
Inflammatory Mechanisms Contributing to Endothelial Dysfunction

 ,
, 
 ,
, 
Abstract
:1. Introduction
2. Physiology of the Vascular Endothelium
2.1. Endothelial Cell Anatomy and Function
2.1.1. Regulation of Vascular Tone
2.1.2. The Role of Nitric Oxide
3. Assessment of Endothelial Function
4. Pathophysiology of Endothelial Dysfunction
4.1. eNOS Uncoupling
4.2. Cardiovascular Risk Factors and Endothelial Dysfunction
4.2.1. Smoking
4.2.2. Diabetes Mellitus
4.2.3. Arterial Hypertension
4.2.4. Hypercholesterolemia
5. The Role of Inflammation in Endothelial Dysfunction
5.1. TLRs and Endothelial Dysfunction
5.2. NLRP3 Inflammasome and Endothelial Dysfunction
5.3. The Role of NF-κB and Adhesion Molecules
5.4. The Pro-Inflammatory Effect of NOX
5.5. Neutrophil Extracellular Traps
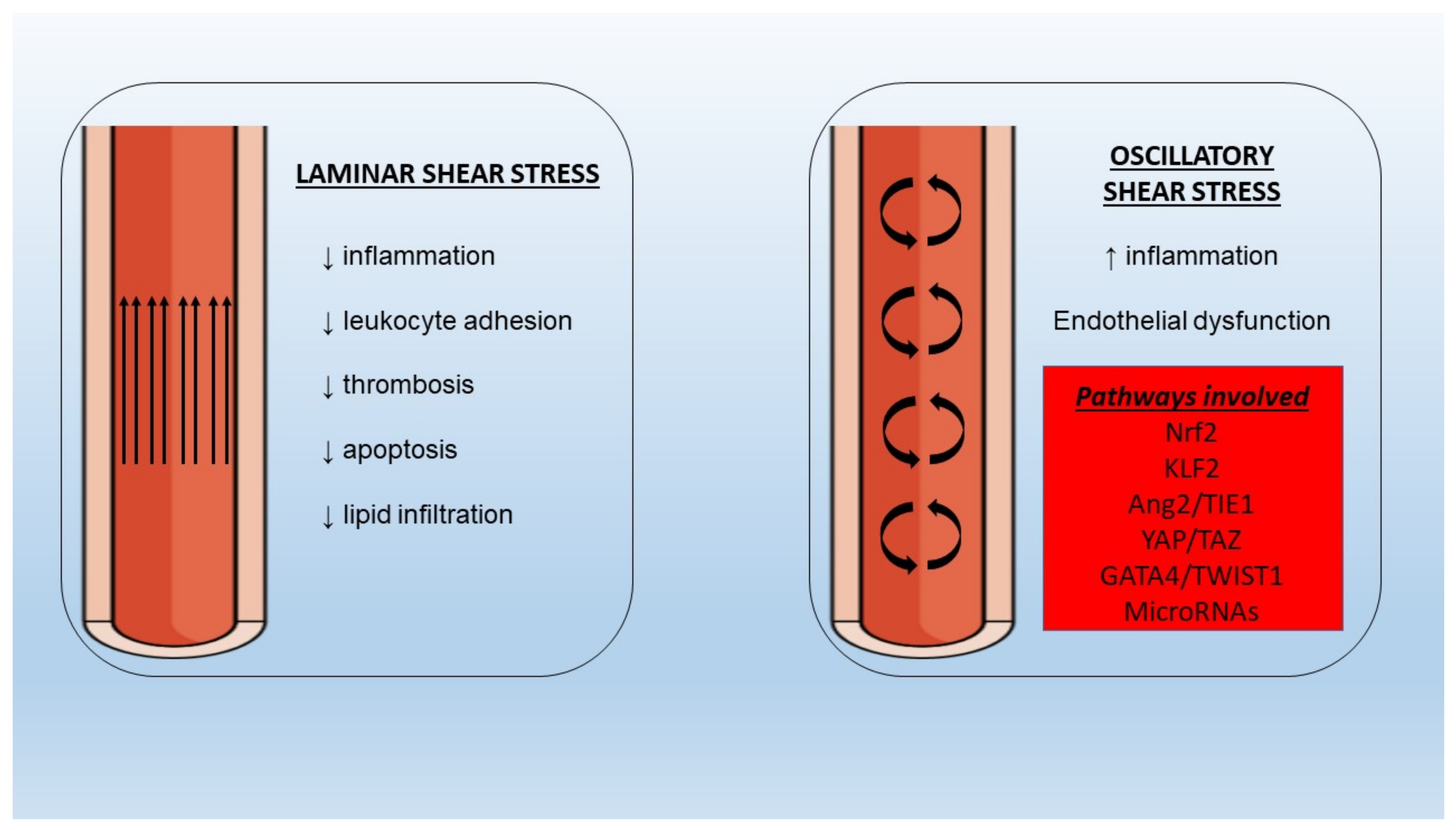
5.6. Shear Stress
5.7. Endothelial Dysfunction in Chronic Inflammatory Diseases
6. A Link between Inflammation and Thrombosis in Endothelial Dysfunction
7. Clinical Implications and Future Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yeboah, J.; Folsom, A.R.; Burke, G.L.; Johnson, C.; Polak, J.F.; Post, W.; Lima, J.A.; Crouse, J.R.; Herrington, D.M. Predictive Value of Brachial Flow-Mediated Dilation for Incident Cardiovascular Events in a Population-Based Study. Circulation 2009, 120, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shechter, M.; Shechter, A.; Koren-Morag, N.; Feinberg, M.S.; Hiersch, L. Usefulness of Brachial Artery Flow-Mediated Dilation to Predict Long-Term Cardiovascular Events in Subjects without Heart Disease. Am. J. Cardiol. 2014, 113, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Rubinshtein, R.; Kuvin, J.T.; Soffler, M.; Lennon, R.J.; Lavi, S.; Nelson, R.E.; Pumper, G.M.; Lerman, L.O.; Lerman, A. Assessment of endothelial function by non-invasive peripheral arterial tonometry predicts late cardiovascular adverse events. Eur. Heart J. 2010, 31, 1142–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeboah, J.; Crouse, J.R.; Hsu, F.-C.; Burke, G.L.; Herrington, D.M. Brachial Flow-Mediated Dilation Predicts Incident Cardiovascular Events in Older Adults. Circulation 2007, 115, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Antoniades, C.; Stefanadis, C. Evaluating endothelial function in humans: A guide to invasive and non-invasive techniques. Heart 2005, 91, 553–558. [Google Scholar] [CrossRef]
- Alexander, Y.; Osto, E.; Schmidt-Trucksäss, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Bäck, M.; Badimon, L.; Cosentino, F.; et al. Endothelial function in cardiovascular medicine: A consensus paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Moncada, S.; Higgs, E.; Vane, J. Human Arterial and Venous Tissues Generate Prostacyclin (Prostaglandin X), a Potent Inhibitor of Platelet Aggregation. Lancet 1977, 309, 18–21. [Google Scholar] [CrossRef]
- Weksler, B.B.; Marcus, A.J.; Jaffe, E.A. Synthesis of prostaglandin I2 (prostacyclin) by cultured human and bovine endothelial cells. Proc. Natl. Acad. Sci. USA 1977, 74, 3922–3926. [Google Scholar] [CrossRef] [Green Version]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.M.; Edwards, D.; Lewis, M.J.; Newby, A.C.; Henderson, A.H. The nature of endothelium-derived vascular relaxant factor. Nature 1984, 308, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Lin, M.I.; Murata, T.; Landskroner-Eiger, S.; Schleicher, M.; Kothiya, M.; Iwakiri, Y.; Yu, J.; Huang, P.; Sessa, W.C. eNOS derived nitric oxide regulates endothelial barrier function via VE cadherin and Rho GTPases. J. Cell Sci. 2013, 126, 5541–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourd’Heuil, D.; Jourd’Heuil, F.L.; Kutchukian, P.S.; Musah, R.A.; Wink, D.A.; Grisham, M.B. Reaction of Superoxide and Nitric Oxide with Peroxynitrite. Implications for peroxynitrite-mediated oxidation reactions in vivo. J. Biol. Chem. 2001, 276, 28799–28805. [Google Scholar] [CrossRef] [Green Version]
- Selemidis, S.; Dusting, G.J.; Peshavariya, H.; Kemp-Harper, B.K.; Drummond, G. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc. Res. 2007, 75, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Flammer, A.; Anderson, T.; Celermajer, D.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.; Lüscher, T.F.; Shechter, M.; Taddei, S.; et al. The Assessment of Endothelial Function. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Flammer, A.J.; Lüscher, T.F. Human endothelial dysfunction: EDRFs. Pflügers Arch. Eur. J. Physiol. 2010, 459, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Petrie, J.R.; Ueda, S.; Morris, A.D.; Murray, L.S.; Elliott, H.L.; Connell, J.M.C. How reproducible is bilateral forearm plethysmography? Br. J. Clin. Pharmacol. 1998, 45, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, D.H.J.; Bruno, R.M.; Van Mil, A.C.C.M.; Holder, S.M.; Faita, F.; Greyling, A.; Zock, P.L.; Taddei, S.; Deanfield, J.; Luscher, T.; et al. Expert consensus and evidence-based recommendations for the assessment of flow-mediated dilation in humans. Eur. Heart J. 2019, 40, 2534–2547. [Google Scholar] [CrossRef]
- Greyling, A.; Van Mil, A.C.; Zock, P.L.; Green, D.J.; Ghiadoni, L.; Thijssen, D.H. Adherence to guidelines strongly improves reproducibility of brachial artery flow-mediated dilation. Atherosclerosis 2016, 248, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Takishima, I.; Nakamura, T.; Hirano, M.; Kitta, Y.; Kobayashi, T.; Fujioka, D.; Saito, Y.; Watanabe, K.; Watanabe, Y.; Mishina, H.; et al. Predictive value of serial assessment of endothelial function in chronic heart failure. Int. J. Cardiol. 2012, 158, 417–422. [Google Scholar] [CrossRef]
- Kitta, Y.; Obata, J.-E.; Nakamura, T.; Hirano, M.; Kodama, Y.; Fujioka, D.; Saito, Y.; Kawabata, K.-I.; Sano, K.; Kobayashi, T.; et al. Persistent Impairment of Endothelial Vasomotor Function Has a Negative Impact on Outcome in Patients with Coronary Artery Disease. J. Am. Coll. Cardiol. 2009, 53, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Dimitropoulos, S.; Mystakidi, V.C.; Oikonomou, E.; Siasos, G.; Tsigkou, V.; Athanasiou, D.; Gouliopoulos, N.; Bletsa, E.; Kalampogias, A.; Charalambous, G.; et al. Association of Soluble Suppression of Tumorigenesis-2 (ST2) with Endothelial Function in Patients with Ischemic Heart Failure. Int. J. Mol. Sci. 2020, 21, 9385. [Google Scholar] [CrossRef]
- Mourouzis, K.; Siasos, G.; Oikonomou, E.; Zaromitidou, M.; Tsigkou, V.; Antonopoulos, A.; Bletsa, E.; Stampouloglou, P.; Vlasis, K.; Vavuranakis, M.; et al. Lipoprotein-associated phospholipase A2 levels, endothelial dysfunction and arterial stiffness in patients with stable coronary artery disease. Lipids Health Dis. 2021, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kuvin, J.T.; Patel, A.; A Sliney, K.; Pandian, N.G.; Sheffy, J.; Schnall, R.P.; Karas, R.H.; E Udelson, J. Assessment of peripheral vascular endothelial function with finger arterial pulse wave amplitude. Am. Heart J. 2003, 146, 168–174. [Google Scholar] [CrossRef]
- Nohria, A.; Gerhard-Herman, M.; Creager, M.A.; Hurley, S.; Mitra, D.; Ganz, P. Role of nitric oxide in the regulation of digital pulse volume amplitude in humans. J. Appl. Physiol. 2006, 101, 545–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, P.O.; Pumper, G.M.; Higano, S.T.; Holmes, D.R.; Kuvin, J.T.; Lerman, A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J. Am. Coll. Cardiol. 2004, 44, 2137–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamburg, N.M.; Palmisano, J.; Larson, M.; Sullivan, L.; Lehman, B.T.; Vasan, R.S.; Levy, D.; Mitchell, G.F.; Vita, J.; Benjamin, E. Relation of Brachial and Digital Measures of Vascular Function in the Community: The Framingham heart study. Hypertension 2011, 57, 390–396. [Google Scholar] [CrossRef]
- Schnabel, R.B.; Schulz, A.; Wild, P.S.; Sinning, C.R.; Wilde, S.; Eleftheriadis, M.; Herkenhoff, S.; Zeller, T.; Lubos, E.; Lackner, K.J.; et al. Noninvasive Vascular Function Measurement in the Community. Circ. Cardiovasc. Imaging 2011, 4, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Sharifizad, M.; Witkowska, K.J.; Aschinger, G.C.; Sapeta, S.; Rauch, A.; Schmidl, D.; Werkmeister, R.M.; Garhöfer, G.; Schmetterer, L. Factors Determining Flicker-Induced Retinal Vasodilation in Healthy Subjects. Investig. Opthalmol. Vis. Sci. 2016, 57, 3306–3312. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Kawasaki, R.; Wang, J.J.; Kreis, A.J.; Shaw, J.; Vilser, W.; Wong, T.Y. Flicker Light-Induced Retinal Vasodilation in Diabetes and Diabetic Retinopathy. Diabetes Care 2009, 32, 2075–2080. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.R.; Bellary, S.; Karimzad, S.; Gherghel, D. Overweight status is associated with extensive signs of microvascular dysfunction and cardiovascular risk. Sci. Rep. 2016, 6, 32282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nägele, M.P.; Barthelmes, J.; Ludovici, V.; Cantatore, S.; Von Eckardstein, A.; Enseleit, F.; Lüscher, T.F.; Ruschitzka, F.; Sudano, I.; Flammer, A.J. Retinal microvascular dysfunction in heart failure. Eur. Heart J. 2018, 39, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anna, M.; Ewa, P.-S.; Katarzyna, B.; Anna, S.; Krzysztof, S.; Piotr, K.M.; Bogusław, M. Correlation between Flicker-Induced Retinal Vessel Vasodilatation and Plasma Biomarkers of Endothelial Dysfunction in Hypertensive Patients. Curr. Eye Res. 2018, 43, 128–134. [Google Scholar] [CrossRef]
- Leite, A.R.; Borges-Canha, M.; Cardoso, R.; Neves, J.S.; Castro-Ferreira, R.; Leite-Moreira, A. Novel Biomarkers for Evaluation of Endothelial Dysfunction. Angiology 2020, 71, 397–410. [Google Scholar] [CrossRef]
- Balta, S.; Mikhailidis, D.P.; Demirkol, S.; Ozturk, C.; Celik, T.; Iyisoy, A. Endocan: A novel inflammatory indicator in cardiovascular disease? Atherosclerosis 2015, 243, 339–343. [Google Scholar] [CrossRef]
- Liaudet, L.; Vassalli, G.; Pacher, P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front. Biosci. Landmark 2009, 14, 4809–4814. [Google Scholar] [CrossRef] [Green Version]
- Diers, A.R.; Broniowska, K.A.; Hogg, N. Nitrosative stress and redox-cycling agents synergize to cause mitochondrial dysfunction and cell death in endothelial cells. Redox Biol. 2013, 1, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alp, N.J.; Channon, K. Regulation of Endothelial Nitric Oxide Synthase by Tetrahydrobiopterin in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Meininger, C.J.; Haynes, T.E.; Hatakeyama, K.; Wu, G. Regulation of Tetrahydrobiopterin Synthesis and Bioavailability in Endothelial Cells. Cell Biochem. Biophys. 2004, 41, 415–434. [Google Scholar] [CrossRef]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef] [Green Version]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Antonopoulos, A.; Warrick, N.; Van-Assche, T.; Cunnington, C.; Tousoulis, D.; Pillai, R.; Ratnatunga, C.; et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: Implications for endothelial function in human atherosclerosis. Eur. Heart J. 2009, 30, 1142–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, M.T.; Clarke, S.G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, S.; Fung, H.-L. Mechanism of Cellular Oxidation Stress Induced by Asymmetric Dimethylarginine. Int. J. Mol. Sci. 2012, 13, 7521–7531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniades, C.; Demosthenous, M.; Tousoulis, D.; Antonopoulos, A.; Vlachopoulos, C.; Toutouza, M.; Marinou, K.; Bakogiannis, C.; Mavragani, K.; Lazaros, G.; et al. Role of Asymmetrical Dimethylarginine in Inflammation-Induced Endothelial Dysfunction in Human Atherosclerosis. Hypertension 2011, 58, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccala, A.; Fiorenza, S.; Rapana, R.; Santoro, A. Hypertension, atherosclerosis and kidney. G. Ital. Nefrol. 2005, 22 (Suppl. 31), S9–S14. [Google Scholar]
- Carnevale, R.; Sciarretta, S.; Violi, F.; Nocella, C.; Loffredo, L.; Perri, L.; Peruzzi, M.; Marullo, A.G.; De Falco, E.; Chimenti, I.; et al. Acute Impact of Tobacco vs Electronic Cigarette Smoking on Oxidative Stress and Vascular Function. Chest 2016, 150, 606–612. [Google Scholar] [CrossRef]
- Sugiura, T.; Dohi, Y.; Takase, H.; Yamashita, S.; Fujii, S.; Ohte, N. Oxidative Stress is Closely Associated with Increased Arterial Stiffness, Especially in Aged Male Smokers without Previous Cardiovascular Events: A Cross-Sectional Study. J. Atheroscler. Thromb. 2017, 24, 1186–1198. [Google Scholar] [CrossRef] [Green Version]
- Vlachopoulos, C.; Aznaouridis, K.; Bratsas, A.; Ioakeimidis, N.; Dima, I.; Xaplanteris, P.; Stefanadis, C.; Tousoulis, D. Arterial stiffening and systemic endothelial activation induced by smoking: The role of COX-1 and COX-2. Int. J. Cardiol. 2015, 189, 293–298. [Google Scholar] [CrossRef]
- Abdelghany, T.M.; Ismail, R.S.; Mansoor, F.A.; Zweier, J.R.; Lowe, F.; Zweier, J.L. Cigarette smoke constituents cause endothelial nitric oxide synthase dysfunction and uncoupling due to depletion of tetrahydrobiopterin with degradation of GTP cyclohydrolase. Nitric Oxide 2018, 76, 113–121. [Google Scholar] [CrossRef]
- Halvorsen, B.; Sagen, L.E.; Ueland, T.; Aukrust, P.; Tonstad, S. Effect of smoking cessation on markers of inflammation and endothelial cell activation among individuals with high risk for cardiovascular disease. Scand. J. Clin. Lab. Investig. 2007, 67, 604–611. [Google Scholar] [CrossRef]
- Tsai, J.-S.; Guo, F.-R.; Chen, S.-C.; Lue, B.-H.; Lee, L.-T.; Huang, K.-C.; Chen, C.-Y.; Hung, S.-H.; Chuang, L.-M.; Chen, C.-Y. Changes of serum adiponectin and soluble intercellular adhesion molecule-1 concentrations after smoking cessation. Clin. Chem. Lab. Med. 2012, 50, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Delgado, G.E.; Krämer, B.K.; Siekmeier, R.; Yazdani, B.; März, W.; Leipe, J.; Kleber, M.E. Influence of smoking and smoking cessation on biomarkers of endothelial function and their association with mortality. Atherosclerosis 2020, 292, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R. Endothelial Dysfunction and Hypertension. Hypertension 2014, 64, 924–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perticone, F.; Ceravolo, R.; Pujia, A.; Ventura, G.; Iacopino, S.; Scozzafava, A.; Ferraro, A.; Chello, M.; Mastroroberto, P.; Verdecchia, P.; et al. Prognostic Significance of Endothelial Dysfunction in Hypertensive Patients. Circulation 2001, 104, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, E.; Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Perticone, F.; Sciacqua, A.; Maio, R.; Perticone, M.; Maas, R.; Boger, R.H.; Tripepi, G.; Sesti, G.; Zoccali, C. Asymmetric Dimethylarginine, L-Arginine, and Endothelial Dysfunction in Essential Hypertension. J. Am. Coll. Cardiol. 2005, 46, 518–523. [Google Scholar] [CrossRef] [Green Version]
- Böhm, F.; Pernow, J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkhorst-Oei, L.T.; Stroes, E.S.G.; Koomans, H.A.; Rabelink, T.J. Acute Simultaneous Stimulation of Nitric Oxide and Oxygen Radicals by Angiotensin II in Humans in Vivo. J. Cardiovasc. Pharmacol. 1999, 33, 420–424. [Google Scholar] [CrossRef]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, A.J.; Irimpen, A.M.; Siebenlist, U.; Chandrasekar, B. OxLDL induces endothelial dysfunction and death via TRAF3IP2: Inhibition by HDL3 and AMPK activators. Free Radic. Biol. Med. 2014, 70, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Dallinga-Thie, G.M.; Kroon, J.; Schnitzler, J.G. The Role of (Modified) Lipoproteins in Vascular Function: A Duet Between Monocytes and the Endothelium. Curr. Med. Chem. 2019, 26, 1594–1609. [Google Scholar] [CrossRef]
- Pober, J.S.; Cotran, R.S. Cytokines and endothelial cell biology. Physiol. Rev. 1990, 70, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2015, 16, 35–50. [Google Scholar] [CrossRef]
- Han, J.; Ulevitch, R.J. Limiting inflammatory responses during activation of innate immunity. Nat. Immunol. 2005, 6, 1198–1205. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Peng, X.; Li, X.; Tu, T.; Yang, H.; Teng, S.; Zhang, W.; Xing, Z.; Tang, J.; Hu, X.; et al. HMGB1 impairs endothelium-dependent relaxation in diabetes through TLR4/eNOS pathway. FASEB J. 2020, 34, 8641–8652. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, H.; Su, S.; Harshfield, G.; Sullivan, J.; Webb, C.; Blumenthal, J.A.; Wang, X.; Huang, Y.; Treiber, F.A.; et al. High-Mobility Group Box-1 Is Associated with Obesity, Inflammation, and Subclinical Cardiovascular Risk among Young Adults: A Longitudinal Cohort Study. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2776–2784. [Google Scholar] [CrossRef]
- Behl, T.; Sharma, E.; Sehgal, A.; Kaur, I.; Kumar, A.; Arora, R.; Pal, G.; Kakkar, M.; Kumar, R.; Bungau, S. Expatiating the molecular approaches of HMGB1 in diabetes mellitus: Highlighting signalling pathways via RAGE and TLRs. Mol. Biol. Rep. 2021, 48, 1869–1881. [Google Scholar] [CrossRef]
- Singh, G.B.; Zhang, Y.; Boini, K.M.; Koka, S. High Mobility Group Box 1 Mediates TMAO-Induced Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, 3570. [Google Scholar] [CrossRef] [Green Version]
- Hernanz, R.; Martínez-Revelles, S.; Palacios, R.; Martín, A.; Cachofeiro, V.; Aguado, A.; García-Redondo, L.; Barrús, M.T.; De Batista, P.R.; Briones, A.M.; et al. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br. J. Pharmacol. 2015, 172, 3159–3176. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef]
- Loukovaara, S.; Piippo, N.; Kinnunen, K.; Hytti, M.; Kaarniranta, K.; Kauppinen, A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. 2017, 95, 803–808. [Google Scholar] [CrossRef] [Green Version]
- De Miguel, C.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. Emerging Role of the Inflammasome and Pyroptosis in Hypertension. Int. J. Mol. Sci. 2021, 22, 1064. [Google Scholar] [CrossRef]
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 2017, 74, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, E.; Leopoulou, M.; Theofilis, P.; Antonopoulos, A.; Siasos, G.; Latsios, G.; Mystakidi, V.C.; Antoniades, C.; Tousoulis, D. A link between inflammation and thrombosis in atherosclerotic cardiovascular diseases: Clinical and therapeutic implications. Atherosclerosis 2020, 309, 16–26. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.; Rapino, F.; Robertson, A.; Cooper, M.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Galea, J.; Armstrong, J.; Gadsdon, P.; Holden, H.; Francis, S.; Holt, C.M. Interleukin-1β in Coronary Arteries of Patients With Ischemic Heart Disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1000–1006. [Google Scholar] [CrossRef]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of Interleukin-18 in Human Atherosclerotic Plaques and Relation to Plaque Instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Fazal, F. Blocking NF- B: An Inflammatory Issue. Proc. Am. Thorac. Soc. 2011, 8, 497–503. [Google Scholar] [CrossRef]
- Rollins, B.J.; Yoshimura, T.; Leonard, E.J.; Pober, J.S. Cytokine-activated human endothelial cells synthesize and secrete a monocyte chemoattractant, MCP-1/JE. Am. J. Pathol. 1990, 136, 1229–1233. [Google Scholar]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Thippegowda, P.B.; Hu, G.; Bachmaier, K.; Christman, J.W.; Malik, A.B.; Tiruppathi, C. NF-κB regulates thrombin-induced ICAM-1 gene expression in cooperation with NFAT by binding to the intronic NF-κB site in the ICAM-1 gene. Physiol. Genomics 2009, 38, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrhof, F.B.; Schmidt-Ullrich, R.; Dietz, R.; Scheidereit, C. Regulation of Vascular Smooth Muscle Cell Proliferation: Role of NF-κB Revisited. Circ. Res. 2005, 96, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Hu, S.-Y.; Wu, S.-H.; Karmenyan, A.; Chiou, A. The Interaction Affinity between Vascular Cell Adhesion Molecule-1 (VCAM-1) and Very Late Antigen-4 (VLA-4) Analyzed by Quantitative FRET. PLoS ONE 2015, 10, e0121399. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Shimaoka, M.; Kurokawa, M. Essential roles of VLA-4 in the hematopoietic system. Int. J. Hematol. 2010, 91, 569–575. [Google Scholar] [CrossRef]
- Frank, P.G.; Lisanti, M.P. ICAM-1: Role in inflammation and in the regulation of vascular permeability. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H926–H927. [Google Scholar] [CrossRef]
- Silva, M.; Videira, P.A.; Sackstein, R. E-Selectin Ligands in the Human Mononuclear Phagocyte System: Implications for Infection, Inflammation, and Immunotherapy. Front. Immunol. 2018, 8, 1878. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.M.J.; Gordon, J.L.; Gearing, A.J.H.; Pigott, R.; Woolf, N.; Katz, D.; Kyriakopoulos, A. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J. Pathol. 1993, 171, 223–229. [Google Scholar] [CrossRef]
- Collins, R.G.; Velji, R.; Guevara, N.V.; Hicks, M.J.; Chan, L.; Beaudet, A.L. P-Selectin or Intercellular Adhesion Molecule (Icam)-1 Deficiency Substantially Protects against Atherosclerosis in Apolipoprotein E–Deficient Mice. J. Exp. Med. 2000, 191, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.M.; Chapman, S.M.; A Brown, A.; Frenette, P.S.; Hynes, R.O.; Wagner, D.D. The combined role of P- and E-selectins in atherosclerosis. J. Clin. Investig. 1998, 102, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Kolattukudy, P.E. Role of MCP-1 in cardiovascular disease: Molecular mechanisms and clinical implications. Clin. Sci. 2009, 117, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of Monocyte Chemoattractant Protein-1 Reduces Atherosclerosis in Low Density Lipoprotein Receptor–Deficient Mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Boring, L.; Gosling, J.; Cleary, M.L.; Charo, I.F. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef]
- Aiello, R.J.; Bourassa, P.-A.K.; Lindsey, S.; Weng, W.; Natoli, E.; Rollins, B.J.; Milos, P.M. Monocyte Chemoattractant Protein-1 Accelerates Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1518–1525. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, X.; Shang, H.; Xu, Y.; Qian, M. Monocyte chemoattractant protein-1 induces endothelial cell apoptosis in vitro through a p53-dependent mitochondrial pathway. Acta Biochim. Biophys. Sin. 2011, 43, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Tousoulis, D.; Antoniades, C.; Bosinakou, E.; Kotsopoulou, M.; Tsoufis, C.; Marinou, K.; Charakida, M.; Stefanadi, E.; Vavuranakis, M.; Latsios, G.; et al. Differences in inflammatory and thrombotic markers between unstable angina and acute myocardial infarction. Int. J. Cardiol. 2007, 115, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Stefanadi, E.; Tousoulis, D.; Androulakis, E.S.; Papageorgiou, N.; Charakida, M.; Siasos, G.; Tsioufis, C.; Stefanadis, C. Inflammatory Markers in Essential Hypertension: Potential Clinical Implications. Curr. Vasc. Pharmacol. 2010, 8, 509–516. [Google Scholar] [CrossRef]
- Tousoulis, D.; Daves, G.J.M.D.; Asimakopoulos, M.D.; Homaei, H.; Zouridakis, E.M.D.; Ahmed, N.; Kaski, J.C. Vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 serum level in patients with chest pain and normal coronary arteries (syndrome X). Clin. Cardiol. 2001, 24, 301–304. [Google Scholar] [CrossRef]
- Tousoulis, D.; Homaei, H.; Ahmed, N.; Asimakopoulos, G.; Zouridakis, E.; Toutouzas, P.; Davies, G.J. Increased plasma adhesion molecule levels in patients with heart failure who have ischemic heart disease and dilated cardiomyopathy. Am. Heart J. 2001, 141, 277–280. [Google Scholar] [CrossRef]
- Tousoulis, D.; Papageorgiou, N.; Androulakis, E.; Siasos, G.; Latsios, G.; Tentolouris, K.; Stefanadis, C. Diabetes Mellitus-Associated Vascular Impairment: Novel Circulating Biomarkers and Therapeutic Approaches. J. Am. Coll. Cardiol. 2013, 62, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pititto, B.D.A.; Ribeiro-Filho, F.F.; Bittencourt, M.S.; Lotufo, P.A.; Bensenor, I.; Ferreira, S.R.G. Usefulness of circulating E-selectin to early detection of the atherosclerotic process in the Brazilian Longitudinal Study of Adult Health (ELSA-Brasil). Diabetol. Metab. Syndr. 2016, 8, 19. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Rifai, N. Soluble P-Selectin and the Risk of Future Cardiovascular Events. Circulation 2001, 103, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-J.; Ballantyne, C.M.; Sharrett, A.R.; Smith, L.C.; Davis, C.E.; Gotto, A.M.; Boerwinkle, E. Circulating Adhesion Molecules VCAM-1, ICAM-1, and E-selectin in Carotid Atherosclerosis and Incident Coronary Heart Disease Cases. Circulation 1997, 96, 4219–4225. [Google Scholar] [CrossRef]
- Schmidt, C.; Hulthe, J.; Fagerberg, B. Baseline ICAM-1 and VCAM-1 are Increased in Initially Healthy Middle-Aged Men who Develop Cardiovascular Disease during 6.6 Years of Follow-Up. Angiology 2009, 60, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Hillis, G.S.; Terregino, C.; Taggart, P.; Killian, A.; Zhao, N.; Dalsey, W.C.; Mangione, A. Elevated soluble P-selectin levels are associated with an increased risk of early adverse events in patients with presumed myocardial ischemia. Am. Heart J. 2002, 143, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Hoogeveen, R.C.; Morrison, A.; Boerwinkle, E.; Miles, J.S.; Rhodes, C.E.; Sharrett, A.R.; Ballantyne, C.M. Plasma MCP-1 level and risk for peripheral arterial disease and incident coronary heart disease: Atherosclerosis Risk in Communities study. Atherosclerosis 2005, 183, 301–307. [Google Scholar] [CrossRef]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef]
- Xia, F.; Wang, C.; Jin, Y.; Liu, Q.; Meng, Q.; Liu, K.; Sun, H. Luteolin Protects HUVECs from TNF-α-induced Oxidative Stress and Inflammation via its Effects on the Nox4/ROS-NF-κB and MAPK Pathways. J. Atheroscler. Thromb. 2014, 21, 768–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Lu, M.; Lin, T.Y.; Chen, Z.; Chen, G.; Wang, W.-C.; Marin, T.; Shentu, T.-P.; Wen, L.; Gongol, B.; et al. Sterol Regulatory Element Binding Protein 2 Activation of NLRP3 Inflammasome in Endothelium Mediates Hemodynamic-Induced Atherosclerosis Susceptibility. Circulation 2013, 128, 632–642. [Google Scholar] [CrossRef]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil extracellular traps participate in cardiovascular diseases: Recent experimental and clinical insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.; Berden, J.H.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, C.; Zhao, M.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2019, 9, 3076. [Google Scholar] [CrossRef]
- Hirota, T.; Levy, J.H.; Iba, T. The influence of hyperglycemia on neutrophil extracellular trap formation and endothelial glycocalyx damage in a mouse model of type 2 diabetes. Microcirculation 2020, 27, 12617. [Google Scholar] [CrossRef] [PubMed]
- Obama, T.; Ohinata, H.; Takaki, T.; Iwamoto, S.; Sawada, N.; Aiuchi, T.; Kato, R.; Itabe, H. Cooperative Action of Oxidized Low-Density Lipoproteins and Neutrophils on Endothelial Inflammatory Responses Through Neutrophil Extracellular Trap Formation. Front. Immunol. 2019, 10, 1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, Q.; Venugopal, J.; Wang, J.; Kleiman, K.; Guo, C.; Eitzman, D.T. Obesity-induced Endothelial Dysfunction is Prevented by Neutrophil Extracellular Trap Inhibition. Sci. Rep. 2018, 8, 4881. [Google Scholar] [CrossRef] [PubMed]
- Jalali, S.; del Pozo, M.A.; Chen, K.-D.; Miao, H.; Li, Y.-S.; Schwartz, M.A.; Shyy, J.Y.-J.; Chien, S. Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ECM) ligands. Proc. Natl. Acad. Sci. USA 2001, 98, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Kuchan, M.J.; Jo, H.; Frangos, J.A. Role of G proteins in shear stress-mediated nitric oxide production by endothelial cells. Am. J. Physiol. Cell Physiol. 1994, 267, C753–C758. [Google Scholar] [CrossRef]
- Pan, S. Molecular Mechanisms Responsible for the Atheroprotective Effects of Laminar Shear Stress. Antioxid. Redox Signal. 2009, 11, 1669–1682. [Google Scholar] [CrossRef]
- Boo, Y.C.; Kim, H.J.; Song, H.; Fulton, D.; Sessa, W.; Jo, H. Coordinated regulation of endothelial nitric oxide synthase activity by phosphorylation and subcellular localization. Free Radic. Biol. Med. 2006, 41, 144–153. [Google Scholar] [CrossRef]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Passerini, A.; Polacek, D.C.; Shi, C.; Francesco, N.M.; Manduchi, E.; Grant, G.; Pritchard, W.F.; Powell, S.; Chang, G.Y.; Stoeckert, C.J.; et al. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc. Natl. Acad. Sci. USA 2004, 101, 2482–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warabi, E.; Takabe, W.; Minami, T.; Inoue, K.; Itoh, K.; Yamamoto, M.; Ishii, T.; Kodama, T.; Noguchi, N. Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: Role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 2007, 42, 260–269. [Google Scholar] [CrossRef]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 Is a Novel Transcriptional Regulator of Endothelial Proinflammatory Activation. J. Exp. Med. 2004, 199, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.-I.; Rakugi, H.; Morishita, R. Insight into the Role of Angiopoietins in Ageing-Associated Diseases. Cells 2020, 9, 2636. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, J.-Y.; Li, B.; Tian, X.Y.; Chen, L.-J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [Green Version]
- Serbanovic-Canic, J.; de Luca, A.; Warboys, C.; Ferreira, P.F.; Luong, L.A.; Hsiao, S.; Gauci, I.; Mahmoud, M.; Feng, S.; Souilhol, C.; et al. Zebrafish Model for Functional Screening of Flow-Responsive Genes. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.M.; Kim, H.R.; Xing, R.; Hsiao, S.; Mammmoto, A.; Chen, J.; Serbanovic-Canic, J.; Feng, S.; Bowden, N.P.; Maguire, R.; et al. TWIST1 Integrates Endothelial Responses to Flow in Vascular Dysfunction and Atherosclerosis. Circ. Res. 2016, 119, 450–462. [Google Scholar] [CrossRef]
- Kim, C.W.; Song, H.; Kumar, S.; Nam, D.; Kwon, H.S.; Chang, K.H.; Son, D.J.; Kang, D.-W.; Brodie, S.A.; Weiss, D.; et al. Anti-Inflammatory and Antiatherogenic Role of BMP Receptor II in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1350–1359. [Google Scholar] [CrossRef] [Green Version]
- Gelfand, B.D.; Meller, J.; Pryor, A.W.; Kahn, M.; Bortz, P.D.S.; Wamhoff, B.R.; Blackman, B.R. Hemodynamic Activation of β-Catenin and T-Cell-Specific Transcription Factor Signaling in Vascular Endothelium Regulates Fibronectin Expression. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1625–1633. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The Endothelial-Specific MicroRNA miR-126 Governs Vascular Integrity and Angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.; Heyll, K.; Noels, H.; Hristov, M.; et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Yang, T.-L. MicroRNA-126 alleviates endothelial cells injury in atherosclerosis by restoring autophagic flux via inhibiting of PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 495, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-T.; Wang, F.; Shao, M.; Wang, Y.; Zhu, H.-Q. MicroRNA-126 suppresses inflammation in endothelial cells under hyperglycemic condition by targeting HMGB1. Vasc. Pharmacol. 2017, 88, 48–55. [Google Scholar] [CrossRef]
- Chen, H.; Li, X.; Liu, S.; Gu, L.; Zhou, X. MircroRNA-19a promotes vascular inflammation and foam cell formation by targeting HBP-1 in atherogenesis. Sci. Rep. 2017, 7, 12089. [Google Scholar] [CrossRef]
- Loyer, X.; Potteaux, S.; Vion, A.-C.; Guérin, C.L.; Boulkroun, S.; Rautou, P.-E.; Ramkhelawon, B.; Esposito, B.; Dalloz, M.; Paul, J.-L.; et al. Inhibition of MicroRNA-92a Prevents Endothelial Dysfunction and Atherosclerosis in Mice. Circ. Res. 2014, 114, 434–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, C.-W.; Qiu, H.; Jo, H. MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1762–H1769. [Google Scholar] [CrossRef] [Green Version]
- Son, D.J.; Kumar, S.; Takabe, W.; Kim, C.W.; Ni, C.-W.; Alberts-Grill, N.; Jang, I.-H.; Kim, S.; Kim, W.; Kang, S.W.; et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat. Commun. 2013, 4, 3000. [Google Scholar] [CrossRef]
- Xu, X.; Yang, Y.; Wang, G.; Yin, Y.; Han, S.; Zheng, D.; Zhou, S.; Zhao, Y.; Chen, Y.; Jin, Y. Low shear stress regulates vascular endothelial cell pyroptosis through miR-181b-5p/STAT-3 axis. J. Cell. Physiol. 2021, 236, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Steyers, C.M., III; Miller, F.J., Jr. Endothelial Dysfunction in Chronic Inflammatory Diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Minno, M.N.D.; Ambrosino, P.; Lupoli, R.; Di Minno, A.; Tasso, M.; Peluso, R.; Tremoli, E. Clinical assessment of endothelial function in patients with rheumatoid arthritis: A meta-analysis of literature studies. Eur. J. Intern. Med. 2015, 26, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Bordy, R.; Totoson, P.; Prati, C.; Marie, C.; Wendling, D.; Demougeot, C. Microvascular endothelial dysfunction in rheumatoid arthritis. Nat. Rev. Rheumatol. 2018, 14, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Nebro, A.; Rúa-Figueroa, Í.; López-Longo, F.J.; Galindo, M.; Calvo-Alén, J.; Olivé-Marqués, A.; Cañizares, M.D.C.O.; Martín-Martínez, M.A.; Blanco, R.; Melero-González, R.; et al. Cardiovascular Events in Systemic Lupus Erythematosus: A Nationwide Study in Spain from the RELESSER Registry. Medicine 2015, 94, e1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Pinto, C.; Rojas-Villarraga, A.; Molano-González, N.; García-Carrasco, M.; Munguía-Realpozo, P.; Etchegaray-Morales, I.; Morales-Sánchez, H.; Berra-Romani, R.; Cervera, R. Endothelial dysfunction and arterial stiffness in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Atherosclerosis 2020, 297, 55–63. [Google Scholar] [CrossRef]
- Alba, B.K.; Greaney, J.L.; Ferguson, S.B.; Alexander, L.M. Endothelial function is impaired in the cutaneous microcirculation of adults with psoriasis through reductions in nitric oxide-dependent vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H343–H349. [Google Scholar] [CrossRef]
- Weber, B.N.; Stevens, E.; Perez-Chada, L.M.; Brown, J.M.; Divakaran, S.; Bay, C.; Bibbo, C.; Hainer, J.; Dorbala, S.; Blankstein, R.; et al. Impaired Coronary Vasodilator Reserve and Adverse Prognosis in Patients with Systemic Inflammatory Disorders. JACC Cardiovasc. Imaging 2021. [Google Scholar] [CrossRef]
- Gravina, A.G.; Dallio, M.; Masarone, M.; Rosato, V.; Aglitti, A.; Persico, M.; Loguercio, C.; Federico, A. Vascular Endothelial Dysfunction in Inflammatory Bowel Diseases: Pharmacological and Nonpharmacological Targets. Oxidative Med. Cell. Longev. 2018, 2018, 2568569. [Google Scholar] [CrossRef] [Green Version]
- Borissoff, J.I.; Spronk, H.M.; Cate, H.T. The Hemostatic System as a Modulator of Atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760. [Google Scholar] [CrossRef]
- Del Turco, S.; Basta, G.; Lazzerini, G.; Chancharme, L.; Lerond, L.; De Caterina, R. Involvement of the TP receptor in TNF-α-induced endothelial tissue factor expression. Vasc. Pharmacol. 2014, 62, 49–56. [Google Scholar] [CrossRef]
- Bavendiek, U.; Libby, P.; Kilbride, M.; Reynolds, R.; Mackman, N.; Schönbeck, U. Induction of Tissue Factor Expression in Human Endothelial Cells by CD40 Ligand Is Mediated via Activator Protein 1, Nuclear Factor κB, and Egr-1. J. Biol. Chem. 2002, 277, 25032–25039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szotowski, B.; Antoniak, S.; Poller, W.; Schultheiss, H.-P.; Rauch, U. Procoagulant Soluble Tissue Factor Is Released from Endothelial Cells in Response to Inflammatory Cytokines. Circ. Res. 2005, 96, 1233–1239. [Google Scholar] [CrossRef] [Green Version]
- Cohen, C.T.; Turner, N.A.; Moake, J.L. Production and control of coagulation proteins for factor X activation in human endothelial cells and fibroblasts. Sci. Rep. 2020, 10, 2005. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M. A Nuclear Attack on Thrombosis and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 221–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, T.E.; Huntington, J.A. Thrombin-Cofactor Interactions: Structural Insights into Regulatory Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1738–1745. [Google Scholar] [CrossRef]
- Anastasiou, G.; Gialeraki, A.; Merkouri, E.; Politou, M.; Travlou, A. Thrombomodulin as a regulator of the anticoagulant pathway: Implication in the development of thrombosis. Blood Coagul. Fibrinolysis 2012, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Wei, X.; Zhang, J.; Yi, B.; Zhang, G.-X.; Yin, L.; Yang, X.-F.; Sun, J. Antithrombotic Effects of Nur77 and Nor1 Are Mediated Through Upregulating Thrombomodulin Expression in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Nan, B.; Yang, H.; Yan, S.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. C-reactive protein decreases expression of thrombomodulin and endothelial protein C receptor in human endothelial cells. Surgery 2005, 138, 212–222. [Google Scholar] [CrossRef]
- Ishii, H.; Tezuka, T.; Ishikawa, H.; Takada, K.; Oida, K.; Horie, S. Oxidized phospholipids in oxidized low-density lipoprotein down-regulate thrombomodulin transcription in vascular endothelial cells through a decrease in the binding of RARβ-RXRα heterodimers and Sp1 and Sp3 to their binding sequences in the TM promoter. Blood 2003, 101, 4765–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanesha, N.; Prakash, P.; Doddapattar, P.; Khanna, I.; Pollpeter, M.J.; Nayak, M.K.; Staber, J.M.; Chauhan, A.K. Endothelial Cell–Derived von Willebrand Factor Is the Major Determinant That Mediates von Willebrand Factor–Dependent Acute Ischemic Stroke by Promoting Postischemic Thrombo-Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1829–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doddapattar, P.; Dhanesha, N.; Chorawala, M.R.; Tinsman, C.; Jain, M.; Nayak, M.K.; Staber, J.M.; Chauhan, A.K. Endothelial Cell–Derived Von Willebrand Factor, But Not Platelet-Derived, Promotes Atherosclerosis in Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Vischer, U.M.; Barth, H.; Wollheim, C.B. Regulated von Willebrand Factor Secretion Is Associated with Agonist-Specific Patterns of Cytoskeletal Remodeling in Cultured Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, A.; Ball, C.; Nolasco, L.; Moake, J.F.; Dong, J.-F. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell–derived ultralarge von Willebrand factor multimers under flow. Blood 2004, 104, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gragnano, F.; Sperlongano, S.; Golia, E.; Natale, F.; Bianchi, R.; Crisci, M.; Fimiani, F.; Pariggiano, I.; Diana, V.; Carbone, A.; et al. The Role of von Willebrand Factor in Vascular Inflammation: From Pathogenesis to Targeted Therapy. Mediat. Inflamm. 2017, 2017, 5620314. [Google Scholar] [CrossRef]
- Delabranche, X.; Helms, J.; Meziani, F. Immunohaemostasis: A new view on haemostasis during sepsis. Ann. Intensiv. Care 2017, 7, 117. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2012, 13, 34–45. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Green, D.J.; Jones, H.; Thijssen, D.; Cable, N.T.; Atkinson, G. Flow-Mediated Dilation and Cardiovascular Event Prediction: Does Nitric Oxide Matter? Hypertension 2011, 57, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y.; Kwon, T.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Prognostic Value of Flow-Mediated Vasodilation in Brachial Artery and Fingertip Artery for Cardiovascular Events: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2015, 4, e002270. [Google Scholar] [CrossRef] [Green Version]
- Fuchsjäger-Mayrl, G.; Pleiner, J.; Wiesinger, G.F.; Sieder, A.E.; Quittan, M.; Nuhr, M.J.; Francesconi, C.; Seit, H.-P.; Francesconi, M.; Schmetterer, L.; et al. Exercise training improves vascular endothelial function in patients with type 1 diabetes. Diabetes Care 2002, 25, 1795–1801. [Google Scholar] [CrossRef] [Green Version]
- Pedralli, M.L.; Marschner, R.A.; Kollet, D.P.; Neto, S.G.; Eibel, B.; Tanaka, H.; Lehnen, A.M. Different exercise training modalities produce similar endothelial function improvements in individuals with prehypertension or hypertension: A randomized clinical trial. Sci. Rep. 2020, 10, 7628. [Google Scholar] [CrossRef] [PubMed]
- A Brown, A.; Hu, F.B. Dietary modulation of endothelial function: Implications for cardiovascular disease. Am. J. Clin. Nutr. 2001, 73, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.; Gossett, L.K.; Piper, M.E.; Aeschlimann, S.E.; Korcarz, C.; Baker, T.B.; Fiore, M.C.; Stein, J.H. Effects of Smoking and Smoking Cessation on Endothelial Function: 1-Year Outcomes from a Randomized Clinical Trial. J. Am. Coll. Cardiol. 2010, 55, 1988–1995. [Google Scholar] [CrossRef] [Green Version]
- Oikonomou, E.; Siasos, G.; Zaromitidou, M.; Hatzis, G.; Mourouzis, K.; Chrysohoou, C.; Zisimos, K.; Mazaris, S.; Tourikis, P.; Athanasiou, D.; et al. Atorvastatin treatment improves endothelial function through endothelial progenitor cells mobilization in ischemic heart failure patients. Atherosclerosis 2015, 238, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Reriani, M.K.; Dunlay, S.M.; Gupta, B.; West, C.P.; Rihal, C.S.; O Lerman, L.; Lerman, A. Effects of statins on coronary and peripheral endothelial function in humans: A systematic review and meta-analysis of randomized controlled trials. Eur. J. Cardiovasc. Prev. Rehabil. 2011, 18, 704–716. [Google Scholar] [CrossRef]
- Gounari, P.; Tousoulis, D.; Antoniades, C.; Kampoli, A.M.; Stougiannos, P.; Papageorgiou, N.; Roulia, G.; Stefanadi, E.; Siasos, G.; Tsioufis, C.; et al. Rosuvastatin but not ezetimibe improves endothelial function in patients with heart failure, by mechanisms independent of lipid lowering. Int. J. Cardiol. 2010, 142, 87–91. [Google Scholar] [CrossRef]
- Alshnbari, A.S.; Millar, S.A.; O’Sullivan, S.E.; Idris, I. Effect of Sodium-Glucose Cotransporter-2 Inhibitors on Endothelial Function: A Systematic Review of Preclinical Studies. Diabetes Ther. 2020, 11, 1947–1963. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xu, H.; Zhang, L.; Liu, L.; Wang, L. GLP-1 receptor agonist lixisenatide protects against high free fatty acids-induced oxidative stress and inflammatory response. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2325–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumaran, V.; Tsuchimochi, H.; Sonobe, T.; Waddingham, M.T.; Shirai, M.; Pearson, J.T. Liraglutide treatment improves the coronary microcirculation in insulin resistant Zucker obese rats on a high salt diet. Cardiovasc. Diabetol. 2020, 19, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajikawa, M.; Higashi, Y.; Tomiyama, H.; Maruhashi, T.; Kurisu, S.; Kihara, Y.; Mutoh, A.; Ueda, S.-I. Effect of short-term colchicine treatment on endothelial function in patients with coronary artery disease. Int. J. Cardiol. 2019, 281, 35–39. [Google Scholar] [CrossRef]
- Yang, M.; Lv, H.; Liu, Q.; Zhang, L.; Zhang, R.; Huang, X.; Wang, X.; Han, B.; Hou, S.; Liu, D.; et al. Colchicine Alleviates Cholesterol Crystal-Induced Endothelial Cell Pyroptosis through Activating AMPK/SIRT1 Pathway. Oxidative Med. Cell. Longev. 2020, 2020, 9173530. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P.; Wei, P.; Milbauer, L.; Corban, M.T.; Solovey, A.; Kiley, J.; Pattee, J.; Lerman, L.O.; Pan, W.; Lerman, A. Abnormal Endothelial Gene Expression Associated with Early Coronary Atherosclerosis. J. Am. Heart Assoc. 2020, 9, e016134. [Google Scholar] [CrossRef] [PubMed]
- LaLonde, S.; Codina-Fauteux, V.-A.; De Bellefon, S.M.; Leblanc, F.; Beaudoin, M.; Simon, M.-M.; Dali, R.; Kwan, T.; Lo, K.S.; Pastinen, T.; et al. Integrative analysis of vascular endothelial cell genomic features identifies AIDA as a coronary artery disease candidate gene. Genome Biol. 2019, 20, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolze, L.K.; Conklin, A.C.; Whalen, M.B.; Rodríguez, M.L.; Õunap, K.; Selvarajan, I.; Toropainen, A.; Örd, T.; Li, J.; Eshghi, A.; et al. Systems Genetics in Human Endothelial Cells Identifies Non-coding Variants Modifying Enhancers, Expression, and Complex Disease Traits. Am. J. Hum. Genet. 2020, 106, 748–763. [Google Scholar] [CrossRef]
- Li, Y.; Yan, H.; Guo, J.; Han, Y.; Zhang, C.; Liu, X.; Du, J.; Tian, X.-L. Down-regulated RGS5 by genetic variants impairs endothelial cell function and contributes to coronary artery disease. Cardiovasc. Res. 2021, 117, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Yang, Y.; Ji, S.; Wang, S.; Peng, X.; Tian, C.; Sun, R.; Yu, T.; Chu, X. MicroRNA-302c-3p inhibits endothelial cell pyroptosis via directly targeting NOD-, LRR- and pyrin domain-containing protein 3 in atherosclerosis. J. Cell. Mol. Med. 2021, 25, 4373–4386. [Google Scholar] [CrossRef]
- Zhou, W.; Xi, D.; Shi, Y.; Wang, L.; Zhong, H.; Huang, Z.; Liu, Y.; Tang, Y.; Lu, N.; Wang, Y.; et al. MicroRNA-1929-3p participates in murine cytomegalovirus-induced hypertensive vascular remodeling through Ednra/NLRP3 inflammasome activation. Int. J. Mol. Med. 2020, 47, 719–731. [Google Scholar] [CrossRef]
- Zhou, T.; Xiang, D.-K.; Li, S.-N.; Yang, L.-H.; Gao, L.-F.; Feng, C. MicroRNA-495 Ameliorates Cardiac Microvascular Endothelial Cell Injury and Inflammatory Reaction by Suppressing the NLRP3 Inflammasome Signaling Pathway. Cell. Physiol. Biochem. 2018, 49, 798–815. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Dong, S.; Liang, Y.; Wang, K.; Qin, Y.; Zhao, X. miR-20b inhibits the senescence of human umbilical vein endothelial cells through regulating the Wnt/β-catenin pathway via the TXNIP/NLRP3 axis. Int. J. Mol. Med. 2020, 45, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhao, D.; Gao, F.; Hu, X.; Hu, X.; Li, M.; Cui, Y.; Wei, X.; Xie, C.; Zhao, Y.; et al. MicroRNA-520c-3p suppresses vascular endothelium dysfunction by targeting RELA and regulating the AKT and NF-κB signaling pathways. J. Physiol. Biochem. 2021, 77, 47–61. [Google Scholar] [CrossRef]
- Yang, S.; Chen, Y.; Mi, X.; Zhang, S.; Yang, Y.; Hui, R.; Zhang, W. MicroRNA-216a Promotes Endothelial Inflammation by Smad7/IκBα Pathway in Atherosclerosis. Dis. Markers 2020, 2020, 8864322. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yao, Y.; Shi, S.; Zhou, M.; Zhou, Y.; Wang, M.; Chiu, J.; Huang, Z.; Zhang, W.; Liu, M.; et al. Inhibition of miR-21 alleviated cardiac perivascular fibrosis via repressing EndMT in T1DM. J. Cell. Mol. Med. 2020, 24, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Zhang, Y.; Chen, H.; Sun, X.-H.; Zhang, P.; Zhang, L.; Liao, M.-Y.; Zhang, F.; Xia, Z.-Y.; Man, R.Y.-K.; et al. MicroRNA-17-3p suppresses NF-κB-mediated endothelial inflammation by targeting NIK and IKKβ binding protein. Acta Pharmacol. Sin. 2021, 1–12. [Google Scholar] [CrossRef]
- Yang, X.; Li, D.; Qi, Y.-Z.; Chen, W.; Yang, C.-H.; Jiang, Y.-H. MicroRNA-217 ameliorates inflammatory damage of endothelial cells induced by oxidized LDL by targeting EGR1. Mol. Cell. Biochem. 2020, 475, 41–51. [Google Scholar] [CrossRef]
- Wang, J.; Li, P.; Xu, X.; Zhang, B.; Zhang, J. MicroRNA-200a Inhibits Inflammation and Atherosclerotic Lesion Formation by Disrupting EZH2-Mediated Methylation of STAT3. Front. Immunol. 2020, 11, 907. [Google Scholar] [CrossRef]
- Jiang, Z.; Wu, J.; Ma, F.; Jiang, J.; Xu, L.; Du, L.; Huang, W.; Wang, Z.; Jia, Y.; Lu, L.; et al. MicroRNA-200a improves diabetic endothelial dysfunction by targeting KEAP1/NRF2. J. Endocrinol. 2020, 245, 129–140. [Google Scholar] [CrossRef]
- Hu, B.; Gong, Z.; Bi, Z. Inhibition of miR-383 suppresses oxidative stress and improves endothelial function by increasing sirtuin 1. Braz. J. Med Biol. Res. 2020, 53, e8616. [Google Scholar] [CrossRef]
- Wu, J.; Liang, W.; Tian, Y.; Ma, F.; Huang, W.; Jia, Y.; Jiang, Z.; Wu, H. Inhibition of P53/miR-34a improves diabetic endothelial dysfunction via activation of SIRT1. J. Cell. Mol. Med. 2019, 23, 3538–3548. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Kim, Y.-R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-Induced MicroRNA-34a Causes Diabetic Endothelial Dysfunction by Downregulating Sirtuin1. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2394–2403. [Google Scholar] [CrossRef] [Green Version]
- Kassan, M.; Vikram, A.; Li, Q.; Kim, Y.-R.; Kumar, S.; Gabani, M.; Liu, J.; Jacobs, J.S.; Irani, K. MicroRNA-204 promotes vascular endoplasmic reticulum stress and endothelial dysfunction by targeting Sirtuin1. Sci. Rep. 2017, 7, 9308. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, J.; Zhang, F.; Lei, Q.; Zhang, T.; Li, K.; Guo, J.; Hong, Y.; Bu, G.; Lv, X.; et al. MicroRNA-181a-5p and microRNA-181a-3p cooperatively restrict vascular inflammation and atherosclerosis. Cell Death Dis. 2019, 10, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, W.-Y.; Yang, W.-K.; Peng, C.-T.; Pai, W.-Y.; Wang, H.-J. MicroRNA-200a/200b Modulate High Glucose-Induced Endothelial Inflammation by Targeting O-linked N-Acetylglucosamine Transferase Expression. Front. Physiol. 2018, 9, 355. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Adhesion Molecule | Ligand | Role | Clinical Significance |
|---|---|---|---|
| ICAM-1 | LFA-1 Mac-1 | Leukocyte adhesion | ICAM-1 correlates with the incidence of CAD and carotid atherosclerosis independently of known cardiovascular risk factors [115]. |
| VCAM-1 | VLA-4 | Leukocyte adhesion | Baseline VCAM-1 is increased in initially healthy middle-aged men who develop cardiovascular disease [116]. |
| E-Selectin | ESL PSGL-1 | Leukocyte adhesion | E-Selectin correlates with the incidence of CAD and carotid atherosclerosis independently of known cardiovascular risk factors [115]. |
| P-Selectin | PSGL-1 | Leukocyte adhesion | Elevated P-selectin levels predict early adverse events in patients with presumed CAD [117]. |
| MCP-1 | CCR2 | Monocyte chemotaxis | Association of MCP-1 with risk of incident PAD and CAD independently of traditional cardiovascular risk factors [118]. |
| MicroRNA | Intervention | Target | Endothelial Effect |
|---|---|---|---|
| 302c-3p [199] | ↑ | NLRP3 inflammasome | ↓ EC pyroptosis |
| 1929-3p [200] | ↑ | endothelin A receptor, NLRP3 inflammasome | ↓ EC injury and vascular remodeling |
| 181b-5p [153] | ↑ | STAT3/NLRP3 inflammasome | ↓ EC pyroptosis |
| 495 [201] | ↑ | NLRP3 inflammasome | ↓ EC inflammation, apoptosis and ↑ EC proliferation |
| 20b [202] | ↑ | TXNIP/NLRP3 | ↑ EC viability |
| 520c-3p [203] | ↑ | NF-κB/Akt pathway | ↓ EC apoptosis |
| 216a [204] | ↓ | Smad7 | ↓ EC adhesive ability to monocytes |
| 21 [205] | ↓ | Smad7 | ↓ endothelial-to-mesenchymal transition |
| 17–3p [206] | ↑ | NIK and IKKβ binding protein | ↓ monocyte adhesion to EC |
| 217 [207] | ↑ | Early growth response protein-1 | Relieve of EC growth inhibition, ↓ endothelial inflammation |
| 200a [208] | ↑ | EZH2-Mediated Methylation of STAT3 | ↓ EC injury, apoptosis, and inflammation |
| 200a [209] | ↑ | KEAP1/NRF2 | ↓ oxidative stress, inflammation, and endothelial dysfunction |
| 383 [210] | ↓ | Sirtuin 1 | ↓ EC apoptosis and ROS production |
| 34a [211] | ↓ | Sirtuin 1 | ↓ EC inflammation, oxidative stress, and endothelial dysfunction |
| 34a [212] | ↓ | Sirtuin 1 | Preservation of endothelium-dependent vasorelaxation |
| 204 [213] | ↓ | Sirtuin 1 | Mitigation of EC dysfunction |
| 181a/181b [214] | ↑ | TAB2, NEMO | ↓ adhesion molecules expression and monocyte-EC interaction |
| 200a/200b [215] | ↑ | O-linked N-acetylglucosamine transferase | ↓ EC inflammation and monocyte adhesion to EC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. https://doi.org/10.3390/biomedicines9070781
Theofilis P, Sagris M, Oikonomou E, Antonopoulos AS, Siasos G, Tsioufis C, Tousoulis D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines. 2021; 9(7):781. https://doi.org/10.3390/biomedicines9070781
Chicago/Turabian StyleTheofilis, Panagiotis, Marios Sagris, Evangelos Oikonomou, Alexios S. Antonopoulos, Gerasimos Siasos, Costas Tsioufis, and Dimitris Tousoulis. 2021. "Inflammatory Mechanisms Contributing to Endothelial Dysfunction" Biomedicines 9, no. 7: 781. https://doi.org/10.3390/biomedicines9070781
APA StyleTheofilis, P., Sagris, M., Oikonomou, E., Antonopoulos, A. S., Siasos, G., Tsioufis, C., & Tousoulis, D. (2021). Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines, 9(7), 781. https://doi.org/10.3390/biomedicines9070781