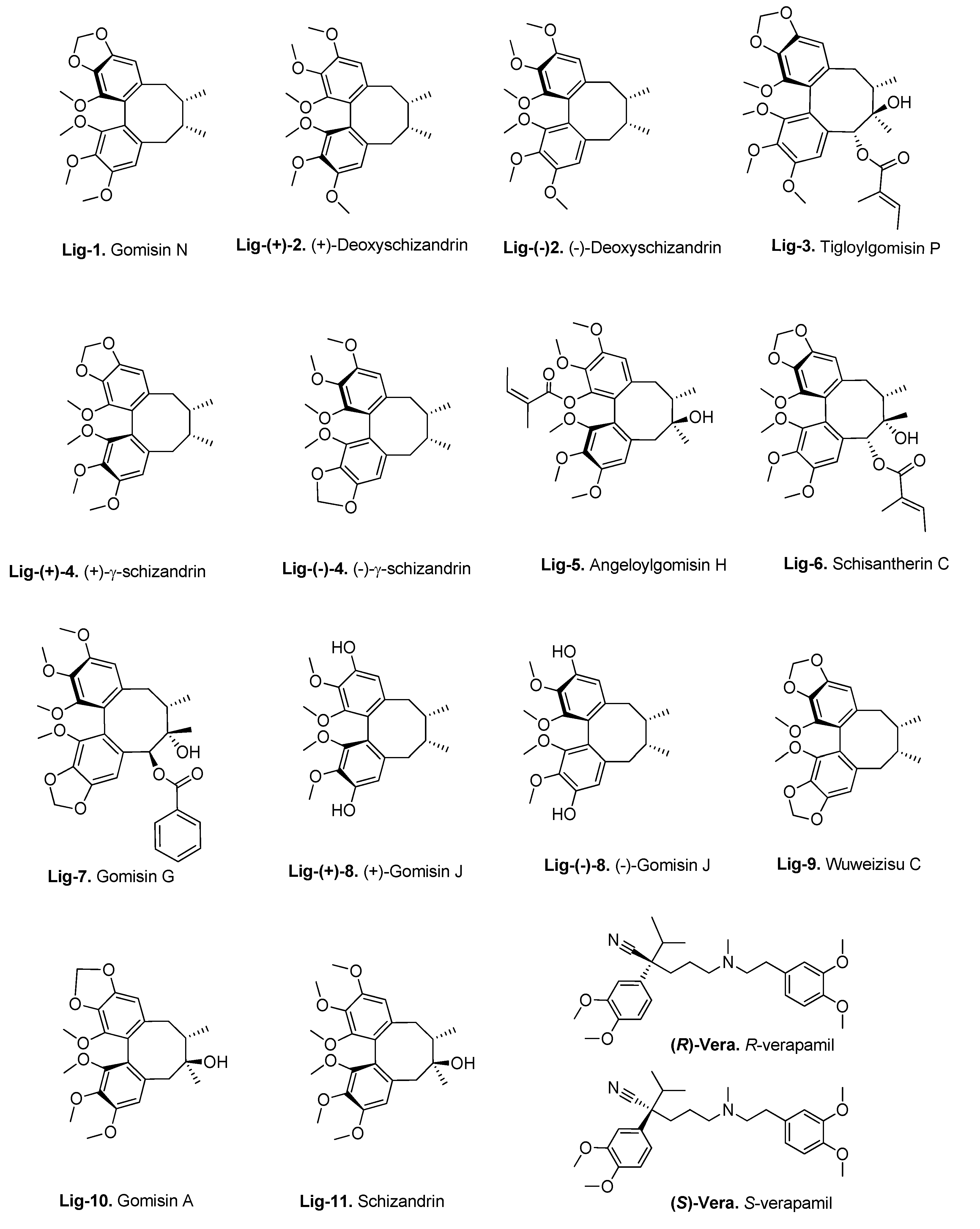{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Activity b | AutoDock Vina | SMINA | NNScore | RF-Score-VS | |
|---|---|---|---|---|---|
| Compound | (% Accumulation) | ΔGbind (kcal/mol) | ΔGbind (kcal/mol) | Kd (nM) | Kd (nM) |
| Lig-1 | 283 | −7.6 | −7.60 | 28.3 | 702.6 |
| Lig-(-)-2 | 277 | −7.6 | −7.57 | 71.4 | 567.8 |
| Lig-(+)-2 | 264 | −7.3 | −7.30 | 144.1 | 631.9 |
| Lig-3 | 234 | −7.3 | −8.32 | 48.3 | 678.0 |
| Lig-(+)-4 | 232 | −7.8 | −7.81 | 17.5 | 626.9 |
| Lig-(-)-4 | 152 | −7.8 | −7.90 | 50.7 | 723.8 |
| Lig-5 | 202 | −7.2 | −7.41 | 46.7 | 737.1 |
| Lig-6 | 201 | −8.9 | −9.08 | 67.1 | 583.8 |
| Lig-7 | 199 | −8.0 | −9.02 | 17.9 | 637.3 |
| Lig-(-)-8 | 191 | −7.2 | −7.61 | 283.2 | 704.3 |
| Lig-(+)-8 | 156 | −7.2 | −7.55 | 181.0 | 814.2 |
| Lig-9 | 187 | −8.4 | −8.45 | 41.2 | 742.7 |
| Lig-10 | 162 | −7.3 | −7.85 | 257.0 | 554.0 |
| Lig-11 | 125 | −7.3 | −7.40 | 359.8 | 609.8 |
| Pearson RP b | −0.0308 | 0.0808 | −0.5448 | −0.2009 | |
| Spearman RS c | −0.0850 | 0.1033 | −0.4505 | −0.2088 | |
| (R)-vera | 239 | −7.9 | −8.06 | 0.74 | 834.5 |
| (S)-vera | 239 | −7.3 | −8.15 | 0.46 | 845.8 |
| Virtual Screening a | Biological Activity b | ||||
|---|---|---|---|---|---|
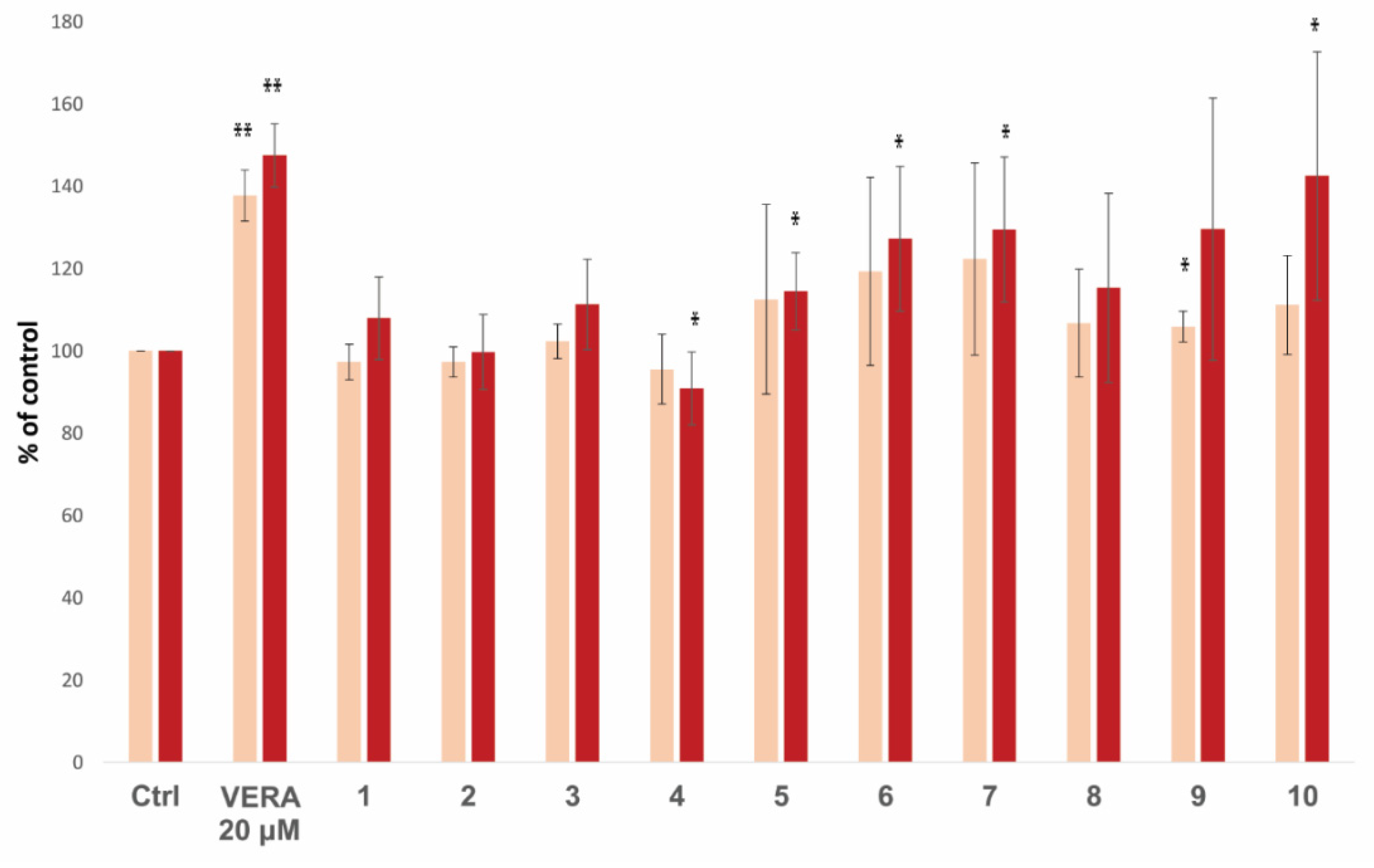
| Ranking | Compound | Kd (nM) | ΔGbind (kcal/mol) | % Accumulation (10 µM) | % Accumulation (20 µM) |
| 1 | 25 | 1.16 | −7.7 | n.d. | n.d. |
| 2 | 37 | 1.27 | −9.0 | n.d. | n.d. |
| … | … | ||||
| 14 | 10 | 3.49 | −9.5 | 111.2 ± 12.0 | 142.5 ± 30.2 |
| 15 | 3 | 3.80 | −9.9 | 102.3 ± 4.2 | 111.3 ± 11.0 |
| 25 | 4 | 9.62 | −8.2 | 95.5 ± 8.5 | 90.9 ± 8.9 |
| 31 | 7 | 14.3 | −10.1 | 122.3 ± 23.4 | 129.5 ± 17.6 |
| 32 | 5 | 15.3 | −9.4 | 112.5 ± 23.1 | 114.5 ± 9.4 |
| 33 | 1 | 16.8 | −9.5 | 97.3 ± 4.3 | 107.9 ± 10.1 |
| 34 | 2 | 17.8 | −9.3 | 97.3 ± 3.7 | 99.7 ± 9.1 |
| 40 | 9 | 26.0 | −9.4 | 105.9 ± 3.7 | 129.6 ± 31.9 |
| 42 | 8 | 28.4 | −9.4 | 106.7 ± 13.1 | 115.3 ± 23.0 |
| 44 | 6 | 36.6 | −9.9 | 119.3 ± 22.9 | 127.2 ± 17.6 |
| Best lignan c | Lig-(+)-4 | 17.5 | −7.6 | n.d. | 283 ± 33 (Lig-1) d |
| Worst lignan c | Lig-11 | 359.8 | −7.3 | n.d. | 125 ± 17 (Lig-11) d |
| (R)-vera (S)-vera | 0.74 0.46 | −7.9 −7.3 | 137.7 ± 6.2 | 147.5 ± 7.7 239 ± 30 d | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, S.M.; Šupolíková, L.; Molčanová, L.; Šmejkal, K.; Bednar, D.; Slaninová, I. Screening of Natural Compounds as P-Glycoprotein Inhibitors against Multidrug Resistance. Biomedicines 2021, 9, 357. https://doi.org/10.3390/biomedicines9040357
Marques SM, Šupolíková L, Molčanová L, Šmejkal K, Bednar D, Slaninová I. Screening of Natural Compounds as P-Glycoprotein Inhibitors against Multidrug Resistance. Biomedicines. 2021; 9(4):357. https://doi.org/10.3390/biomedicines9040357
Chicago/Turabian StyleMarques, Sérgio M., Lucie Šupolíková, Lenka Molčanová, Karel Šmejkal, David Bednar, and Iva Slaninová. 2021. "Screening of Natural Compounds as P-Glycoprotein Inhibitors against Multidrug Resistance" Biomedicines 9, no. 4: 357. https://doi.org/10.3390/biomedicines9040357
APA StyleMarques, S. M., Šupolíková, L., Molčanová, L., Šmejkal, K., Bednar, D., & Slaninová, I. (2021). Screening of Natural Compounds as P-Glycoprotein Inhibitors against Multidrug Resistance. Biomedicines, 9(4), 357. https://doi.org/10.3390/biomedicines9040357