Caudal Regression Syndrome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
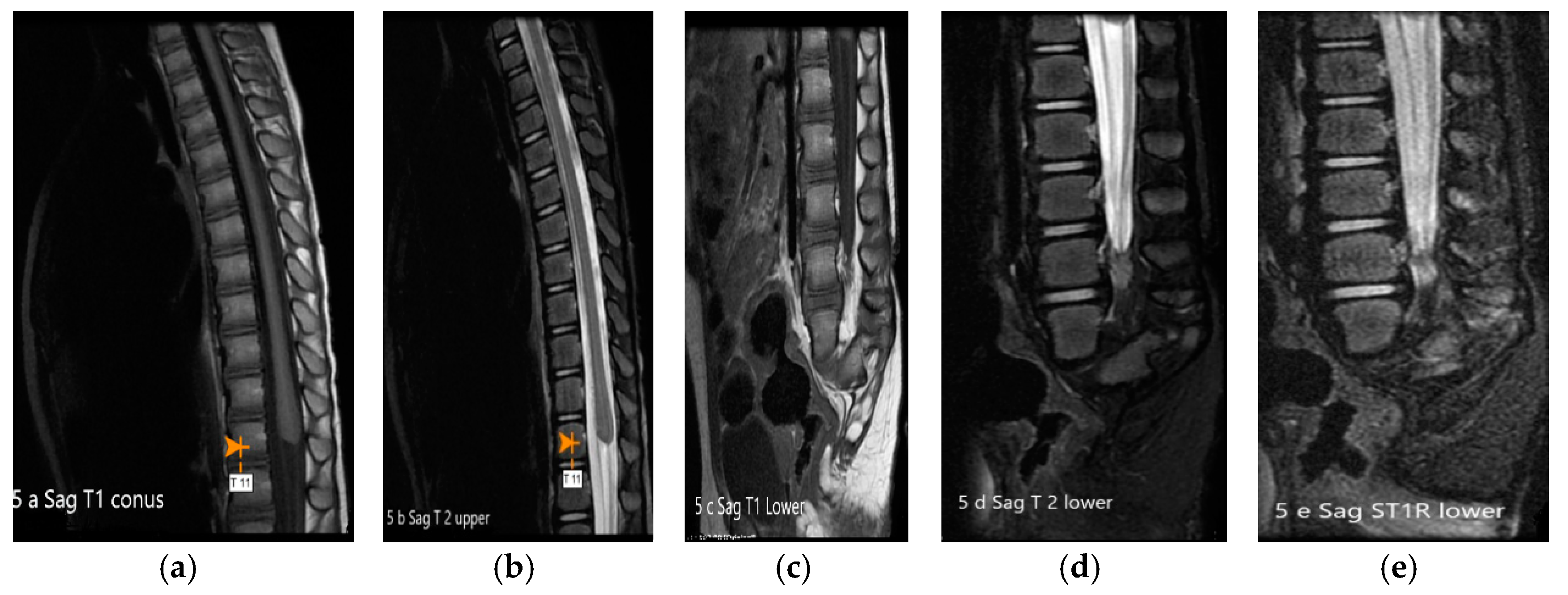
2. Case Presentation
3. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Duhamel, B. From the mermaid to anal imperforation: The syndrome of caudal regression. Arch. Dis. Child. 1961, 36, 152–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Vaquer, A.; Hadjantonakis, A.-K. Birth defects associated with perturbations in preimplantation, gastrulation, and axis extension: From conjoined twinning to caudal dysgenesis. Wiley Interdiscip Rev. Dev. Biol. 2012, 2, 427–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Singh, R.D.; Sharma, A. Caudal regression syndrome—Case report and review of literature. Pediatr. Surg. Int. 2005, 21, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Thottungal, A.D.; Charles, A.K.; Dickinson, J.E.; Bower, C. Caudal dysgenesis and sirenomelia-single centre experience suggests common pathogenic basis. Am. J. Med. Genet. Part A 2010, 152, 2578–2587. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Allepuz, C.; Haro, E.; González-Lamuño, M.; Martínez-Frías, M.L.; Bertocchini, F.; Ros, M.A. A clinical and experimental overview of sirenomelia: Insight into the mechanisms of congenital limb malformations. Dis. Model. Mech. 2011, 4, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boer, L.L.; Morava, E.; Klein, W.M.; Schepens-Franke, A.N.; Oostra, R.-J. Sirenomelia: A multi-systemic polytopic field defect with ongoing controversies. Birth Defects Res. 2017, 109, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Orioli, I.M.; Amar, E.; Arteaga-Vazquez, J.; Bakker, M.K.; Bianca, S.; Botto, L.D.; Clementi, M.; Correa, A.; Csáky-Szunyogh, M.; Leoncini, E.; et al. Sirenomelia: An epidemiologic study in a large dataset from the International Clearinghouse of Birth Defects Surveillance and Research, and literature review. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.A.; Wang, Y.; Strachan, T.; Burn, J.; Lindsay, S. Autosomal dominant sacral agenesis: Currarino syndrome. J. Med. Genet. 2000, 37, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Martucciello, G.; Torre, M.; Belloni, E.; Lerone, M.; Prato, A.; Cama, A.; Jasonni, V. Currarino syndrome: Proposal of a diagnostic and therapeutic protocol. J. Pediatr. Surg. 2004, 39, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Sood, V.; Deswal, S.; Aggarwal, K.C. Caudal regression syndrome with bilateral popliteal webbing without maternal diabetes: A rare entity. Child’s Nerv. Syst. 2012, 28, 1819–1821. [Google Scholar] [CrossRef]
- Lynch, S.A.; Wright, C. Sirenomelia, limb reduction defects, cardiovascular malformation, renal agenesis in an infant born to a diabetic mother. Clin. Dysmorphol. 1997, 6, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kaygusuz, E.I.; Eken, M.K.; Sivrikoz, O.N.; Cetiner, H. Sirenomelia: A review of embryogenic theories and discussion of the differences from caudal regression syndrome. J. Matern. Neonatal Med. 2015, 29, 949–953. [Google Scholar] [CrossRef]
- Porsch, R.M.; Merello, E.; De Marco, P.; Cheng, G.; Rodríguez, L.; So, M.; Sham, P.C.; Tam, P.K.; Capra, V.; Cherny, S.S.; et al. Sacral agenesis: A pilot whole exome sequencing and copy number study. BMC Med. Genet. 2016, 17, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marco, P.; Merello, E.; Piatelli, G.; Cama, A.; Kibar, Z.; Capra, V. Planar cell polarity gene mutations contribute to the etiology of human neural tube defects in our population. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Torban, E.; McDearmid, J.R.; Reynolds, A.; Berghout, J.; Mathieu, M.; Kirillova, I.; De Marco, P.; Merello, E.; Hayes, J.M.; et al. Mutations inVANGL1Associated with Neural-Tube Defects. N. Engl. J. Med. 2007, 356, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Barceló, M.M.; Lui, V.C.H.; So, M.-T.; Miao, X.; Leon, T.Y.-Y.; Yuan, Z.-W.; Ngan, E.S.-W.; Ehsan, T.; Chung, P.H.-Y.; Khong, P.-L.; et al. MNX1 (HLXB9) mutations in Currarino patients. J. Pediatr. Surg. 2009, 44, 1892–1898. [Google Scholar] [CrossRef]
- Pang, D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery 1993, 32, 778–779. [Google Scholar] [CrossRef]
- Kjaer, K.W.; Keeling, J.W.; Opitz, J.M.; Gilbert-Barness, E.; Hartling, U.; Hansen, B.F.; Kjaer, I. Sirenomelia sequence according to the distance between the first sacral vertebra and the ilia. Am. J. Med. Genet. Part A 2003, 503–508. [Google Scholar] [CrossRef]
- Stocker, J.T.; Heifetz, S.A. A morphological study of 33 cases and review of the literature. Perspect. Pediatr. Pathol. 1987, 10, 7–50. [Google Scholar]
- Renshaw, T.S. Sacral agenesis. J. Bone Joint Surg. Am. 1978, 60, 373–383. [Google Scholar] [CrossRef]
- Devesa, J.; Alonso, A.; López, N.; García, J.; Puell, C.I.; Pablos, T.; Devesa, P. Growth Hormone (GH) and rehabilitation promoted distal innervation in a child affected by caudal regression syndrome. Int. J. Mol. Sci. 2017, 18, 230. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kylat, R.I.; Bader, M. Caudal Regression Syndrome. Children 2020, 7, 211. https://doi.org/10.3390/children7110211
Kylat RI, Bader M. Caudal Regression Syndrome. Children. 2020; 7(11):211. https://doi.org/10.3390/children7110211
Chicago/Turabian StyleKylat, Ranjit I., and Mohammad Bader. 2020. "Caudal Regression Syndrome" Children 7, no. 11: 211. https://doi.org/10.3390/children7110211
APA StyleKylat, R. I., & Bader, M. (2020). Caudal Regression Syndrome. Children, 7(11), 211. https://doi.org/10.3390/children7110211