
Endothelial Dysfunction and Cardiovascular Disease: History and Analysis of the Clinical Utility of the Relationship




{kind=link}
Abstract
:1. Introduction
2. History of Endothelial Dysfunction
3. Endothelial Dysfunction and Cardiovascular Disease
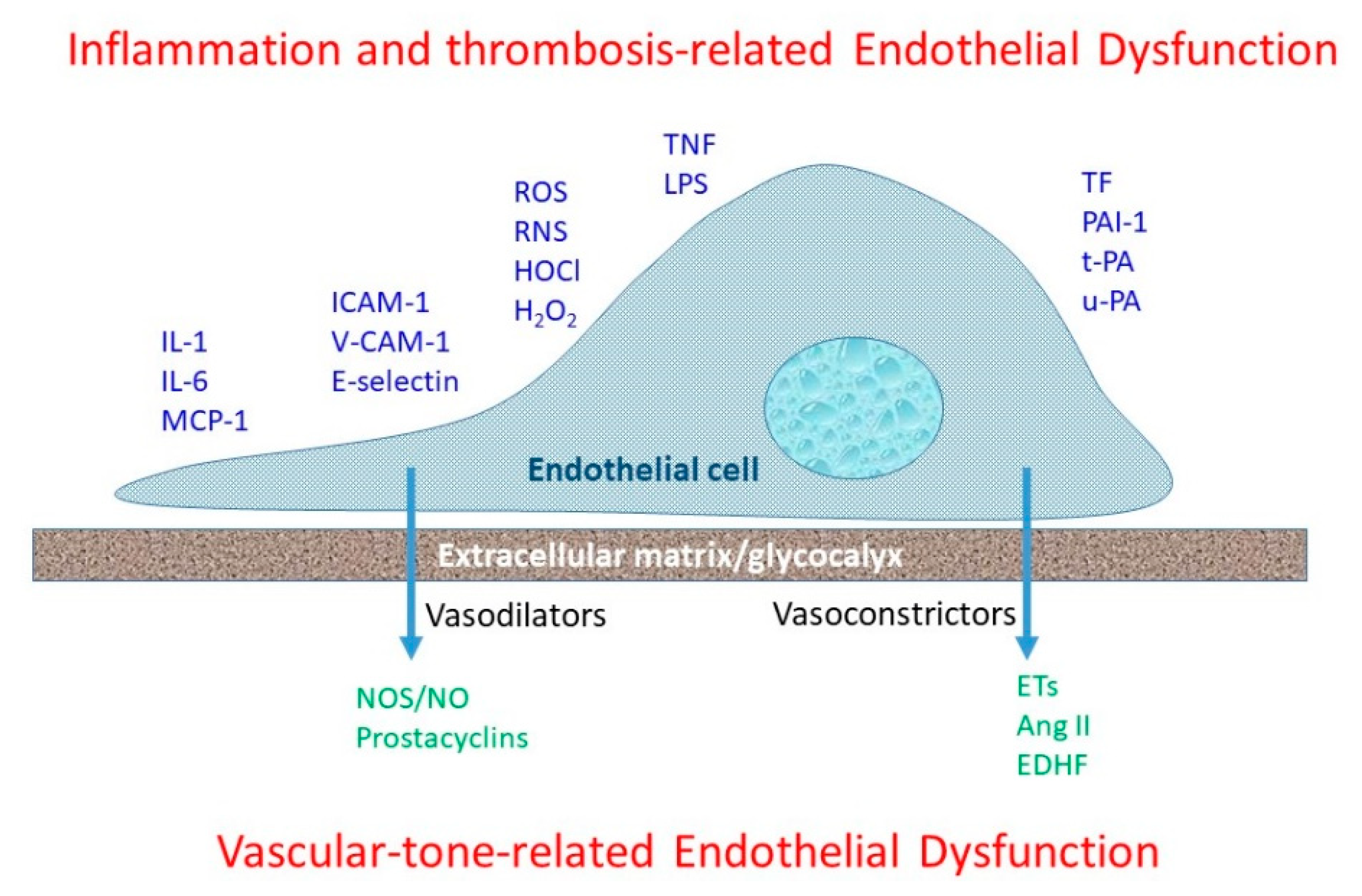
4. Endothelial Dysfunction and Mechanisms of Atherosclerosis
5. Endothelial Dysfunction—Clinical Aspects
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gimbrone, M.A. Endothelial Dysfunction and the Pathogenesis of Atherosclerosis. In Atherosclerosis, V; Springer: New York, NY, USA, 1980. [Google Scholar]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sade, R.M.; Folkman, J. En face stripping of vascular endothelium. Microvasc. Res. 1972, 4, 77–80. [Google Scholar] [CrossRef]
- Weibel, E.R.; Palade, G.E. New Cytoplasmic Components in Arterial Endothelia. J. Cell Biol. 1964, 23, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Topper, J.N.; Gimbrone, M.A., Jr. Blood flow and vascular gene expression: Fluid shear stress as a modulator of endothelial phenotype. Mol. Med. Today 1999, 5, 40–46. [Google Scholar] [CrossRef]
- Furchgott, R.F. The Discovery of Endothelium-Derived Relaxing Factor and Its Importance in the Identification of Nitric Oxide. JAMA 1996, 276, 1186–1188. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. The pathogenesis of atherosclerosis. N. Engl. J. Med. 1986, 314, 488–500. [Google Scholar] [CrossRef]
- Libby, P.; Aikawa, M.; Jain, M. Vascular endothelium and atherosclerosis. In The Vascular Endothelium II; Springer: Berlin/Heidelberg, Germany, 2006; pp. 285–306. [Google Scholar]
- Williams, S.L.; Milne, I.R.; Bagley, C.J.; Gamble, J.R.; Vadas, M.A.; Pitson, S.M.; Khew-Goodall, Y. A proinflammatory role for proteolytically cleaved annexin A1 in neutrophil transendothelial migration. J. Immunol. 2010, 185, 3057–3063. [Google Scholar] [CrossRef] [Green Version]
- Vadas, M.A.; Gamble, J.R. Regulation of the adhesion of neutrophils to endothelium. Biochem. Pharmacol. 1990, 40, 1683–1687. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Browder, T.; Butterfield, C.E.; Kraling, B.M.; Shi, B.; Marshall, B.; O’Reilly, M.S.; Folkman, J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000, 60, 1878–1886. [Google Scholar]
- Browder, T.; Folkman, J.; Pirie-Shepherd, S. The hemostatic system as a regulator of angiogenesis. J. Biol. Chem. 2000, 275, 1521–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, T.J.; Gerhard, M.D.; Meredith, I.T.; Charbonneau, F.; Delagrange, D.; Creager, M.A.; Selwyn, A.P.; Ganz, P. Systemic nature of endothelial dysfunction in atherosclerosis. Am. J. Cardiol. 1995, 75, 71b–74b. [Google Scholar] [CrossRef]
- Laurent, S.; Vanhoutte, P.; Cavero, I.; Chabrier, P.E.; Dupuis, B.; Elghozi, J.L.; Hamon, G.; Janiak, P.; Juillet, Y.; Kher, A.; et al. The arterial wall: A new pharmacological and therapeutic target. Fundam. Clin. Pharmacol. 1996, 10, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Clapp, B.R.; Hirschfield, G.M.; Storry, C.; Gallimore, J.R.; Stidwill, R.P.; Singer, M.; Deanfield, J.E.; MacAllister, R.J.; Pepys, M.B.; Vallance, P.; et al. Inflammation and endothelial function: Direct vascular effects of human C-reactive protein on nitric oxide bioavailability. Circulation 2005, 111, 1530–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furchgott, R.; Zawadzki, J. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ferrige, A.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, R.M.; Murad, F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ. Res. 1983, 52, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Little, P.J.; Chait, A.; Bobik, A. Cellular and cytokine-based inflammatory processes as novel therapeutic targets for the prevention and treatment of atherosclerosis. Pharmacol. Ther. 2011, 131, 255–268. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Z.; Ilyas, I.; Little, P.J.; Kamato, D.; Sahebka, A.; Chen, Z.; Luo, S.; Zheng, X.; Weng, J.; et al. GLP-1 receptor agonists (GLP-1RAs): Cardiovascular actions and therapeutic potential. Int. J. Biol. Sci. 2021, 17, 2050–2068. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, X.; Ilyas, I.; Zheng, X.; Luo, S.; Little, P.J.; Kamato, D.; Sahebkar, A.; Wu, W.; Weng, J.; et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: From pharmacology to pre-clinical and clinical therapeutics. Theranostics 2021, 11, 4502–4515. [Google Scholar] [CrossRef] [PubMed]
- Bocchio, M.; Desideri, G.; Scarpelli, P.; Necozione, S.; Properzi, G.; Spartera, C.; Francavilla, F.; Ferri, C.; Francavilla, S. Endothelial cell activation in men with erectile dysfunction without cardiovascular risk factors and overt vascular damage. J. Urol. 2004, 171, 1601–1604. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Briganti, A.; Jackson, G.; Kloner, R.A.; Montorsi, F.; Montorsi, P.; Vlachopoulos, C. A systematic review of the association between erectile dysfunction and cardiovascular disease. Eur. Urol. 2014, 65, 968–978. [Google Scholar] [CrossRef]
- Cartledge, J.; Minhas, S.; Eardley, I. The role of nitric oxide in penile erection. Expert Opin. Pharmacother. 2001, 2, 95–107. [Google Scholar] [CrossRef]
- Weng, W.; Kong, S.X.; Ganguly, R.; Hersloev, M.; Brett, J.; Hobbs, T.; Baeres, F.M.M. The prevalence of cardiovascular disease by vascular bed and impact on healthcare costs in a large, real-world population with type 2 diabetes. Endocrinol. Diabetes Metab. 2020, 3, e00106. [Google Scholar] [CrossRef] [Green Version]
- Ibanez, B.; Fuster, V. CANTOS: A Gigantic Proof-of-Concept Trial. Circ. Res. 2017, 121, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. Mortality Differences Associated with Treatment Responses in CANTOS and FOURIER: Insights and Implications. Circulation 2017, 137, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Lee, S. Update on SGLT2 Inhibitors-New Data Released at the American Diabetes Association. Crit. Pathw. Cardiol. 2017, 16, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Effect of Empagliflozin on Endothelial Function in Patients with Type 2 Diabetes and Cardiovascular Disease: Results from the Multicenter, Randomized, Placebo-Controlled, Double-Blind EMBLEM Trial. Diabetes Care 2019, 42, e159–e161. [Google Scholar] [CrossRef] [Green Version]
- Falk, E. Morphologic features of unstable atherothrombotic plaques underlying acute coronary syndromes. Am. J. Cardiol. 1989, 63, 114E–120E. [Google Scholar] [CrossRef]
- Falk, E.; Fernandez-Ortiz, A. Role of thrombosis in atherosclerosis and its complications. Am. J. Cardiol. 1995, 75, 3B–11B. [Google Scholar] [CrossRef]
- Nigro, J.; Osman, N.; Dart, A.M.; Little, P.J. Insulin Resistance and Atherosclerosis. Endocr. Rev. 2006, 27, 242–259. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.A. The pathogenesis of atherosclerosis (second of two parts). N. Engl. J. Med. 1976, 295, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Glomset, J.A. The pathogenesis of atherosclerosis (first of two parts). N. Engl. J. Med. 1976, 295, 369–377. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of atherogenesis reinforced. Curr. Opin. Lipidol. 1998, 9, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Little, P.J.; Osman, N.; O’Brien, K.D. Hyperelongated biglycan: The surreptitious initiator of atherosclerosis. Curr. Opin. Lipidol. 2008, 19, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Fujii, H.; Sumiyoshi, S.; Wight, T.N.; Sueishi, K. Early human atherosclerosis: Accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1159–1165. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, M.L.; Osman, N.; Hashimura, K.; de Hann, J.; Jandeleit-Dahm, K.; Allen, T.J.; Tannock, L.R.; Rutledge, J.C.; Little, P.J. Imatinib inhibits vascular smooth muscle proteoglycan synthesis and reduces LDL binding in vitro and aortic lipid deposition in vivo. J. Cell Mol. Med. 2010, 14, 1408–1418. [Google Scholar] [CrossRef]
- Little, P.J.; Tannock, L.; Olin, K.L.; Chait, A.; Wight, T.N. Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to LDLs. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalen, K.; Gustafsson, M.; Rydberg, E.K.; Hulten, L.M.; Wiklund, O.; Innerarity, T.L.; Boren, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Little, P.J.; Ballinger, M.L.; Burch, M.L.; Osman, N. Biosynthesis of natural and hyperelongated chondroitin sulfate glycosaminoglycans: New insights into an elusive process. Open Biochem. J. 2008, 2, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Afroz, R.; Cao, Y.; Rostam, M.A.; Ta, H.; Xu, S.; Zheng, W.; Osman, N.; Kamato, D.; Little, P.J. Signalling pathways regulating galactosaminoglycan synthesis and structure in vascular smooth muscle: Implications for lipoprotein binding and atherosclerosis. Pharmacol. Ther. 2018, 187, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Little, P.J.; Burch, M.L.; Getachew, R.; Al-aryahi, S.; Osman, N. Endothelin-1 stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by endothelin receptor transactivation of the transforming growth factor-[beta] type I receptor. J. Cardiovasc. Pharmacol. 2010, 56, 360–368. [Google Scholar] [CrossRef]
- Afroz, R.; Zhou, Y.; Little, P.J.; Xu, S.; Mohamed, R.; Stow, J.; Kamato, D. Toll-like Receptor 4 Stimulates Gene Expression via Smad2 Linker Region Phosphorylation in Vascular Smooth Muscle Cells. ACS Pharmacol. Transl. Sci. 2020, 3, 524–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostam, M.A.; Shajimoon, A.; Kamato, D.; Mitra, P.; Piva, T.; Getachew, R.; Cao, Y.; Zheng, W.; Osman, N.; Little, P.J. Flavopiridol inhibits TGF-beta-stimulated biglycan synthesis by blocking linker region phosphorylation and nuclear translocation of Smad2. J. Pharmacol. Exp. Ther. 2018, 365, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, R.; Dayati, P.; Mehr, R.N.; Kamato, D.; Seif, F.; Babaahmadi-Rezaei, H.; Little, P.J. Transforming growth factor-beta1 mediated CHST11 and CHSY1 mRNA expression is ROS dependent in vascular smooth muscle cells. J. Cell Commun. Signal. 2019, 13, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Do, B.H.; Osman, N.; Ross, B.P.; Mohamed, R.; Xu, S.; Little, P.J. Smad linker region phosphorylation is a signalling pathway in its own right and not only a modulator of canonical TGF-beta signalling. Cell Mol. Life Sci. 2020, 77, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Ta, H.; Afroz, R.; Xu, S.; Osman, N.; Little, P.J. Mechanisms of PAR-1 mediated kinase receptor transactivation: Smad linker region phosphorylation. J. Cell Commun. Signal. 2019, 13, 539–548. [Google Scholar] [CrossRef]
- Kamato, D.; Burch, M.; Zhou, Y.; Mohamed, R.; Stow, J.L.; Osman, N.; Zheng, W.; Little, P.J. Individual Smad2 linker region phosphorylation sites determine the expression of proteoglycan and glycosaminoglycan synthesizing genes. Cell Signal. 2019, 53, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostam, M.A.; Kamato, D.; Piva, T.J.; Zheng, W.; Little, P.J.; Osman, N. The role of specific Smad linker region phosphorylation in TGF-beta mediated expression of glycosaminoglycan synthesizing enzymes in vascular smooth muscle. Cell Signal. 2016, 28, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Getachew, R.; Ballinger, M.L.; Burch, M.L.; Reid, J.J.; Khachigian, L.M.; Wight, T.N.; Little, P.J.; Osman, N. PDGF beta-receptor kinase activity and ERK1/2 mediate glycosaminoglycan elongation on biglycan and increases binding to LDL. Endocrinology 2010, 151, 4356–4367. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Tak, B.T.; Balci, K.G.; Erken, H.; Gerede, D.M.; Tak, S.; Goksuluk, H.; Turhan, S.; Erol, C. Evaluation of endothelial dysfunction with flow-mediated dilatation after transradial coronary angiography. Acta Cardiol. 2017, 72, 305–310. [Google Scholar] [CrossRef]
- Mortensen, S.P.; Nyberg, M.; Thaning, P.; Saltin, B.; Hellsten, Y. Adenosine contributes to blood flow regulation in the exercising human leg by increasing prostaglandin and nitric oxide formation. Hypertension 2009, 53, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Anderson, T.J.; Uehata, A.; Gerhard, M.D.; Meredith, I.T.; Knab, S.; Delagrange, D.; Lieberman, E.H.; Ganz, P.; Creager, M.A.; Yeung, A.C.; et al. Close relation of endothelial function in the human coronary and peripheral circulations. J. Am. Coll. Cardiol. 1995, 26, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Celermajer, D.S.; Sorensen, K.E.; Gooch, V.M.; Spiegelhalter, D.J.; Miller, O.I.; Sullivan, I.D.; Lloyd, J.K.; Deanfield, J.E. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 1992, 340, 1111–1115. [Google Scholar] [CrossRef]
- Joannides, R.; Haefeli, W.E.; Linder, L.; Richard, V.; Bakkali, E.H.; Thuillez, C.; Luscher, T.F. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries In Vivo. Circulation 1995, 91, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, E.H.; Gerhard, M.D.; Uehata, A.; Selwyn, A.P.; Ganz, P.; Yeung, A.C.; Creager, M.A. Flow-induced vasodilation of the human brachial artery is impaired in patients <40 years of age with coronary artery disease. Am. J. Cardiol. 1996, 78, 1210–1214. [Google Scholar]
- Green, D. Point: Flow-mediated dilation does reflect nitric oxide-mediated endothelial function. J. Appl. Physiol. 2005, 99, 1233–1234; discussion 1237–1238. [Google Scholar] [CrossRef] [PubMed]
- Charakida, M.; Masi, S.; Luscher, T.F.; Kastelein, J.J.; Deanfield, J.E. Assessment of atherosclerosis: The role of flow-mediated dilatation. Eur. Heart J. 2010, 31, 2854–2861. [Google Scholar] [CrossRef] [PubMed]
- Halcox, J.P.; Donald, A.E.; Ellins, E.; Witte, D.R.; Shipley, M.J.; Brunner, E.J.; Marmot, M.G.; Deanfield, J.E. Endothelial function predicts progression of carotid intima-media thickness. Circulation 2009, 119, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.G.; Perissiou, M.; Windsor, M.T.; Schulze, K.; Nam, M.; Magee, R.; Leicht, A.S.; Green, D.J.; Greaves, K.; Golledge, J.; et al. Effects of acute exercise on endothelial function in patients with abdominal aortic aneurysm. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H19–H30. [Google Scholar] [CrossRef] [Green Version]
- Grenon, S.M.; Chong, K.; Alley, H.; Nosova, E.; Gasper, W.; Hiramoto, J.; Boscardin, W.J.; Owens, C.D. Walking disability in patients with peripheral artery disease is associated with arterial endothelial function. J. Vasc. Surg. 2014, 59, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y.; Kwon, T.G.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Prognostic Value of Flow-Mediated Vasodilation in Brachial Artery and Fingertip Artery for Cardiovascular Events: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Green, D.J.; Jones, H.; Thijssen, D.; Cable, N.T.; Atkinson, G. Flow-mediated dilation and cardiovascular event prediction: Does nitric oxide matter? Hypertension 2011, 57, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, S.P.; Askew, C.D.; Walker, M.; Nyberg, M.; Hellsten, Y. The hyperaemic response to passive leg movement is dependent on nitric oxide: A new tool to evaluate endothelial nitric oxide function. J. Physiol. 2012, 590, 4391–4400. [Google Scholar] [CrossRef]
- Trinity, J.D.; Groot, H.J.; Layec, G.; Rossman, M.J.; Ives, S.J.; Runnels, S.; Gmelch, B.; Bledsoe, A.; Richardson, R.S. Nitric oxide and passive limb movement: A new approach to assess vascular function. J. Physiol. 2012, 590, 1413–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, P.O.; Pumper, G.M.; Higano, S.T.; Holmes, D.R.; Kuvin, J.T., Jr.; Lerman, A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J. Am. Coll. Cardiol. 2004, 44, 2137–2141. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, R.B.; Schulz, A.; Wild, P.S.; Sinning, C.R.; Wilde, S.; Eleftheriadis, M.; Herkenhoff, S.; Zeller, T.; Lubos, E.; Lackner, K.J.; et al. Noninvasive vascular function measurement in the community: Cross-sectional relations and comparison of methods. Circ. Cardiovasc. Imaging 2011, 4, 371–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidis, J.P. Prediction of cardiovascular disease outcomes and established cardiovascular risk factors by genome-wide association markers. Circ. Cardiovasc. Genet. 2009, 2, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Holder, S.M.; Bruno, R.M.; Shkredova, D.A.; Dawson, E.A.; Jones, H.; Hopkins, N.D.; Hopman, M.T.E.; Bailey, T.G.; Coombes, J.S.; Askew, C.D.; et al. Reference Intervals for Brachial Artery Flow-Mediated Dilation and the Relation with Cardiovascular Risk Factors. Hypertension 2021, 77, 1469–1480. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Little, P.J.; Askew, C.D.; Xu, S.; Kamato, D. Endothelial Dysfunction and Cardiovascular Disease: History and Analysis of the Clinical Utility of the Relationship. Biomedicines 2021, 9, 699. https://doi.org/10.3390/biomedicines9060699
Little PJ, Askew CD, Xu S, Kamato D. Endothelial Dysfunction and Cardiovascular Disease: History and Analysis of the Clinical Utility of the Relationship. Biomedicines. 2021; 9(6):699. https://doi.org/10.3390/biomedicines9060699
Chicago/Turabian StyleLittle, Peter J., Christopher D. Askew, Suowen Xu, and Danielle Kamato. 2021. "Endothelial Dysfunction and Cardiovascular Disease: History and Analysis of the Clinical Utility of the Relationship" Biomedicines 9, no. 6: 699. https://doi.org/10.3390/biomedicines9060699
APA StyleLittle, P. J., Askew, C. D., Xu, S., & Kamato, D. (2021). Endothelial Dysfunction and Cardiovascular Disease: History and Analysis of the Clinical Utility of the Relationship. Biomedicines, 9(6), 699. https://doi.org/10.3390/biomedicines9060699