
A Metabolic Change towards Fermentation Drives Cancer Cachexia in Myotubes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Cell Culture
2.3. Conditioned Media
2.4. Immunoblot Analysis
2.5. Glucose Uptake
2.6. Oxygen Consumption Assay
2.7. PDH Activity
2.8. Lactate Assay
2.9. Confocal Analysis
2.10. Statistical Analysis
3. Results
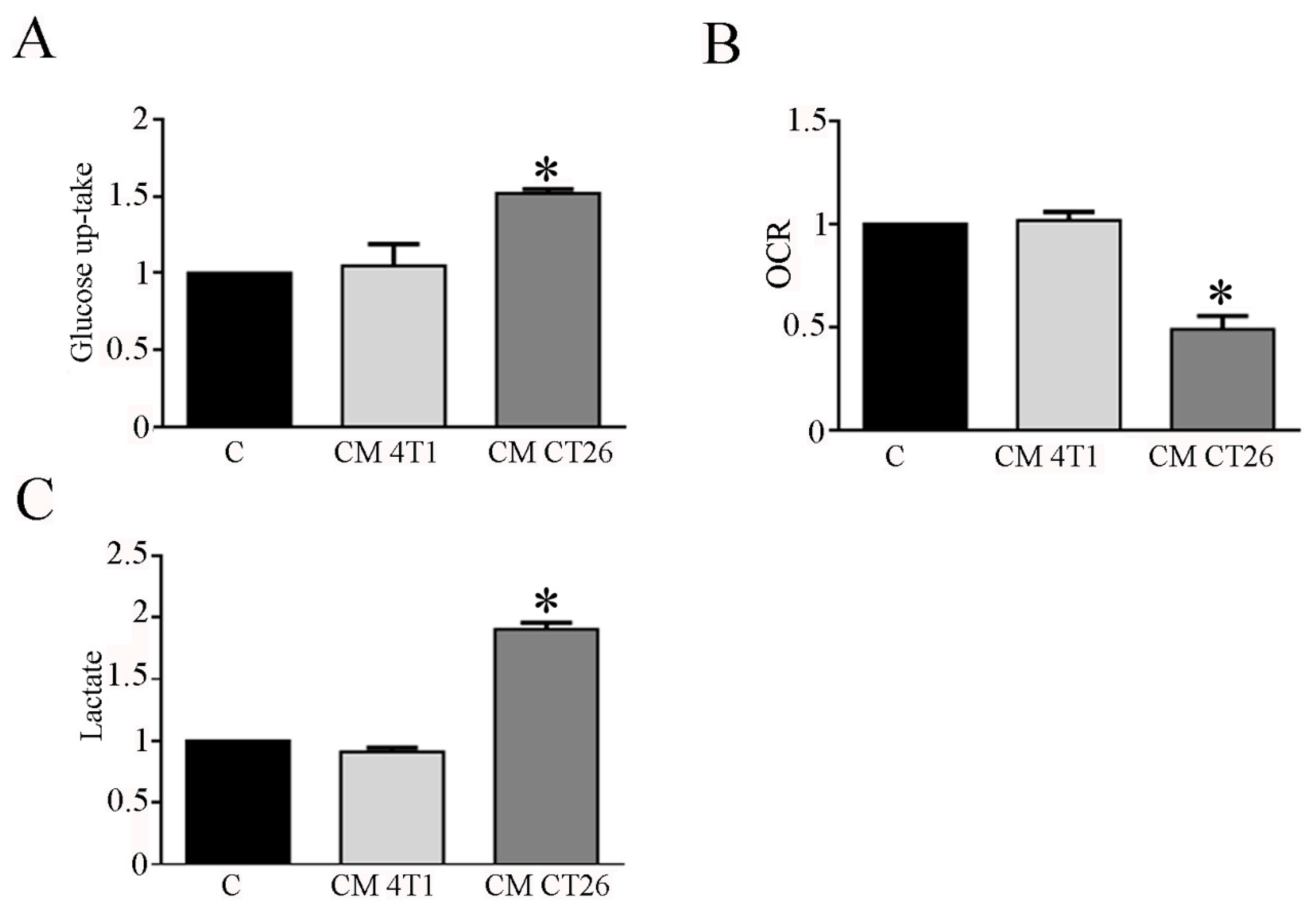
3.1. The Treatment with CM CT26 Induces a Metabolic Modification in Myotubes
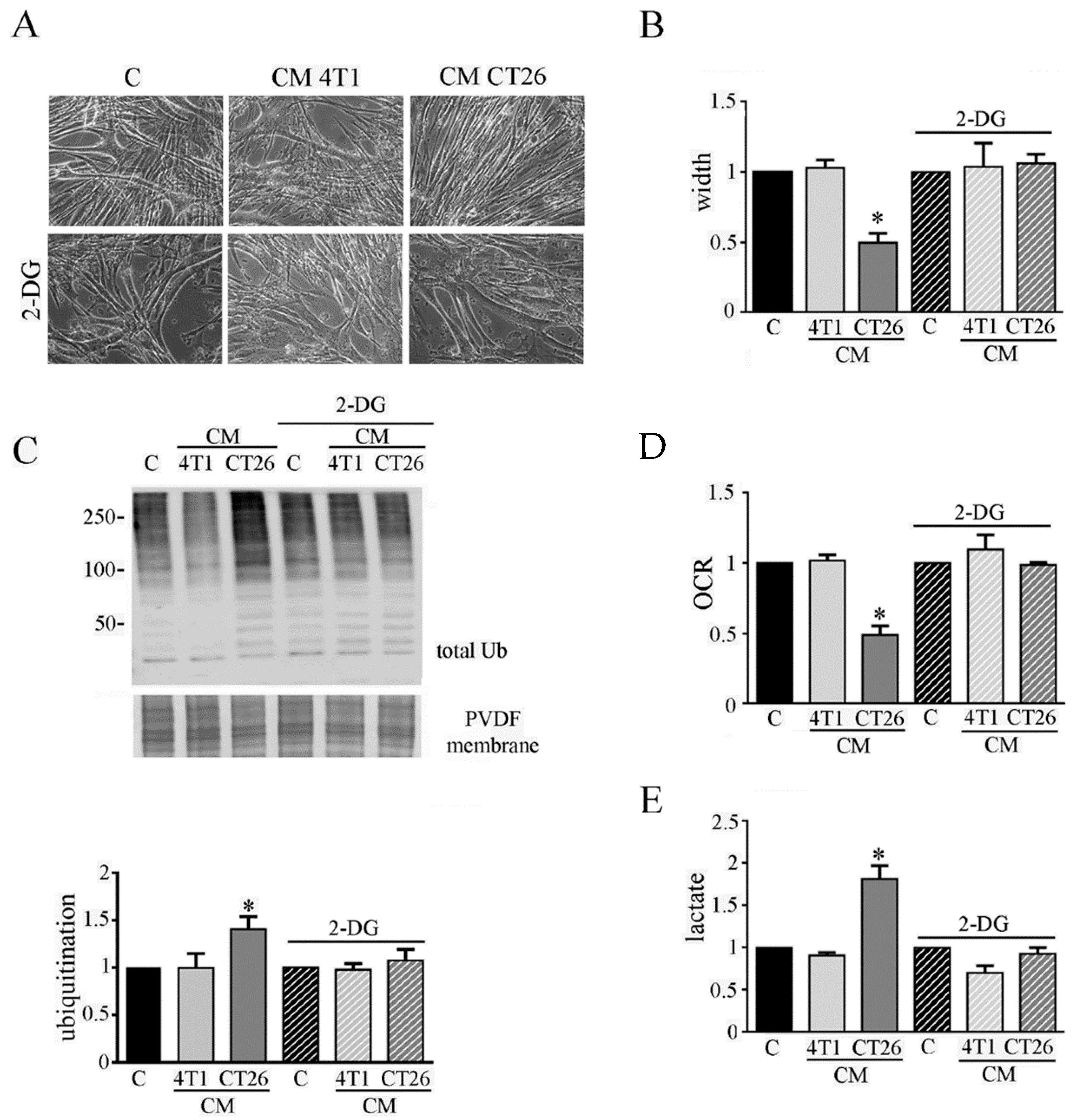
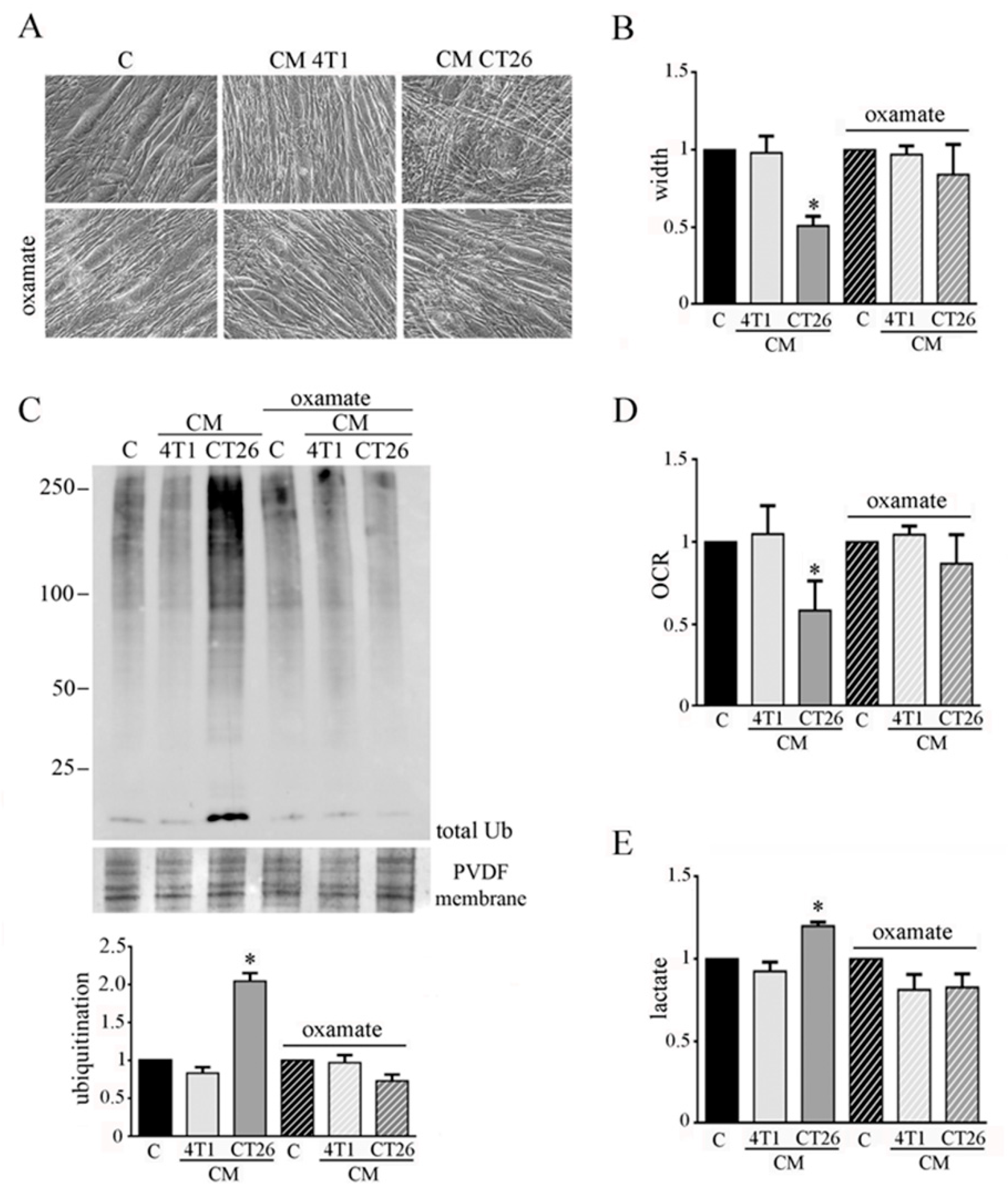
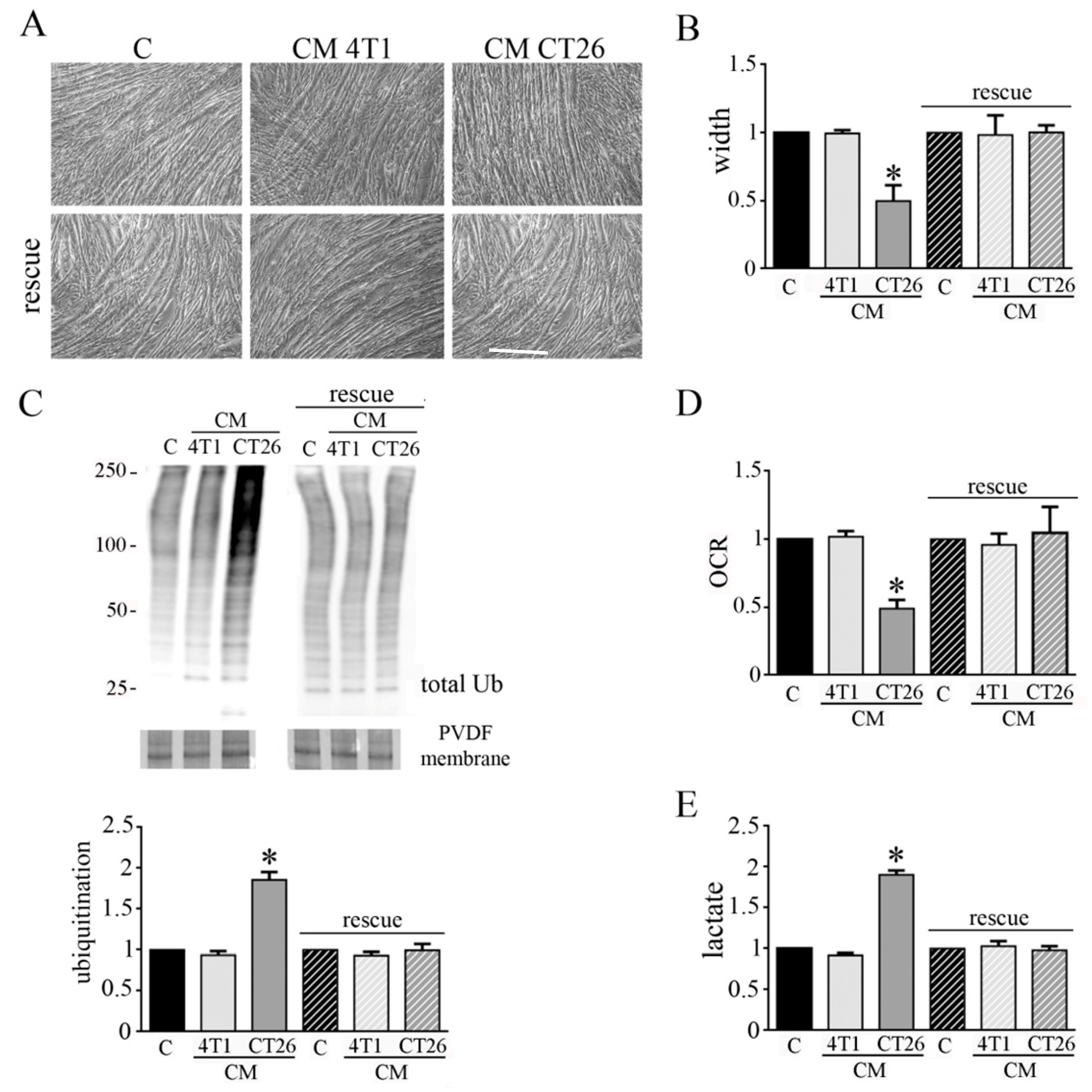
3.2. Inhibition of Glycolysis or Lactate Production Prevents the CM-CT26-Induced Cachexia in Myotubes
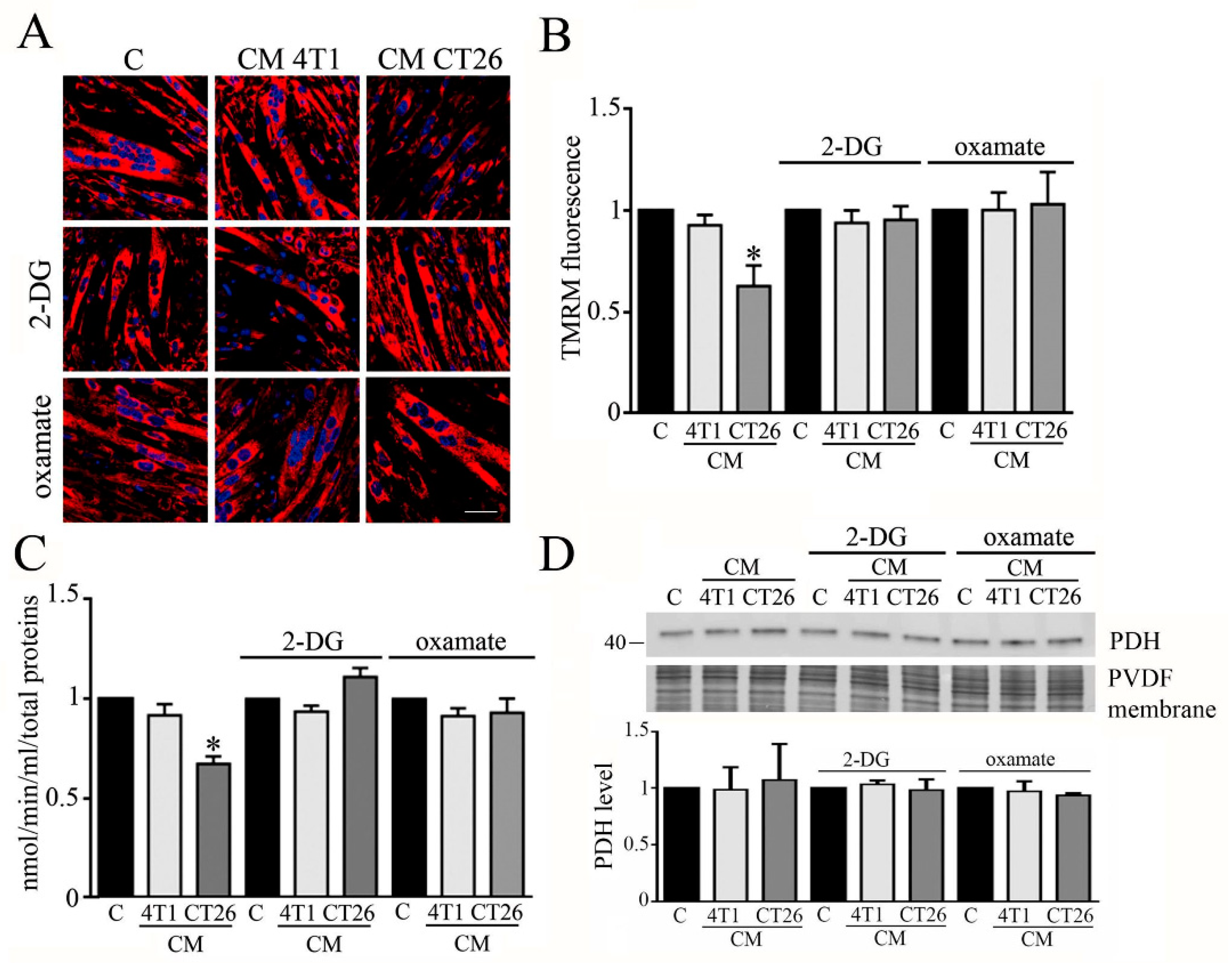
3.3. Inhibition of Glycolysis or Lactate Production Restores PDH Activity and Mitochondrial Membrane Potential in Cachectic Myotubes
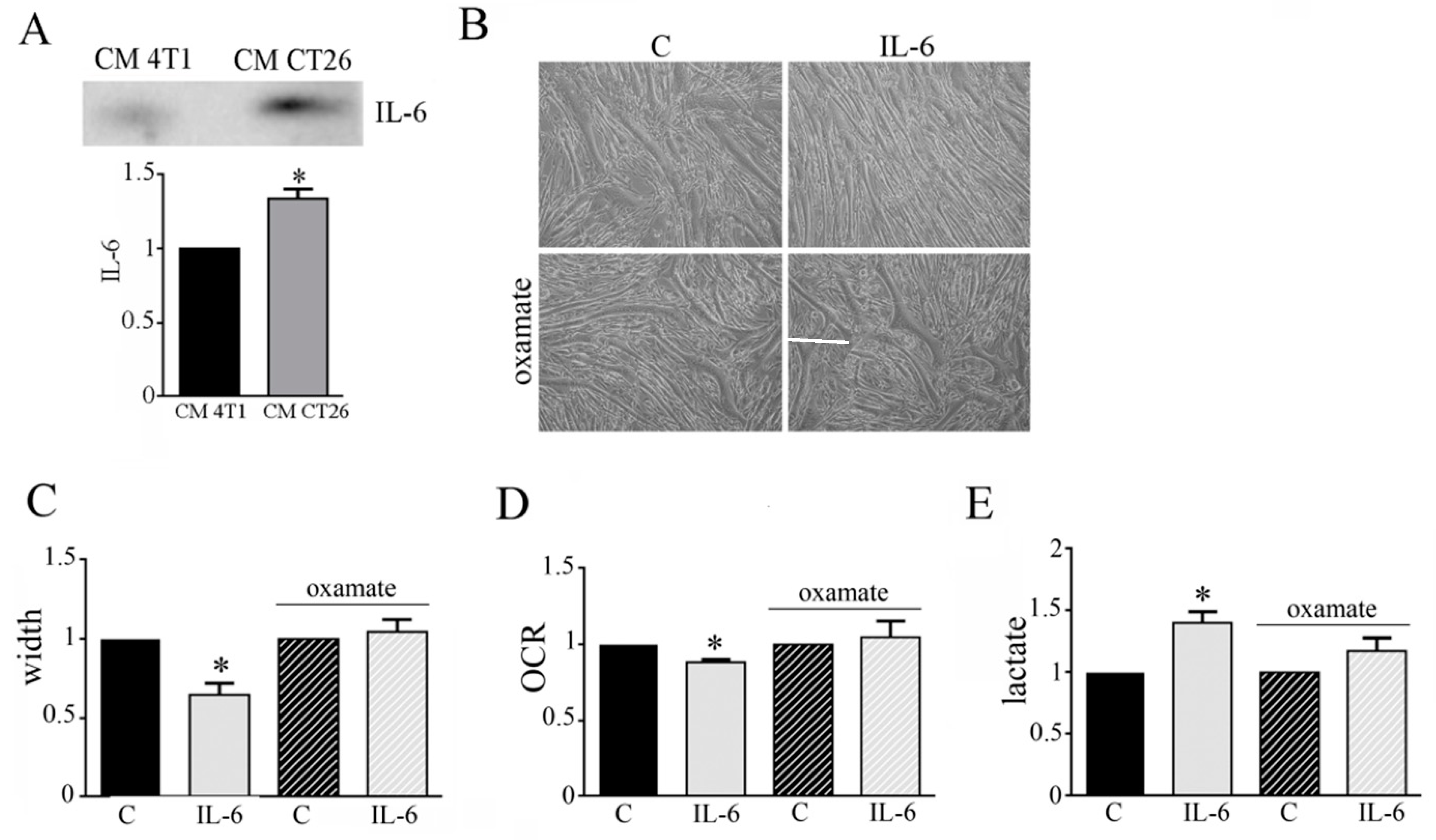
3.4. IL-6 Is Involved in the Metabolic Change Leading to Cachectic Myotubes
3.5. CM CT26 Removal Restores Phenotype and Metabolism of Control Myotubes
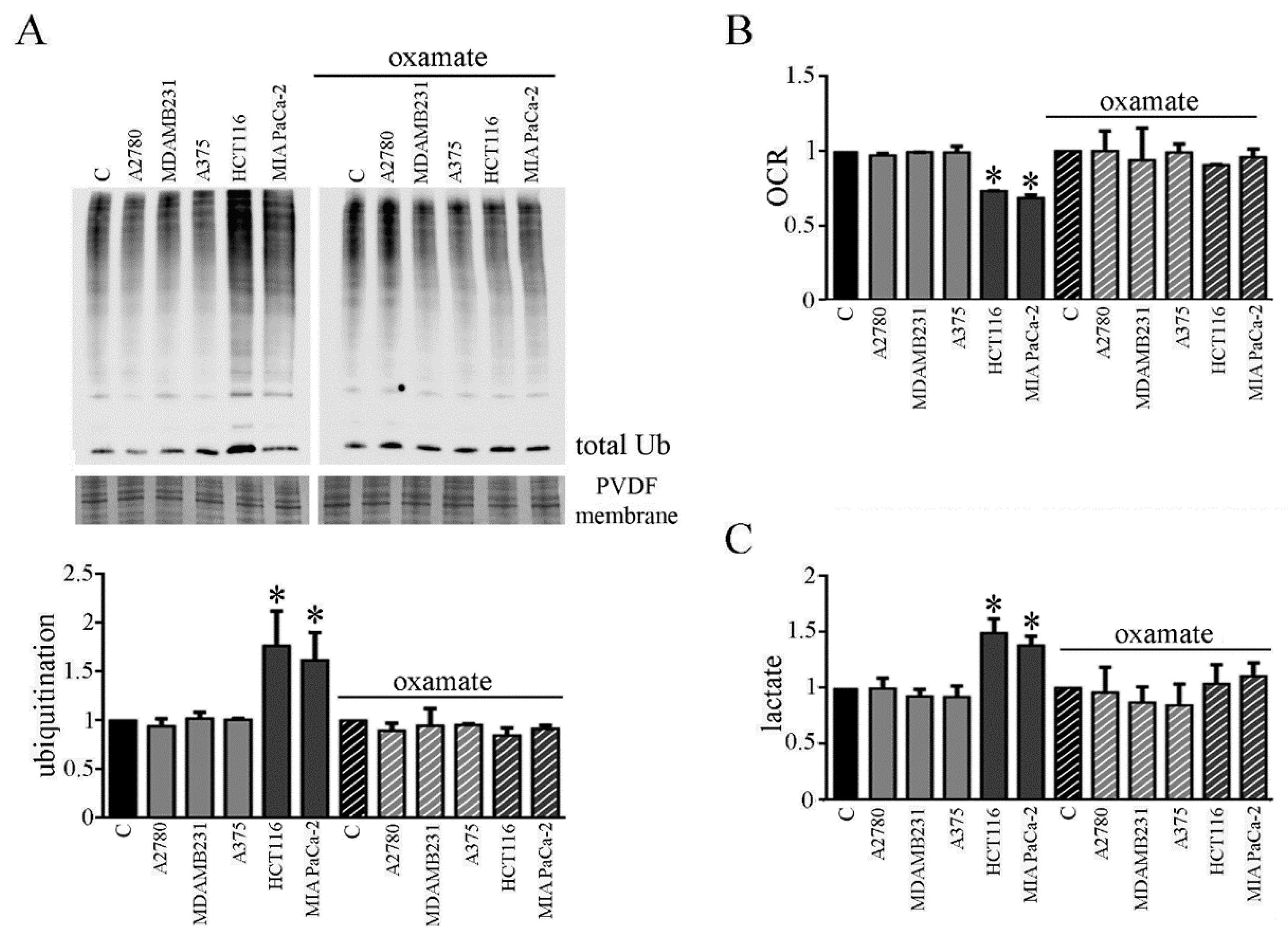
3.6. CMs from Colon and Pancreatic Human Carcinoma Cell Lines Induce Lactate Production by Myotubes Associated to Cachectic Phenotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Schmidt, S.F.; Rohm, M.; Herzig, S.; Diaz, M.B. Cancer Cachexia: More Than Skeletal Muscle Wasting. Trends Cancer 2018, 4, 849–860. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Pin, F.; Couch, M.E.; Bonetto, A. Preservation of muscle mass as a strategy to reduce the toxic effects of cancer chemotherapy on body composition. Curr. Opin. Support. Palliat. Care 2018, 12, 420–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, K.; Arends, J.; Baracos, V. Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 2013, 10, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Hanna, D.L.; Zhang, W.; Baba, H.; Lenz, H. Molecular Pathways: Cachexia Signaling—A Targeted Approach to Cancer Treatment. Clin. Cancer Res. 2016, 22, 3999–4004. [Google Scholar] [CrossRef] [Green Version]
- Argilés, J.M.; López-Soriano, F.J. The ubiquitin-dependent proteolytic pathway in skeletal muscle: Its role in pathological states. Trends Pharm. Sci. 1996, 17, 223–226. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.P.; Schwartz, R.J.; Waddell, I.D.; Holloway, B.R.; Reid, M.B. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. Faseb J. 1998, 12, 871–880. [Google Scholar]
- Fong, Y.; Moldawer, L.L.; Marano, M.; Wei, H.; Barber, A.; Manogue, K.; Tracey, K.J.; Kuo, G.; Fischman, D.A.; Cerami, A.; et al. Cachectin/TNF or IL-1 alpha induces cachexia with redistribution of body proteins. Am. J. Physiol. 1989, 256, R659–R665. [Google Scholar] [CrossRef] [PubMed]
- Narsale, A.A.; Carson, J.A. Role of interleukin-6 in cachexia: Therapeutic implications. Curr. Opin. Support. Palliat. Care 2014, 8, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazeille, C.; Jouinot, A.; Durand, J.P.; Neveux, N.; Boudou-Rouquette, P.; Huillard, O.; Alexandre, J.; Cynober, L.; Goldwasser, F. Relation between hypermetabolism, cachexia, and survival in cancer patients: A prospective study in 390 cancer patients before initiation of anticancer therapy. Am. J. Clin. Nutr. 2017, 105, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.A.; Elliott, S.A.; Baracos, V.E.; Chu, Q.S.C.; Prado, C.M. Key determinants of energy expenditure in cancer and implications for clinical practice. Eur. J. Clin. Nutr. 2016, 70, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Tzika, A.A.; Fontes-Oliveira, C.C.; Shestov, A.A.; Constantinou, C.; Psychogios, N.; Righi, V.; Mintzopoulos, D.; Busquets, S.; Lopez-Soriano, F.J.; Milot, S.; et al. Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int. J. Oncol. 2013, 43, 886–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argilés, J.M.; Fontes-Oliveira, C.C.; Toledo, M.; López-Soriano, F.J.; Busquets, S. Cachexia: A problem of energetic inefficiency. J. Cachexia Sarcopenia Muscle 2014, 5, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.B.; Olivari, D.; Re Cecconi, A.D.; Talamini, L.; Ottoboni, L.; Lecker, S.H.; Stretch, C.; Baracos, V.E.; Bathe, O.F.; Resovi, A.; et al. Activation of the SDF1/CXCR4 pathway retards muscle atrophy during cancer cachexia. Oncogene 2016, 35, 6212–6222. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Sgrignani, J.; Chen, J.; Alimonti, A.; Cavalli, A. How phosphorylation influences E1 subunit pyruvate dehydrogenase: A computational study. Sci. Rep. 2018, 8, 14683. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Novinger, L.J.; Huot, J.R.; Harris, R.A.; Couch, M.E.; O’Connell, T.M.; Bonetto, A. PDK4 drives metabolic alterations and muscle atrophy in cancer cachexia. Faseb J. 2019, 33, 7778–7790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Ladner, K.J.; Nelsen, L.L.; Damrauer, J.; Reiser, P.J.; Swoap, S.; Guttridge, D.C. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J. Clin. Investig. 2004, 114, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, A.; Blenis, J.; Gomes, A.P. Beyond the Warburg Effect: How Do Cancer Cells Regulate One-Carbon Metabolism? Front. Cell Dev. Biol. 2018, 6, 90. [Google Scholar] [CrossRef]
- Der-Torossian, H.; Wysong, A.; Shadfar, S.; Willis, M.S.; McDunn, J.; Couch, M.E. Metabolic derangements in the gastrocnemius and the effect of Compound A therapy in a murine model of cancer cachexia. J. Cachexia Sarcopenia Muscle 2013, 4, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Oliveira, C.C.; Busquets, S.; Toledo, M.; Penna, F.; Paz Aylwin, M.; Sirisi, S.; Silva, A.P.; Orpí, M.; García, A.; Sette, A.; et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: Altered energetic efficiency? Biochim. Biophys. Acta 2013, 1830, 2770–2778. [Google Scholar] [CrossRef] [Green Version]
- Shum, A.M.; Mahendradatta, T.; Taylor, R.J.; Painter, A.B.; Moore, M.M.; Tsoli, M.; Tan, T.C.; Clarke, S.J.; Robertson, G.R.; Polly, P. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging (Albany NY) 2012, 4, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, F.; Ballarò, R.; Martinez-Cristobal, P.; Sala, D.; Sebastian, D.; Busquets, S.; Muscaritoli, M.; Argilés, J.M.; Costelli, P.; Zorzano, A. Autophagy Exacerbates Muscle Wasting in Cancer Cachexia and Impairs Mitochondrial Function. J. Mol. Biol. 2019, 431, 2674–2686. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.; Padrão, A.I.; Maciel, E.; Santinha, D.; Oliveira, P.; Vitorino, R.; Moreira-Gonçalves, D.; Colaço, B.; Pires, M.J.; Nunes, C.; et al. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim. Biophys. Acta 2014, 1841, 896–905. [Google Scholar] [CrossRef]
- Fermoselle, C.; García-Arumí, E.; Puig-Vilanova, E.; Andreu, A.L.; Urtreger, A.J.; Bal de Kier Joffé, E.D.; Tejedor, A.; Puente-Maestu, L.; Barreiro, E. Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp. Physiol. 2013, 98, 1349–1365. [Google Scholar] [CrossRef] [Green Version]
- Padrão, A.I.; Oliveira, P.; Vitorino, R.; Colaço, B.; Pires, M.J.; Márquez, M.; Castellanos, E.; Neuparth, M.J.; Teixeira, C.; Costa, C.; et al. Bladder cancer-induced skeletal muscle wasting: Disclosing the role of mitochondria plasticity. Int. J. Biochem. Cell Biol. 2013, 45, 1399–1409. [Google Scholar] [CrossRef]
- Rohm, M.; Zeigerer, A.; Machado, J.; Herzig, S. Energy metabolism in cachexia. EMBO Rep. 2019, 20, e47258. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Barreto, R.; Couch, M.E.; Bonetto, A.; O’Connell, T.M. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J. Cachexia Sarcopenia Muscle 2019, 10, 140–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukawa, T.; Yan-Jiang, B.C.; Min-Wen, J.C.; Jun-Hao, E.T.; Huang, D.; Qian, C.N.; Ong, P.; Li, Z.; Chen, S.; Mak, S.Y.; et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 2016, 22, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.F.; Sanchez, B.J.; Hall, D.T.; Tremblay, A.M.K.; Di Marco, S.; Gallouzi, I.E. STAT3 promotes IFNγ/TNFα-induced muscle wasting in an NF-κB-dependent and IL-6-independent manner. EMBO Mol. Med. 2017, 9, 622–637. [Google Scholar] [CrossRef] [PubMed]
- VanderVeen, B.N.; Fix, D.K.; Carson, J.A. Disrupted Skeletal Muscle Mitochondrial Dynamics, Mitophagy, and Biogenesis during Cancer Cachexia: A Role for Inflammation. Oxid Med. Cell Longev. 2017, 2017, 3292087. [Google Scholar] [CrossRef]
- Carey, A.L.; Steinberg, G.R.; Macaulay, S.L.; Thomas, W.G.; Holmes, A.G.; Ramm, G.; Prelovsek, O.; Hohnen-Behrens, C.; Watt, M.J.; James, D.J.; et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 2006, 55, 2688–2697. [Google Scholar] [CrossRef] [Green Version]
- Biensø, R.S.; Knudsen, J.G.; Brandt, N.; Pedersen, P.A.; Pilegaard, H. Effects of IL-6 on pyruvate dehydrogenase regulation in mouse skeletal muscle. Pflug. Arch. 2014, 466, 1647–1657. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.A.; Bowker-Kinley, M.M.; Huang, B.; Wu, P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv. Enzym. Regul. 2002, 42, 249–259. [Google Scholar] [CrossRef]
- Ralsera, M.; Wamelinkb, M.M.; Struys, E.A.; Joppich, C.; Krobitsch, S.; Jakobs, C.; Lehrach, H. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc. Natl. Acad. Sci. USA 2008, 46, 17807–17811. [Google Scholar] [CrossRef] [Green Version]
- Argilés, J.M.; López-Soriano, F.J.; Stemmler, B.; Busquets, S. Novel targeted therapies for cancer cachexia. Biochem. J. 2017, 474, 2663–2678. [Google Scholar] [CrossRef]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 2015, 7, 17–29. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Lopez, K.J.; Castro-Munoz, L.J.; Reyes-Hernandes, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and terapeuthic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mannelli, M.; Gamberi, T.; Magherini, F.; Fiaschi, T. A Metabolic Change towards Fermentation Drives Cancer Cachexia in Myotubes. Biomedicines 2021, 9, 698. https://doi.org/10.3390/biomedicines9060698
Mannelli M, Gamberi T, Magherini F, Fiaschi T. A Metabolic Change towards Fermentation Drives Cancer Cachexia in Myotubes. Biomedicines. 2021; 9(6):698. https://doi.org/10.3390/biomedicines9060698
Chicago/Turabian StyleMannelli, Michele, Tania Gamberi, Francesca Magherini, and Tania Fiaschi. 2021. "A Metabolic Change towards Fermentation Drives Cancer Cachexia in Myotubes" Biomedicines 9, no. 6: 698. https://doi.org/10.3390/biomedicines9060698
APA StyleMannelli, M., Gamberi, T., Magherini, F., & Fiaschi, T. (2021). A Metabolic Change towards Fermentation Drives Cancer Cachexia in Myotubes. Biomedicines, 9(6), 698. https://doi.org/10.3390/biomedicines9060698