
Targeting STAT3 Signaling Pathway in Colorectal Cancer




Abstract
:1. Introduction
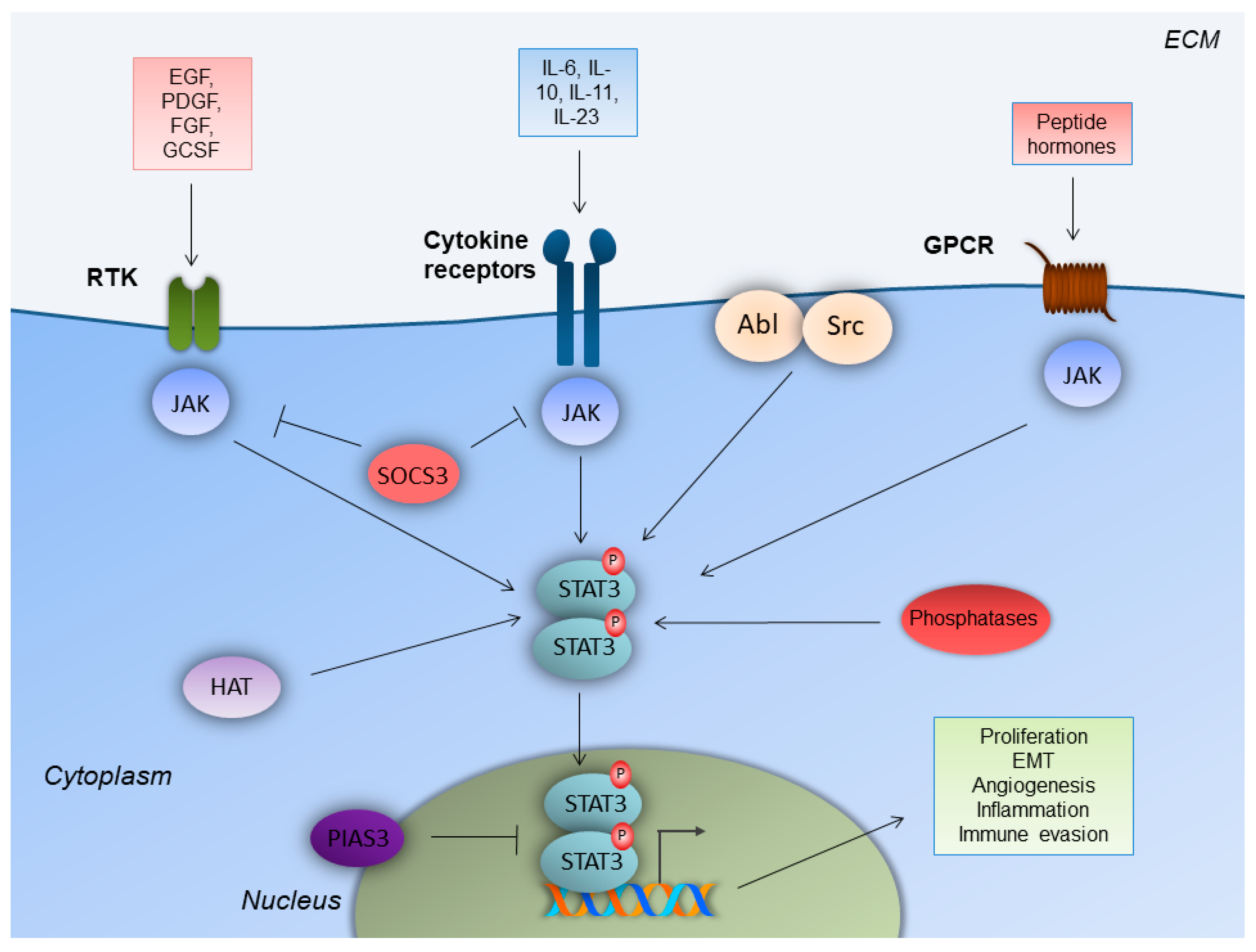
2. STAT3 Structure and Regulation of Activity in Normal Cells and Disease
3. Mechanisms of STAT3 Implication in Tumorigenesis
4. Mechanisms of STAT3 Engagement in Colorectal Tumorigenesis
4.1. STAT3 Expression and Regulation in Colorectal Cancer Cells
4.2. STAT3 in Invasion and Metastasis
4.3. STAT3 in Angiogenesis
4.4. STAT3 in Tumor-Promoting Inflammation
5. STAT3 in Colorectal Cancer Treatment
5.1. STAT3 Engagement in Mechanisms of Resistance to Therapy
5.2. STAT3 in Combination with Immunotherapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Munro, M.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2018, 71, 110–116. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, L.; Li, W.; Zou, D.; Yu, J.; Wang, L.; Wong, C.C. Transcription factors in colorectal cancer: Molecular mechanism and therapeutic implications. Oncogene 2021, 40, 1555–1569. [Google Scholar] [CrossRef]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufman, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 Activation in Colon Cancer Is Associated with Enhanced Cell Proliferation and Tumor Growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 Are Required for Survival of Intestinal Epithelial Cells and Development of Colitis-Associated Cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Zhang, Z.-G.; Tian, X.-Q.; Sun, D.-F.; Liang, Q.-C.; Zhang, Y.-J.; Lu, R.; Chen, Y.-X.; Fang, J.-Y. Inhibition of JAK1, 2/STAT3 Signaling Induces Apoptosis, Cell Cycle Arrest, and Reduces Tumor Cell Invasion in Colorectal Cancer Cells. Neoplasia 2008, 10, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Thilakasiri, P.S.; Dmello, R.S.; Nero, T.L.; Parker, M.; Ernst, M.; Chand, A.L. Repurposing of drugs as STAT3 inhibitors for cancer therapy. Semin. Cancer Biol. 2021, 68, 31–46. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutticken, C.; Wegenka, U.; Yuan, J.; Buschmann, J.; Schindler, C.; Ziemiecki, A.; Harpur, A.; Wilks, A.; Yasukawa, K.; Taga, T.; et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 1994, 263, 89–92. [Google Scholar] [CrossRef]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Varghese, L.N.; Nicola, N. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific Inhibition of Stat3 Signal Transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef]
- Kim, D.J.; Tremblay, M.L.; DiGiovanni, J. Protein Tyrosine Phosphatases, TC-PTP, SHP1, and SHP2, Cooperate in Rapid Dephosphorylation of Stat3 in Keratinocytes Following UVB Irradiation. PLoS ONE 2010, 5, e10290. [Google Scholar] [CrossRef] [Green Version]
- Peyser, N.D.; Du, Y.; Li, H.; Lui, V.; Xiao, X.; Chan, T.A.; Grandis, J.R. Loss-of-Function PTPRD Mutations Lead to Increased STAT3 Activation and Sensitivity to STAT3 Inhibition in Head and Neck Cancer. PLoS ONE 2015, 10, e0135750. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef]
- Liao, W.; Schones, D.E.; Oh, J.; Cui, Y.; Cui, K.; Roh, T.-Y.; Zhao, K.; Leonard, W.J. Priming for T helper type 2 differentiation by interleukin 2–mediated induction of interleukin 4 receptor α-chain expression. Nat. Immunol. 2008, 9, 1288–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stritesky, G.L.; Muthukrishnan, R.; Sehra, S.; Goswami, R.; Pham, D.; Travers, J.; Nguyen, E.T.; Levy, D.; Kaplan, M.H. The Transcription Factor STAT3 Is Required for T Helper 2 Cell Development. Immunity 2011, 34, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Laurence, A.; Kanno, Y.; Pacher-Zavisin, M.; Zhu, B.-M.; Tato, C.; Yoshimura, A.; Hennighausen, L.; O’Shea, J.J. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8137–8142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired TH17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nat. Cell Biol. 2008, 452, 773–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, T.P.; Milner, J.D.; Cooper, M.A. The Ying and Yang of STAT3 in Human Disease. J. Clin. Immunol. 2015, 35, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, A.; Rudra, D.; Treuting, P.; Samstein, R.M.; Liang, Y.; Kas, A.; Rudensky, A.Y. CD4+ Regulatory T Cells Control TH17 Responses in a Stat3-Dependent Manner. Science 2009, 326, 986–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Diamanti-Kandarakis, E.; Piouka, A.; Livadas, S.; Piperi, C.; Katsikis, I.; Papavassiliou, A.G.; Panidis, D. Anti-mullerian hormone is associated with advanced glycosylated end products in lean women with polycystic ovary syndrome. Eur. J. Endocrinol. 2009, 160, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maliqueo, M.; Vanky, E.; Fornes, R.; Benrick, A.; Stridsklev, S.; Jansson, T.; Poromaa, I.S.; Åkerud, H.; Labrie, F.; Stener-Victorin, E. Placental STAT3 signaling is activated in women with polycystic ovary syndrome. Hum. Reprod. 2015, 30, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.L.; Hirpara, J.L.; Pervaiz, S.; Eu, J.-Q.; Sethi, G.; Goh, B.-C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Song, D.; Li, H.; Yang, Y.; Ma, X.; Deng, S.; Ren, C.; Shu, X. Negative regulators of STAT3 signaling pathway in cancers. Cancer Manag. Res. 2019, 11, 4957–4969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Niu, N.; Wei, T.; Tozawa, H.; Chen, X.; Zhang, C.; Zhang, J.; Wada, Y.; Kapron, C.M.; Liu, J. The roles of signal transducer and activator of transcription factor 3 in tumor angiogenesis. Oncotarget 2017, 8, 69139–69161. [Google Scholar] [CrossRef] [Green Version]
- Rébé, C.; Ghiringhelli, F. STAT3, a Master Regulator of Anti-Tumor Immune Response. Cancers 2019, 11, 1280. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, T.; Baba, Y.; Yamauchi, M.; Kuchiba, A.; Nosho, K.; Shima, K.; Tanaka, N.; Huttenhower, C.; Frank, D.A.; Fuchs, C.S.; et al. STAT3 Expression, Molecular Features, Inflammation Patterns, and Prognosis in a Database of 724 Colorectal Cancers. Clin. Cancer Res. 2011, 17, 1452–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusaba, T.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Yoshizaki, A.; Inoue, K.; Nagayasu, T.; Sekine, I. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol. Rep. 2006, 15, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guo, A.; Yu, J.; Possemato, A.; Chen, Y.; Zheng, W.; Polakiewicz, R.D.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.; et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc. Natl. Acad. Sci. USA 2007, 104, 4060–4064. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.; Wang, Z. Tumour suppressor function of protein tyrosine phosphatase receptor-T. Biosci. Rep. 2011, 31, 303–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.; Van Der Heijden, M.S.; et al. Mutational Analysis of the Tyrosine Phosphatome in Colorectal Cancers. Science 2004, 304, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Peyser, N.D.; Freilino, M.L.; Wang, L.; Zeng, Y.; Li, H.; Johnson, D.E.; Grandis, J.R. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene 2016, 35, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, J.C.; Lee, S.-E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal Transducer and Activator of Transcription 3 (STAT3) Protein Suppresses Adenoma-to-carcinoma Transition in Apcmin/+ Mice via Regulation of Snail-1 (SNAI) Protein Stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 Is a Negative Regulator of Intestinal Tumor Progression in ApcMin Mice. Gastroenterology 2010, 138, 1003–1011.e5. [Google Scholar] [CrossRef]
- Liu, H.; Ren, G.; Wang, T.; Chen, Y.; Gong, C.; Bai, Y.; Wang, B.; Qi, H.; Shen, J.; Zhu, L.; et al. Aberrantly expressed Fra-1 by IL-6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial–mesenchymal transition. Carcinogenesis 2015, 36, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Kaller, M.; Rokavec, M.; Kirchner, T.; Horst, D.; Hermeking, H. Characterization of a p53/miR-34a/CSF1R/STAT3 Feedback Loop in Colorectal Cancer. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 391–418. [Google Scholar] [CrossRef]
- Yao, C.; Su, L.; Shan, J.; Zhu, C.; Liu, L.; Liu, C.; Xu, Y.; Yang, Z.; Bian, X.; Shao, J.; et al. IGF/STAT3/NANOG/Slug Signaling Axis Simultaneously Controls Epithelial-Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer. Stem Cells 2016, 34, 820–831. [Google Scholar] [CrossRef]
- Patel, S.A.; Bhambra, U.; Charalambous, M.P.; David, R.M.; Edwards, R.J.; Lightfoot, T.; Boobis, A.R.; Gooderham, N.J. Interleukin-6 mediated upregulation of CYP1B1 and CYP2E1 in colorectal cancer involves DNA methylation, miR27b and STAT3. Br. J. Cancer 2014, 111, 2287–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Shembrey, C.; Smith, J.; Paquet-Fifield, S.; Behrenbruch, C.; Beyit, L.M.; Thomson, B.N.; Heriot, A.G.; Cao, Y.; Hollande, F. Laminin 521 enhances self-renewal via STAT3 activation and promotes tumor progression in colorectal cancer. Cancer Lett. 2020, 476, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Hong, Y.; Dai, L.; Qian, Y.; Zhu, C.; Wu, B.; Li, S. CRH promotes human colon cancer cell proliferation via IL-6/JAK2/STAT3 signaling pathway and VEGF-induced tumor angiogenesis. Mol. Carcinog. 2017, 56, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Zhao, Y.; Liang, Y.; Xiang, C.; Zhou, H.; Zhang, H.; Zhang, Q.; Qing, H.; Jiang, B.; et al. CD24 promoted cancer cell angiogenesis via Hsp90-mediated STAT3/VEGF signaling pathway in colorectal cancer. Oncotarget 2016, 7, 55663–55676. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Sun, L.; Lan, J.; Xu, L.; Zhang, M.; Luo, X.; Gong, J.; Wang, G.; Yuan, X.; Hu, J.; et al. BRG1 targeting STAT3/VEGFC signaling regulates lymphangiogenesis in colorectal cancer. Oncotarget 2016, 7, 36501–36509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Hirayama, D.; Wagatsuma, K.; Yamakawa, T.; Yokoyama, Y.; Nakase, H. Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 3062. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.; Mayberry, J. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-Mediated Stat3 Activation in Enterocytes Regulates Cell Survival and Cell-Cycle Progression during Colitis-Associated Tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Chen, A.; Vamadevan, A.S.; Cohen, J.; Breglio, K.; Krishnareddy, S.; Hsu, D.; Xu, R.; Harpaz, N.; Dannenberg, A.J.; et al. Toll-Like Receptor-4 Promotes the Development of Colitis-Associated Colorectal Tumors. Gastroenterology 2007, 133, 1869–1881. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.; Xavier, R.J.; Gevers, D. The Microbiome in Inflammatory Bowel Disease: Current Status and the Future Ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.C.; Jobin, C. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes 2013, 4, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesselring, R.; Glaesner, J.; Hiergeist, A.; Naschberger, E.; Neumann, H.; Brunner, S.M.; Wege, A.K.; Seebauer, C.; Köhl, G.; Merkl, S.; et al. IRAK-M Expression in Tumor Cells Supports Colorectal Cancer Progression through Reduction of Antimicrobial Defense and Stabilization of STAT3. Cancer Cell 2016, 29, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.; Berg, T. Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Spitzner, M.; Roesler, B.; Bielfeld, C.; Emons, G.; Gaedcke, J.; Wolff, H.A.; Rave-Fränk, M.; Kramer, F.; Beissbarth, T.; Kitz, J.; et al. STAT3 inhibition sensitizes colorectal cancer to chemoradiotherapyin vitroandin vivo. Int. J. Cancer 2014, 134, 997–1007. [Google Scholar] [CrossRef]
- Li, F.; Zhan, L.; Dong, Q.; Wang, Q.; Wang, Y.; Li, X.; Zhang, Y.; Zhang, J. Tumor-Derived Exosome-Educated Hepatic Stellate Cells Regulate Lactate Metabolism of Hypoxic Colorectal Tumor Cells via the IL-6/STAT3 Pathway to Confer Drug Resistance. OncoTargets Ther. 2020, 13, 7851–7864. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, Z.; Liu, Y.; Mei, Z.; Wang, G.; Liang, Z.; Cui, A.; Hu, X.; Cui, L.; Yang, Y.; et al. Interleukin 22 protects colorectal cancer cells from chemotherapy by activating the STAT3 pathway and inducing autocrine expression of interleukin 8. Clin. Immunol. 2014, 154, 116–126. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, H.; Guan, L.; Lai, C.; Yu, W.; Lai, M. LL1, a novel and highly selective STAT3 inhibitor, displays anti-colorectal cancer activities in v itro and in vivo. Br. J. Pharmacol. 2020, 177, 298–313. [Google Scholar] [CrossRef]
- Wei, N.; Li, J.; Fang, C.; Chang, J.; Xirou, V.; Syrigos, N.; Marks, B.J.; Chu, E.; Schmitz, J.C. Targeting colon cancer with the novel STAT3 inhibitor bruceantinol. Oncogene 2019, 38, 1676–1687. [Google Scholar] [CrossRef]
- Gonçalves-Ribeiro, S.; Díaz-Maroto, N.G.; Berdiel-Acer, M.; Soriano, A.; Guardiola, J.; Martínez-Villacampa, M.; Salazar, R.; Capellà, G.; Villanueva, A.; Martínez-Balibrea, E.; et al. Carcinoma-associated fibroblasts affect sensitivity to oxaliplatin and 5FU in colorectal cancer cells. Oncotarget 2016, 7, 59766–59780. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Chen, C.; An, Q.; Yang, T.; Sang, Z.; Yang, Y.; Ju, Y.; Tong, A.; Luo, Y. A novel series of napabucasin derivatives as orally active inhibitors of signal transducer and activator of transcription 3 (STAT3). Eur. J. Med. Chem. 2019, 162, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Jonker, D.J.; Nott, L.; Yoshino, T.; Gill, S.; Shapiro, J.; Ohtsu, A.; Zalcberg, J.; Vickers, M.M.; Wei, A.C.; Gao, Y.; et al. Napabucasin versus placebo in refractory advanced colorectal cancer: A randomised phase 3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 263–270. [Google Scholar] [CrossRef]
- Zhuang, Y.; Bai, Y.; Hu, Y.; Guo, Y.; Xu, L.; Hu, W.; Yang, L.; Zhao, C.; Li, X.; Zhao, H. Rhein sensitizes human colorectal cancer cells to EGFR inhibitors by inhibiting STAT3 pathway. OncoTargets Ther. 2019, 12, 5281–5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef]
- Zhong, Q.; Fang, Y.; Lai, Q.; Wang, S.; He, C.; Li, A.; Liu, S.; Yan, Q. CPEB3 inhibits epithelial-mesenchymal transition by disrupting the crosstalk between colorectal cancer cells and tumor-associated macrophages via IL-6R/STAT3 signaling. J. Exp. Clin. Cancer Res. 2020, 39, 132. [Google Scholar] [CrossRef]
- Wang, D.; Yu, W.; Lian, J.; Wu, Q.; Liu, S.; Yang, L.; Li, F.; Huang, L.; Chen, X.; Zhang, Z.; et al. Th17 cells inhibit CD8+ T cell migration by systematically downregulating CXCR3 expression via IL-17A/STAT3 in advanced-stage colorectal cancer patients. J. Hematol. Oncol. 2020, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Huang, T.; Zou, Q.; Liu, D.; Wang, Y.; Tan, X.; Wei, Y.; Qiu, H. FGFR2 Promotes Expression of PD-L1 in Colorectal Cancer via the JAK/STAT3 Signaling Pathway. J. Immunol. 2019, 202, 3065–3075. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, M.Y.; Zhang, Z.H.; Zuo, H.X.; Wang, J.Y.; Xing, Y.; Ri, M.; Jin, H.L.; Jin, C.H.; Xu, G.H.; et al. Panaxadiol inhibits programmed cell death-ligand 1 expression and tumour proliferation via hypoxia-inducible factor (HIF)-1α and STAT3 in human colon cancer cells. Pharmacol. Res. 2020, 155, 104727. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Ma, Y.; Wang, M.; Shen, J.; Wu, H.; Li, J.; Gao, N.; Gu, Y.; Zhang, X.; Zhang, G.; et al. B7-H3 confers resistance to Vγ9Vδ2 T cell-mediated cytotoxicity in human colon cancer cells via the STAT3/ULBP2 axis. Cancer Immunol. Immunother. 2021, 70, 1213–1226. [Google Scholar] [CrossRef]


{kind=link}
| Identifier | Status | Title | Condition | Phase | Drug |
|---|---|---|---|---|---|
| NCT03647839 | Active, not recruiting | Modulation Of The Tumour Microenvironment Using Either Vascular Disrupting Agents or STAT3 Inhibition in Order to Synergise With PD1 Inhibition in Microsatellite Stable, Refractory Colorectal Cancer | Colorectal Cancer Metastatic | II | BBI608 |
| NCT02983578 | Active, not recruiting | Danvatirsen and Durvalumab in Treating Patients With Advanced and Refractory Pancreatic, non-small cell Lung Cancer, and Mismatch Repair Deficient Colorectal Cancer | Advanced Colorectal Carcinoma Mismatch Repair Deficiency Refractory Colorectal Carcinoma Stage III Colorectal Cancer AJCC v8 Stage IIIA Colorectal Cancer AJCC v8 Stage IIIB Colorectal Cancer AJCC v8 Stage IIIC Colorectal Cancer AJCC v8 Stage IV Colorectal Cancer AJCC v8 Stage IVA Colorectal Cancer AJCC v8 Stage IVB Colorectal Cancer AJCC v8 Stage IVC Colorectal Cancer AJCC v8 | II | Danvatirsen |
| NCT03195699 | Recruiting | Oral STAT3 Inhibitor, TTI-101, in Patients With Advanced Cancers | Colorectal Cancer Advanced Cancer | I | TTI-101 |
| NCT03522649 | Recruiting | A Phase III Clinical Study of Napabucasin (GB201) Plus FOLFIRI in Adult Patients With Metastatic Colorectal Cancer | Previously Treated Metastatic Colorectal Cancer | III | BBI608 |
| NCT02753127 | Completed | A Study of Napabucasin (BBI-608) in Combination With FOLFIRI in Adult Patients With Previously Treated Metastatic Colorectal Cancer (CanStem303C) | Colorectal cancer | III | BBI608 |
| NCT01776307 | Completed | A Study of BBI608 in Adult Patients With Advanced Colorectal Cancer | Colorectal Cancer | II | BBI608 |
| NCT03522649 | Recruiting | A Phase III Clinical Study of Napabucasin (GB201) Plus FOLFIRI in Adult Patients With Metastatic Colorectal Cancer | Previously Treated Metastatic Colorectal Cancer | III | BBI608 |
| NCT02641873 | Completed | A Study of BBI608 Administrated With FOLFIRI + Bevacizumab in Adult Patients With Metastatic Colorectal Cancer | Metastatic Colorectal Cancer | I | BBI608 |
| NCT03647839 | Active, not recruiting | Modulation Of The Tumour Microenvironment Using Either Vascular Disrupting Agents or STAT3 Inhibition in Order to Synergise With PD1 Inhibition in Microsatellite Stable, Refractory Colorectal Cancer (MODULATE) | Colorectal Cancer Metastatic | II | BBI608 |
| NCT01830621 | Completed | BBI608 and Best Supportive Care vs. Placebo and Best Supportive Care in Pretreated Advanced Colorectal Carcinoma | Colorectal Carcinoma | III | BBI608 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargalionis, A.N.; Papavassiliou, K.A.; Papavassiliou, A.G. Targeting STAT3 Signaling Pathway in Colorectal Cancer. Biomedicines 2021, 9, 1016. https://doi.org/10.3390/biomedicines9081016
Gargalionis AN, Papavassiliou KA, Papavassiliou AG. Targeting STAT3 Signaling Pathway in Colorectal Cancer. Biomedicines. 2021; 9(8):1016. https://doi.org/10.3390/biomedicines9081016
Chicago/Turabian StyleGargalionis, Antonios N., Kostas A. Papavassiliou, and Athanasios G. Papavassiliou. 2021. "Targeting STAT3 Signaling Pathway in Colorectal Cancer" Biomedicines 9, no. 8: 1016. https://doi.org/10.3390/biomedicines9081016
APA StyleGargalionis, A. N., Papavassiliou, K. A., & Papavassiliou, A. G. (2021). Targeting STAT3 Signaling Pathway in Colorectal Cancer. Biomedicines, 9(8), 1016. https://doi.org/10.3390/biomedicines9081016