
Drug Carriers: A Review on the Most Used Mathematical Models for Drug Release


Abstract
:1. Introduction
2. Description of Models for Drug Release
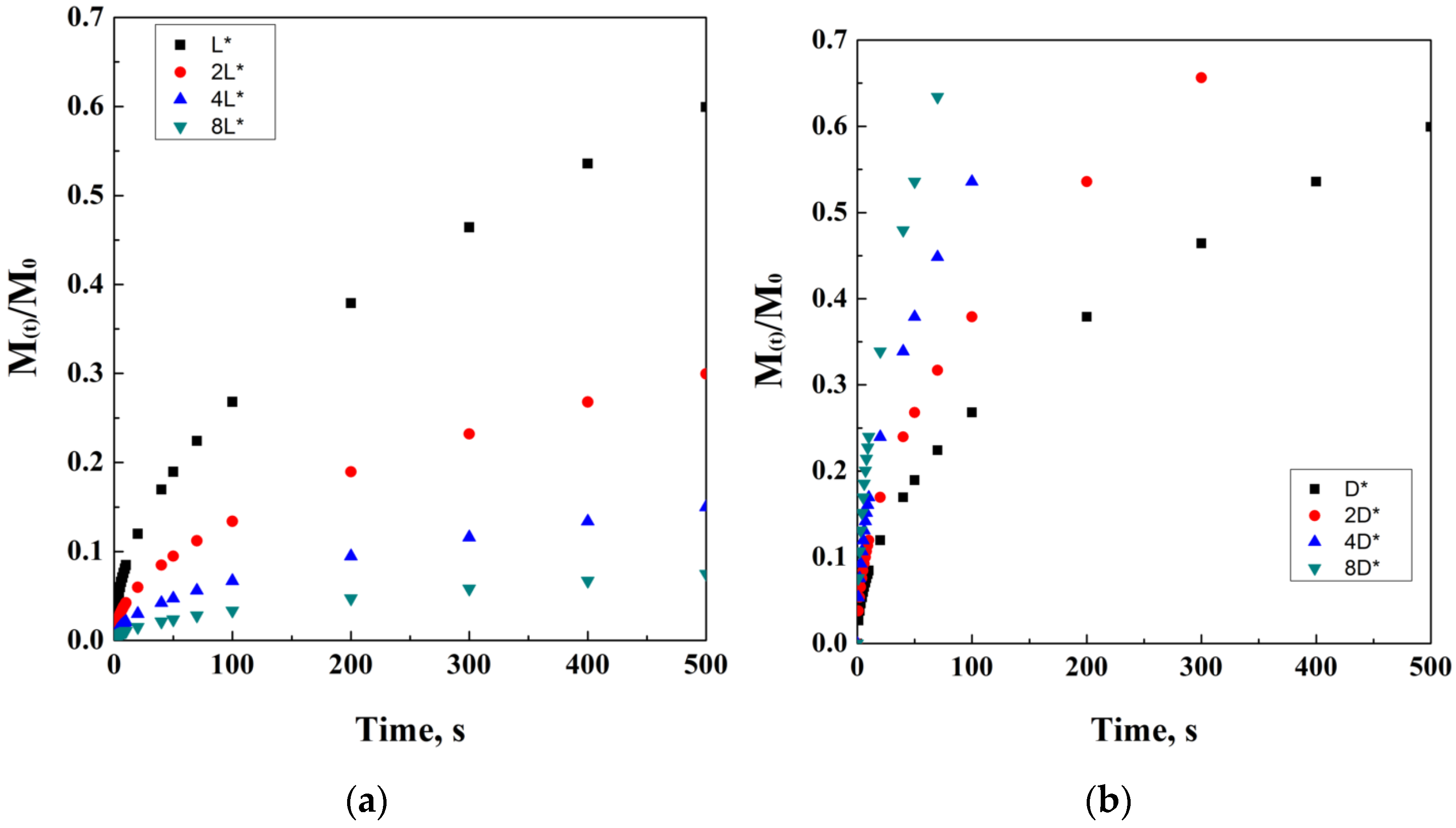
2.1. Fick’s Classic Model
2.2. Higuchi’s Model
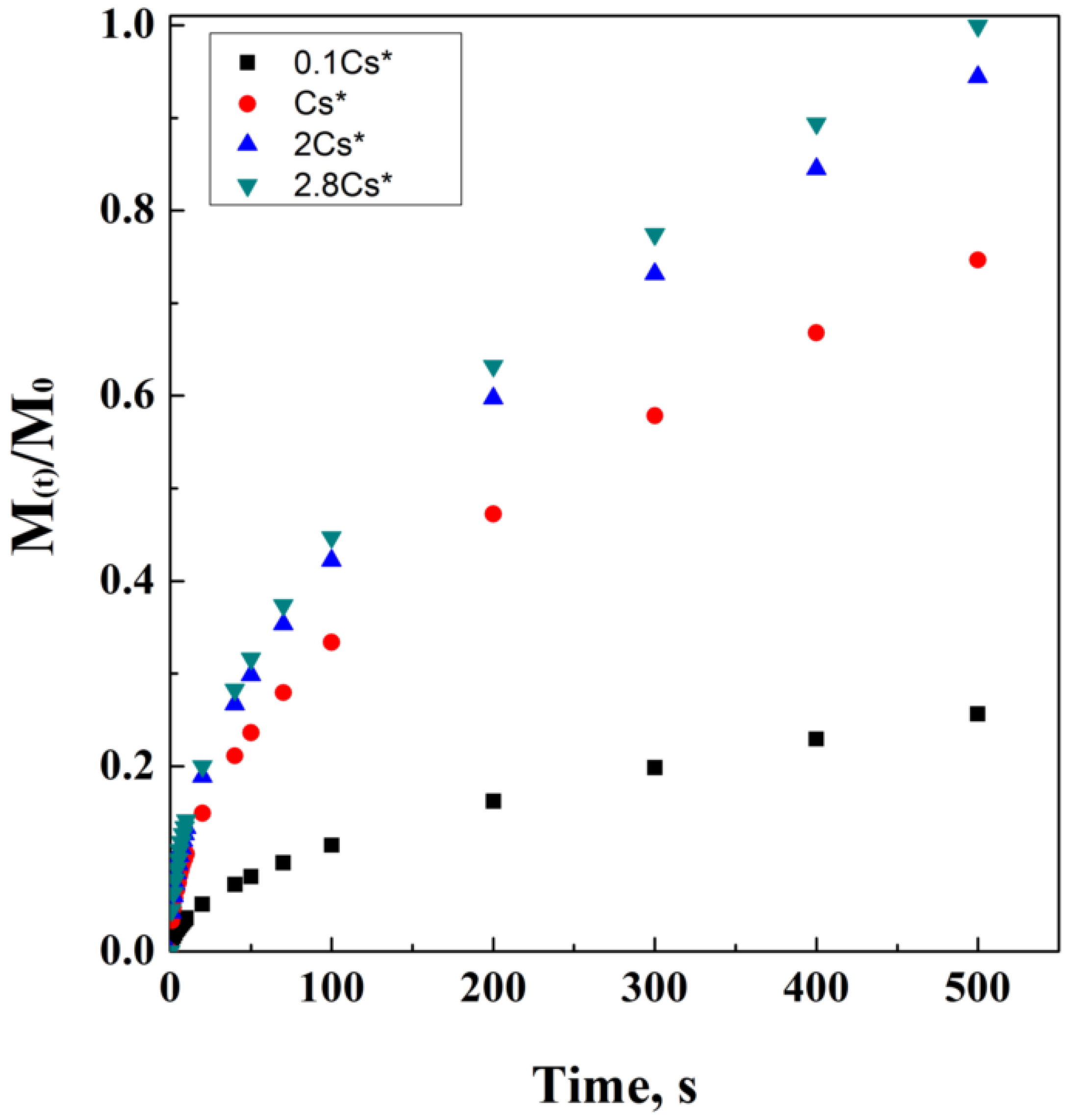
2.3. Hixson-Crowell’s Model
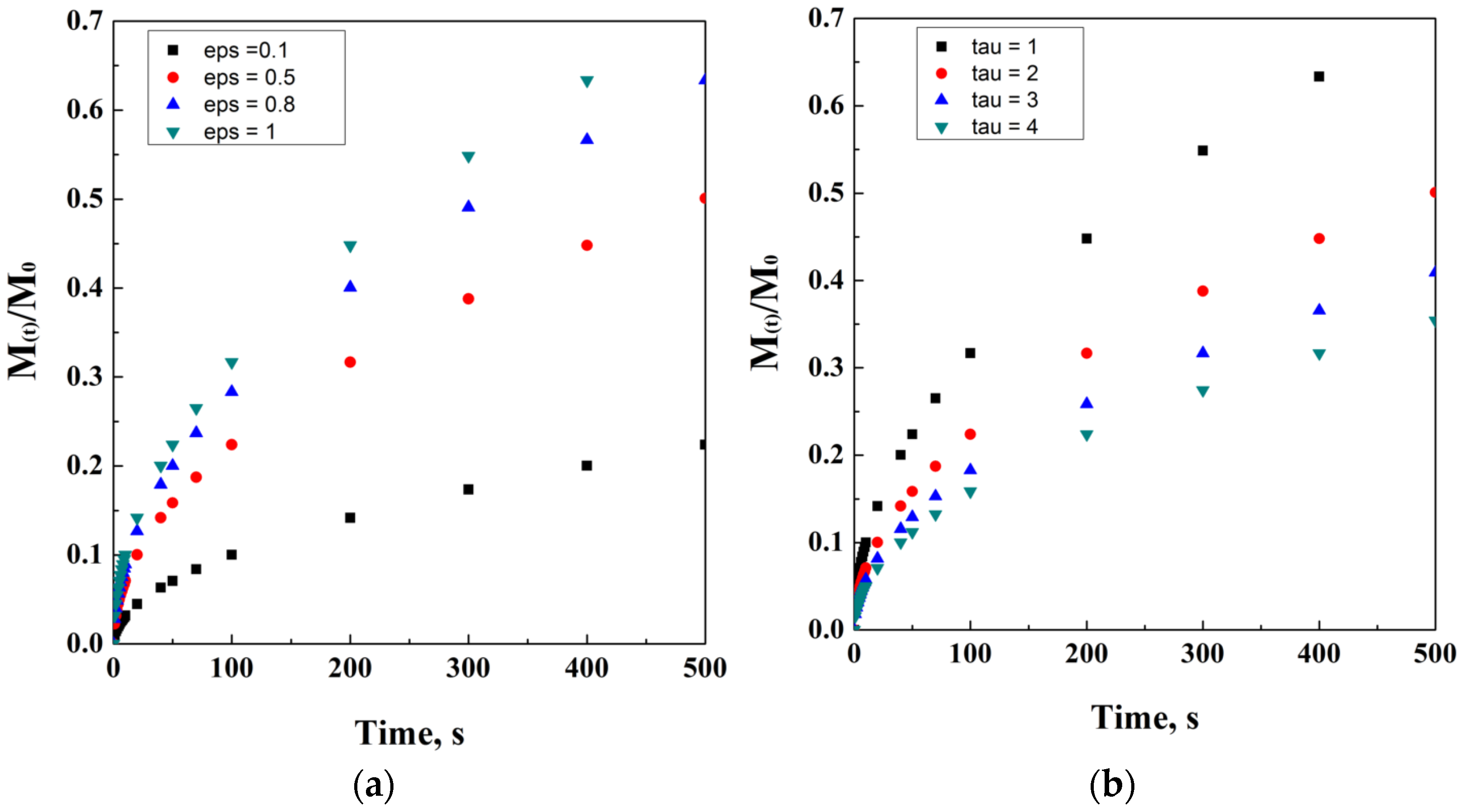
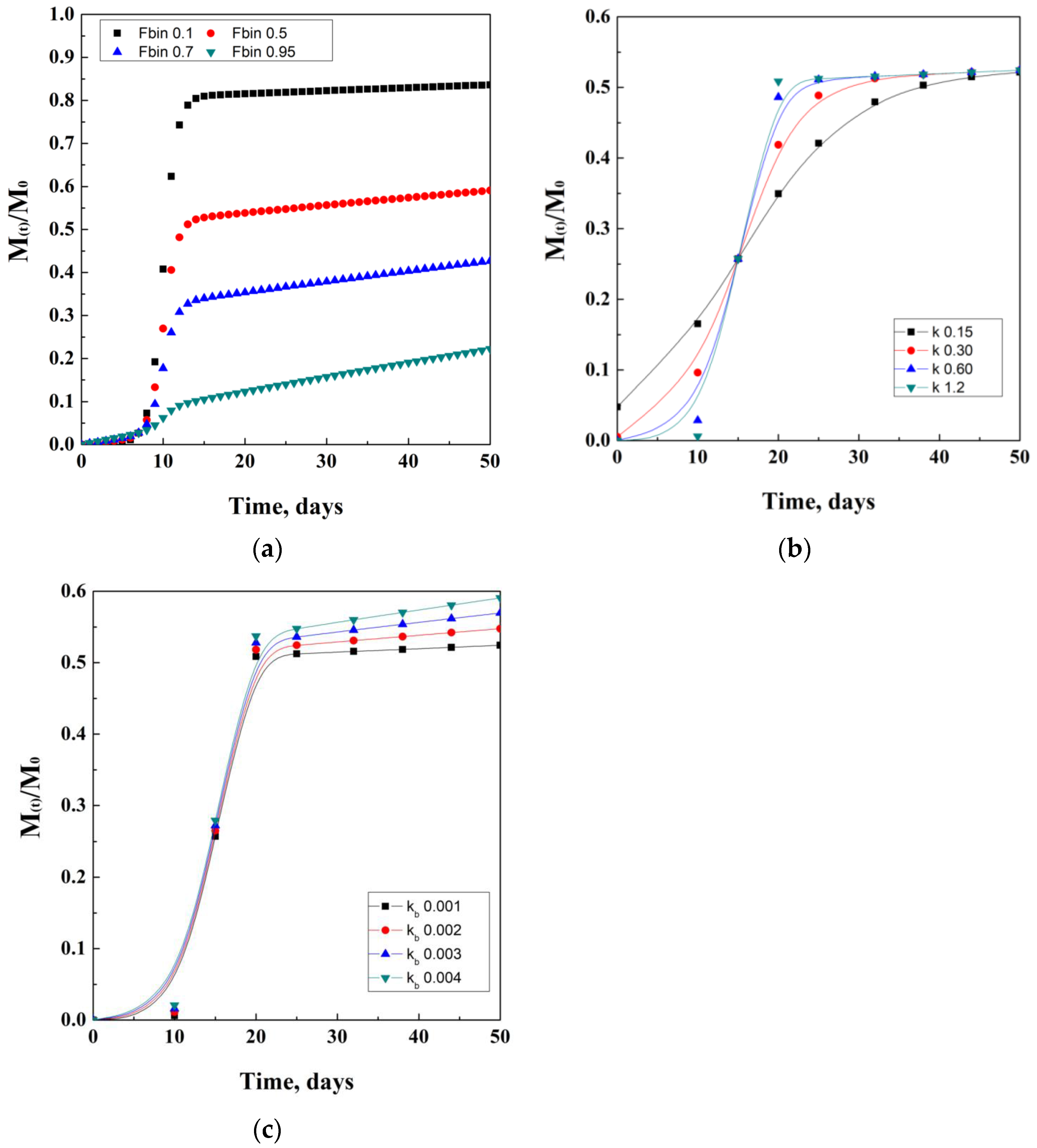
2.4. Peppas’ Models
2.5. Baker’s and Lonsdale’s Model
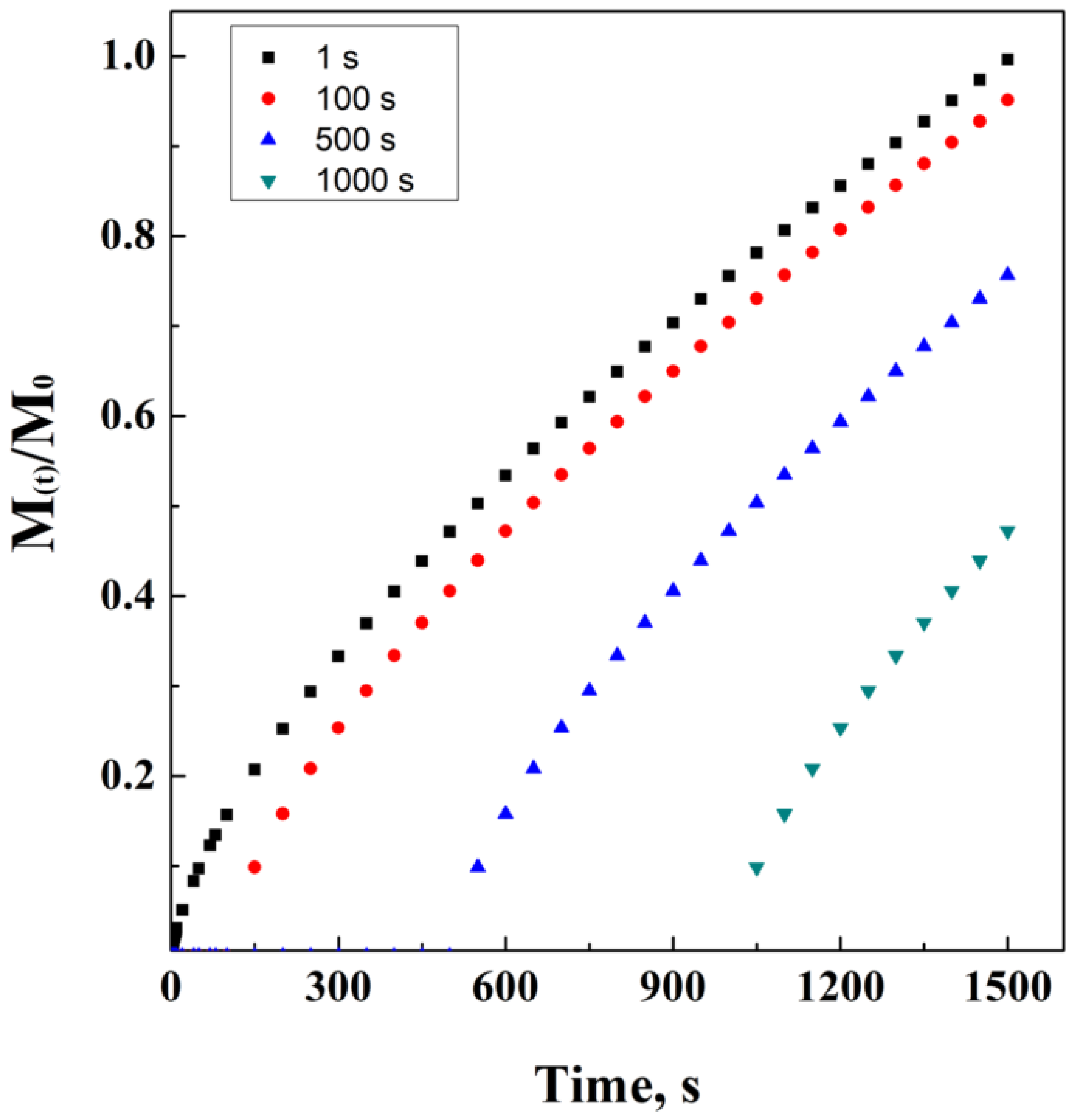
2.6. Hopfenberg’s Model
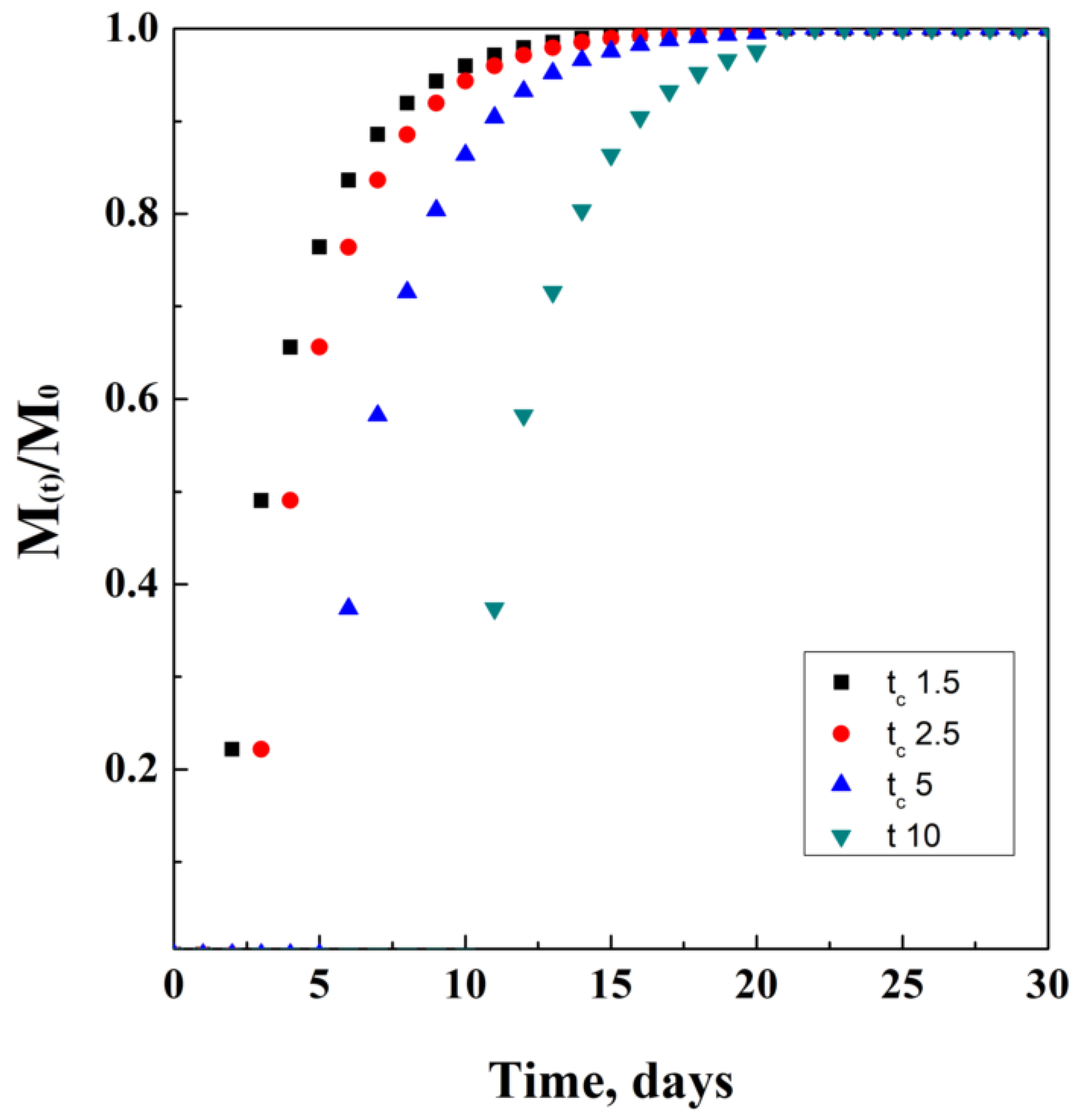
2.7. Corrigan’s Model
2.8. Weibull’s Model
2.9. Sivak Model
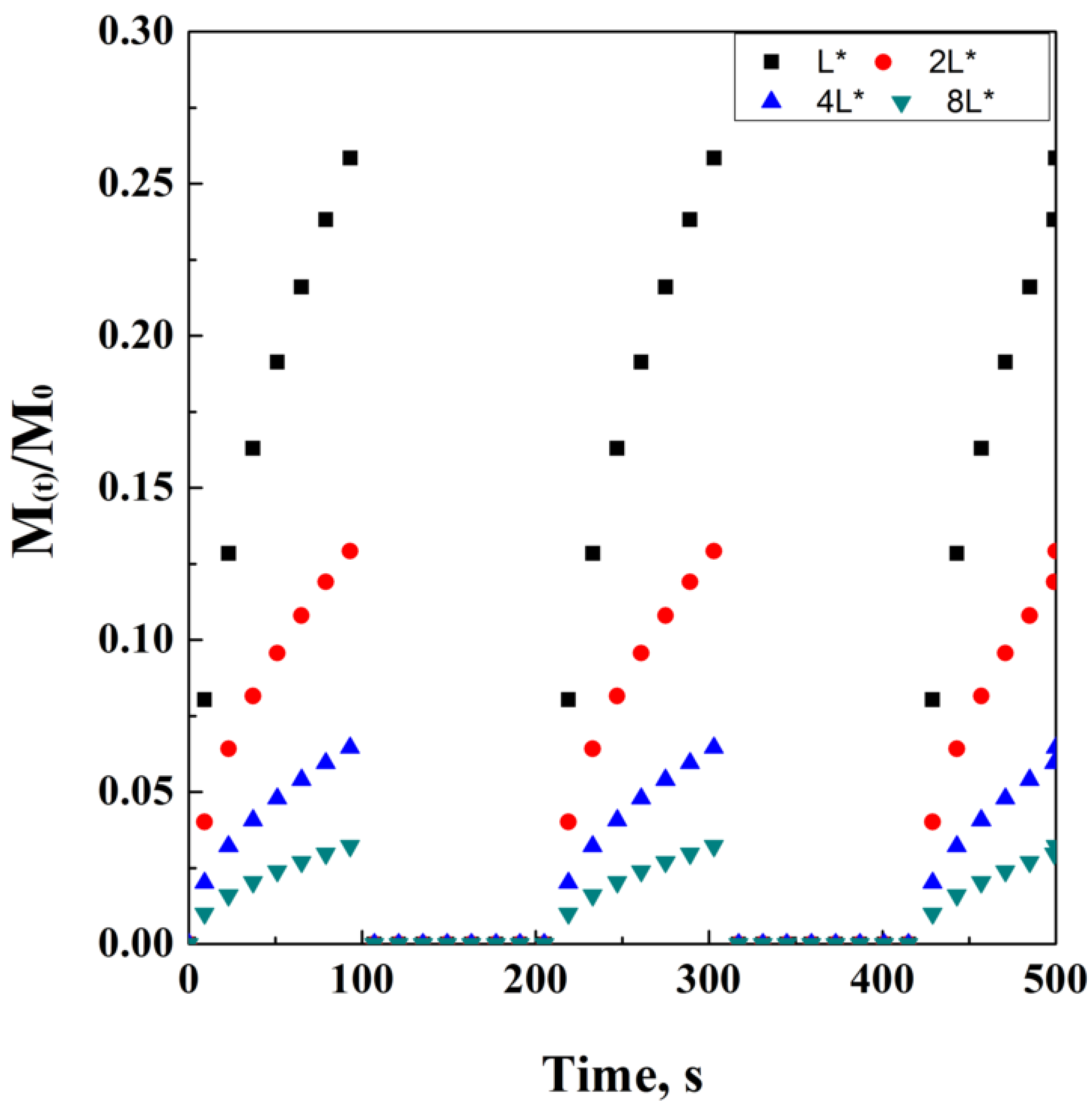
2.10. Pulsatile Drug Delivery
3. Discussion
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| DC | Drug Carriers |
| DDS | Drug Delivery Systems |
| IS | Immune System |
| CPP | Cell-Penetrating-Peptide |
| MEMS | Micro-electro-mechanical Systems |
| UV-Vis | Ultraviolet-Visible |
| SSE | Summed Square Error |
| EE | Encapsulation Efficiency |
| PLGA | poly(lactic-co-glycolic) Acids |
| BSA | Bovine Serum Albumin |
| PLA | Poly-lactic Acid |
| PBS | Phosphate-buffered saline |
References
- Chothe, P.P.; Nakakariya, M.; Rotter, C.J.; Sandoval, P.; Tohyama, K. Recent advances in drug transporter sciences: Highlights from the year 2020. Drug Metab. Rev. 2001, 53, 321–349. [Google Scholar] [CrossRef]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Labhasetwar, V. Nanotech approaches to drug delivery and imaging. Drug Discov. Today 2003, 8, 1112–1120. [Google Scholar]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [Green Version]
- Coelho, J.; Trucillo, P.; Nobre, B.; Palavra, A.F.; Campardelli, R.; Reverchon, E. Extraction and bioprocessing with supercritical fluids. Phys. Sci. Rev. 2020, 5, 1–14. [Google Scholar] [CrossRef]
- Balasubramanian, V.; Onaca, O.; Enea, R.; Hughes, D.W.; Palivan, C.G. Protein delivery: From conventional drug delivery carriers to polymeric nanoreactors. Expert Opin. Drug Deliv. 2010, 7, 63–78. [Google Scholar] [CrossRef]
- D’Angelo, I.; Conte, C.; La Rotonda, M.I.; Miro, A.; Quaglia, F.; Ungaro, F. Improving the efficacy of inhaled drugs in cystic fibrosis: Challenges and emerging drug delivery strategies. Adv. Drug Deliv. Rev. 2014, 75, 92–111. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinís, M.Á.; Rodríguez-Gascón, A.; Almeida, A.J.; Préat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 143–161. [Google Scholar] [CrossRef]
- Jahromi, L.P.; Ghazali, M.; Ashrafi, H.; Azadi, A. A comparison of models for the analysis of the kinetics of drug release from PLGA-based nanoparticles. Heliyon 2020, 6, e03451. [Google Scholar] [CrossRef] [Green Version]
- Patra, D.; Sengupta, S.; Duan, W.; Zhang, H.; Pavlick, R.; Sen, A. Intelligent, self-powered, drug delivery systems. Nanoscale 2013, 5, 1273–1283. [Google Scholar] [CrossRef]
- Dutta, R.C. Drug carriers in pharmaceutical design: Promises and progress. Curr. Pharm. Des. 2007, 13, 761–769. [Google Scholar] [CrossRef]
- Alvarez-Lorenzo, C.; Bromberg, L.; Concheiro, A. Light-sensitive intelligent drug delivery systems. Photochem. Photobiol. 2009, 85, 848–860. [Google Scholar] [CrossRef]
- Langer, R.; Peppas, N.A. Advances in biomaterials, drug delivery, and bionanotechnology. AIChE J. 2003, 49, 2990–3006. [Google Scholar] [CrossRef]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2015, 14, 29–44. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.; Langer, R.; Jia, X. Nanostructured materials for applications in drug delivery and tissue engineering. J. Biomater. Sci. 2007, 18, 241–268. [Google Scholar] [CrossRef] [Green Version]
- Trucillo, P. Drug carriers: Classification, administration, release profiles, and industrial approach. Processes 2021, 9, 470. [Google Scholar] [CrossRef]
- Lutz, J.F. Polymerization of oligo (ethylene glycol)(meth) acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3459–3470. [Google Scholar] [CrossRef]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef]
- Davoodi, P.; Lee, L.Y.; Xu, Q.; Sunil, V.; Sun, Y.; Soh, S.; Wang, C.H. Drug delivery systems for programmed and on-demand release. Adv. Drug Deliv. Rev. 2018, 132, 104–138. [Google Scholar] [CrossRef]
- Champion, J.A.; Katare, Y.K.; Mitragotri, S. Particle shape: A new design parameter for micro-and nanoscale drug delivery carriers. J. Control. Release 2007, 121, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Champion, J.A.; Katare, Y.K.; Mitragotri, S. Making polymeric micro-and nanoparticles of complex shapes. Proc. Natl. Acad. Sci. USA 2007, 104, 11901–11904. [Google Scholar] [CrossRef] [Green Version]
- Holder, S.J.; Sommerdijk, N.A. New micellar morphologies from amphiphilic block copolymers: Disks, toroids and bicontinuous micelles. Polym. Chem. 2011, 2, 1018–1028. [Google Scholar] [CrossRef]
- Trucillo, P.; Campardelli, R.; Scognamiglio, M.; Reverchon, E. Control of liposomes diameter at micrometric and nanometric level using a supercritical assisted technique. J. CO2 Util. 2019, 32, 119–127. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, N.; Feng, X. The role of internal and external stimuli in the rational design of skin-specific drug delivery systems. Int. J. Pharm. 2021, 592, 120081. [Google Scholar] [CrossRef]
- Fuller, E.G.; Sun, H.; Dhavalikar, R.D.; Unni, M.; Scheutz, G.M.; Sumerlin, B.S.; Rinaldi, C. Externally triggered heat and drug release from magnetically controlled nanocarriers. ACS Appl. Polym. Mater. 2019, 1, 211–220. [Google Scholar] [CrossRef]
- Choi, S.W.; Zhang, Y.; Xia, Y. A temperature-sensitive drug release system based on phase-change materials. Angew. Chem. Int. Ed. 2010, 49, 7904–7908. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhang, Q.; Wang, J.; Chen, M.; Li, S.; Lin, Z.; Li, J. Tumor-targeted aggregation of pH-sensitive nanocarriers for enhanced retention and rapid intracellular drug release. Polym. Chem. 2014, 5, 5668–5679. [Google Scholar] [CrossRef]
- Luo, L.; Bian, Y.; Liu, Y.; Zhang, X.; Wang, M.; Xing, S.; Li, L.; Gao, D. Combined near infrared photothermal therapy and chemotherapy using gold nanoshells coated liposomes to enhance antitumor effect. Small 2016, 12, 4103–4112. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, Y.; Wang, H.; Wang, J.; Shin, M.C.; Byun, Y.; He, H.; Liang, Y.; Yang, V.C. Curb challenges of the “Trojan Horse” approach: Smart strategies in achieving effective yet safe cell-penetrating peptide-based drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 1299–1315. [Google Scholar] [CrossRef] [Green Version]
- Casalini, T.; Rossi, F.; Lazzari, S.; Perale, G.; Masi, M. Mathematical modeling of PLGA microparticles: From polymer degradation to drug release. Mol. Pharm. 2014, 11, 4036–4048. [Google Scholar] [CrossRef]
- Yun, Y.H.; Lee, B.K.; Park, K. Controlled drug delivery: Historical perspective for the next generation. J. Control. Release 2015, 219, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Bowers, L.D. Therapeutic monitoring for cyclosporine: Difficulties in establishing a therapeutic window. Clin. Biochem. 1991, 24, 81–87. [Google Scholar] [CrossRef]
- Hampton Atkinson, J.; Patel, S.M.; Meyer, J.M.; Slater, M.A.; Zisook, S.; Capparelli, E. Is there a therapeutic window with some antidepressants for analgesic response? Curr. Pain Headache Rep. 2009, 13, 93–99. [Google Scholar] [CrossRef]
- Khera, E.; Thurber, G.M. Pharmacokinetic and immunological considerations for expanding the therapeutic window of next-generation antibody–drug conjugates. BioDrugs 2018, 32, 465–480. [Google Scholar] [CrossRef]
- Williams, D.F. On the mechanisms of biocompatibility. Biomaterials 2008, 29, 2941–2953. [Google Scholar] [CrossRef]
- Kohane, D.S.; Langer, R. Biocompatibility and drug delivery systems. Chem. Sci. 2010, 1, 441–446. [Google Scholar] [CrossRef]
- Tsai, N.C.; Sue, C.Y. Review of MEMS-based drug delivery and dosing systems. Sens. Actuators A Phys. 2007, 134, 555–564. [Google Scholar] [CrossRef]
- Liepmann, D.; Pisano, A.P.; Sage, B. Microelectromechanical systems technology to deliver insulin. Diabetes Technol. Ther. 1999, 1, 469–476. [Google Scholar] [CrossRef]
- Hultström, M.; Roxhed, N.; Nordquist, L. Intradermal insulin delivery: A promising future for diabetes management. J. Diabetes Sci. Technol. 2014, 8, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Trevitt, S.; Simpson, S.; Wood, A. Artificial pancreas device systems for the closed-loop control of type 1 diabetes: What systems are in development? J. Diabetes Sci. Technol. 2016, 10, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, E.; Ginsburg, G.S.; Silver, M. The personalized medicine coalition. Am. J. Pharm. 2005, 5, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torrez, L.A.; Grillo, R.; Swamy, K.M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Dan, N. Vesicle-based drug carriers: Liposomes, polymersomes, and niosomes. In Design and Development of New Nanocarriers; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 1–55. [Google Scholar]
- Brandl, M. Liposomes as drug carriers: A technological approach. Biotechnol. Annu. Rev. 2001, 7, 59–85. [Google Scholar] [PubMed]
- Torchilin, V.P. Liposomes as targetable drug carriers. Crit. Rev. Ther. Drug Carr. Syst. 1985, 2, 65–115. [Google Scholar]
- Namdeo, A.; Jain, N.K. Niosomes as drug carriers. Indian J. Pharm. Sci. 1996, 58, 41–47. [Google Scholar]
- Shahiwala, A.; Misra, A. Studies in topical application of niosomally entrapped nimesulide. J. Pharm. Pharm. Sci. 2002, 5, 220–225. [Google Scholar] [PubMed]
- Wissing, S.A.; Kayser, O.; Müller, R.H. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Feijen, J. Polymersomes for drug delivery: Design, formation and characterization. J. Control. Release 2012, 161, 473–483. [Google Scholar] [CrossRef]
- Yu, C.Y.; Jia, L.H.; Yin, B.C.; Zhang, X.Z.; Cheng, S.X.; Zhuo, R.X. Fabrication of nanospheres and vesicles as drug carriers by self-assembly of alginate. J. Phys. Chem. C 2008, 112, 16774–16778. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.; Wang, Y.; Zhuo, R.X.; Zhang, X.Z. Controllable exploding microcapsules as drug carriers. Chem. Commun. 2011, 47, 4457–4459. [Google Scholar] [CrossRef]
- Ran, R.; Sun, Q.; Baby, T.; Wibowo, D.; Middelberg, A.P.; Zhao, C.X. Multiphase microfluidic synthesis of micro-and nanostructures for pharmaceutical applications. Chem. Eng. Sci. 2017, 169, 78–96. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, P.; Famili, A.; Nagapudi, K.; Al-Sayah, M.A. Using supercritical fluid technology as a green alternative during the preparation of drug delivery systems. Pharmaceutics 2019, 11, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovskaya, D.D.; Lebedev, A.E.; Menshutina, N.V. Aerogels as drug delivery systems: In vitro and in vivo evaluations. J. Supercrit. Fluids 2015, 106, 115–121. [Google Scholar] [CrossRef]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Hydrogels as drug delivery systems; pros and cons. Trends Pharm. Sci. 2019, 5, 7–24. [Google Scholar]
- Zhao, S.; Malfait, W.J.; Guerrero-Alburquerque, N.; Koebel, M.M.; Nyström, G. Biopolymer aerogels and foams: Chemistry, properties, and applications. Angew. Chem. Int. Ed. 2018, 57, 7580–7608. [Google Scholar] [CrossRef]
- Yousefi, M.; Narmani, A.; Jafari, S.M. Dendrimers as efficient nanocarriers for the protection and delivery of bioactive phytochemicals. Adv. Colloid Interface Sci. 2020, 278, 102125. [Google Scholar] [CrossRef]
- García-González, C.A.; Sosnik, A.; Kalmár, J.; De Marco, I.; Erkey, C.; Concheiro, A.; Alvarez-Lorenzo, C. Aerogels in drug delivery: From design to application. J. Control. Release 2021, 332, 40–63. [Google Scholar] [CrossRef]
- Tamarkin, D.; Friedman, D.; Shemer, A. Emollient foam in topical drug delivery. Expert Opin. Drug Deliv. 2006, 3, 799–807. [Google Scholar] [CrossRef]
- Trucillo, P.; Di Maio, E. Classification and Production of Polymeric Foams among the Systems for Wound Treatment. Polymers 2021, 13, 1608. [Google Scholar] [CrossRef]
- Marrazzo, C.; Maio, E.; Iannace, S. Foaming of synthetic and natural biodegradable polymers. J. Cell. Plast. 2007, 43, 123–133. [Google Scholar] [CrossRef]
- Marrazzo, C.; Di Maio, E.; Iannace, S.; Nicolais, L. Process-structure relationships in PCL foaming. J. Cell. Plast. 2008, 44, 37–52. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Md, S.; Sahni, J.K.; Baboota, S.; Dang, S.; Ali, J. Nanostructured lipid carriers system: Recent advances in drug delivery. J. Drug Target. 2012, 20, 813–830. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.S.; Zunino, P. A mixture model for water uptake, degradation, erosion and drug release from polydisperse polymeric networks. Biomaterials 2010, 31, 3032–3042. [Google Scholar] [PubMed]
- Raza, S.N.; Khan, N.A. Role of mathematical modelling in controlled release drug delivery. Int. J. Med. Res. Pharm. Sci 2017, 4, 84–95. [Google Scholar]
- Grassi, M.; Grassi, G. Mathematical modelling and controlled drug delivery: Matrix systems. Curr. Drug Deliv. 2005, 2, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar]
- Arifin, D.Y.; Lee, L.Y.; Wang, C.H. Mathematical modeling and simulation of drug release from microspheres: Implications to drug delivery systems. Adv. Drug Deliv. Rev. 2006, 58, 1274–1325. [Google Scholar]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef]
- Siepmann, J.; Siegel, R.A.; Siepmann, F. Diffusion controlled drug delivery systems. In Fundamentals and Applications of Controlled Release Drug Delivery; Springer: Boston, MA, USA, 2012; pp. 127–152. [Google Scholar]
- Sripetch, S.; Loftsson, T. Topical drug delivery to the posterior segment of the eye: Thermodynamic considerations. Int. J. Pharm. 2021, 597, 120332. [Google Scholar] [CrossRef]
- Serra, L.; Doménech, J.; Peppas, N.A. Engineering design and molecular dynamics of mucoadhesive drug delivery systems as targeting agents. Eur. J. Pharm. Biopharm. 2009, 71, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2012, 64, 163–174. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Hydrophilic matrices for controlled drug delivery: An improved mathematical model to predict the resulting drug release kinetics (the “sequential layer” model). Pharm. Res. 2000, 17, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Muschert, S.; Siepmann, F.; Leclercq, B.; Carlin, B.; Siepmann, J. Prediction of drug release from ethylcellulose coated pellets. J. Control. Release 2009, 135, 71–79. [Google Scholar] [CrossRef]
- Kedem, O.; Katchalsky, A. Thermodynamic analysis of the permeability of biological membranes to non-electrolytes. Biochim. Et Biophys. Acta 1958, 27, 229–246. [Google Scholar] [CrossRef]
- Peppas, N.A.; Narasimhan, B. Mathematical models in drug delivery: How modeling has shaped the way we design new drug delivery systems. J. Control. Release 2014, 190, 75–81. [Google Scholar] [CrossRef]
- Verma, R.K.; Mishra, B.; Garg, S. Osmotically controlled oral drug delivery. Drug Dev. Ind. Pharm. 2000, 26, 695–708. [Google Scholar] [CrossRef]
- Gupta, B.P.; Thakur, N.; Jain, N.P.; Banweer, J.; Jain, S. Osmotically controlled drug delivery system with associated drugs. J. Pharm. Pharm. Sci. 2010, 13, 571–588. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.R. Elaborations on the Higuchi model for drug delivery. Int. J. Pharm. 2011, 418, 13–17. [Google Scholar] [CrossRef]
- Ramteke, K.H.; Dighe, P.A.; Kharat, A.R.; Patil, S.V. Mathematical models of drug dissolution: A review. Sch. Acad. J. Pharm. 2014, 3, 388–396. [Google Scholar]
- Singhvi, G.; Singh, M. In-vitro drug release characterization models. Int. J. Pharm. Stud. Res. 2011, 2, 77–84. [Google Scholar]
- Rehman, Q.; Akash, M.S.H.; Rasool, M.F.; Rehman, K. Role of Kinetic Models in Drug Stability. In Drug Stability and Chemical Kinetics; Springer: Singapore, 2020; pp. 155–165. [Google Scholar]
- Peppas, N.A. A model of dissolution-controlled solute release from porous drug delivery polymeric systems. J. Biomed. Mater. Res. 1983, 17, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Campelo, P.; Neves-Moreira, F.; Amorim, P.; Almada-Lobo, B. Consistent vehicle routing problem with service level agreements: A case study in the pharmaceutical distribution sector. Eur. J. Oper. Res. 2009, 273, 131–145. [Google Scholar] [CrossRef]
- Colombo, P.; Bettini, R.; Santi, P.; Peppas, N.A. Swellable matrices for controlled drug delivery: Gel-layer behaviour, mechanisms and optimal performance. Pharm. Sci. Technol. Today 2000, 3, 198–204. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Kiortsis, S.; Kachrimanis, K.; Broussali, T.; Malamataris, S. Drug release from tableted wet granulations comprising cellulosic (HPMC or HPC) and hydrophobic component. Eur. J. Pharm. Biopharm. 2005, 59, 73–83. [Google Scholar] [CrossRef]
- Paarakh, M.P.; Jose, P.A.; Setty, C.M.; Christoper, G.P. Release kinetics–concepts and applications. Int. J. Pharm. Res. Technol. 2018, 8, 12–20. [Google Scholar]
- Adrover, M.E.; Pedernera, M.; Bonne, M.; Lebeau, B.; Bucalá, V.; Gallo, L. Synthesis and characterization of mesoporous SBA-15 and SBA-16 as carriers to improve albendazole dissolution rate. Saudi Pharm. J. 2020, 28, 15–24. [Google Scholar] [CrossRef]
- Bhardwaj, S.B.; Shukla, A.J.; Collins, C.C. Effect of varying drug loading on particle size distribution and drug release kinetics of verapamil hydrochloride microspheres prepared with cellulose esters. J. Microencapsul. 1995, 12, 71–81. [Google Scholar] [CrossRef]
- Hopfenberg, H.B. Membranes. In Polymers in Medicine and Surgery; Springer: Boston, MA, USA, 1975; pp. 99–107. [Google Scholar]
- Katzhendler, I.; Hoffman, A.; Goldberger, A.; Friedman, M. Modeling of drug release from erodible tablets. J. Pharm. Sci. 1997, 86, 110–115. [Google Scholar] [CrossRef]
- Corrigan, O.I.; Li, X. Quantifying drug release from PLGA nanoparticulates. Eur. J. Pharm. Sci. 2009, 37, 477–485. [Google Scholar] [CrossRef]
- Coughlan, D.C.; Quilty, F.P.; Corrigan, O.I. Effect of drug physicochemical properties on swelling/deswelling kinetics and pulsatile drug release from thermoresponsive poly (N-isopropylacrylamide) hydrogels. J. Control. Release 2004, 98, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.F.; Corrigan, O.I. Mechanisms governing drug release from poly-α-hydroxy aliphatic esters: Diltiazem base release from poly-lactide-co-glycolide delivery systems. Polym. Deliv. Syst. 1993, 23, 311–326. [Google Scholar]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, K.; Argyrakis, P.; Macheras, P. A reappraisal of drug release laws using Monte Carlo simulations: The prevalence of the Weibull function. Pharm. Res. 2003, 20, 988–995. [Google Scholar] [CrossRef]
- Ghosal, K.; Chandra, A.; Rajabalaya, R.; Chakraborty, S.; Nanda, A. Mathematical modeling of drug release profiles for modified hydrophobic HPMC based gels. Die Pharm.-Int. J. Pharm. Sci. 2012, 67, 147–155. [Google Scholar]
- Sivak, W.N.; Zhang, J.; Petoud, S.; Beckman, E.J. Simultaneous drug release at different rates from biodegradable polyurethane foams. Acta Biomater. 2009, 5, 2398–2408. [Google Scholar] [CrossRef]
- Sivak, W.N. Synthesis and Characterization of Novel Polyurethane Drug Delivery Systems. Ph.D. Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 2007. [Google Scholar]
- Cherng, J.Y.; Hou, T.Y.; Shih, M.F.; Talsma, H.; Hennink, W.E. Polyurethane-based drug delivery systems. Int. J. Pharm. 2013, 450, 145–162. [Google Scholar] [CrossRef]
- De Geest, B.G.; Mehuys, E.; Laekeman, G.; Demeester, J.; De Smedt, S.C. Pulsed drug delivery. Expert Opin. Drug Deliv. 2006, 3, 459–462. [Google Scholar] [CrossRef]
- Marin, A.; Muniruzzaman, M.; Rapoport, N. Acoustic activation of drug delivery from polymeric micelles: Effect of pulsed ultrasound. J. Control. Release 2001, 71, 239–249. [Google Scholar] [CrossRef]
- Kikuchi, A.; Okano, T. Pulsatile drug release control using hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 53–77. [Google Scholar] [CrossRef]
- Göpferich, A. Mechanisms of polymer degradation and erosion. In The Biomaterials: Silver Jubilee Compendium; Elsevier: Amsrerdam, The Netherlands, 1996; pp. 117–128. [Google Scholar]
- Zhu, X.; Braatz, R.D. A mechanistic model for drug release in PLGA biodegradable stent coatings coupled with polymer degradation and erosion. J. Biomed. Mater. Res. Part A 2015, 103, 2269–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göpferich, A.; Tessmar, J. Polyanhydride degradation and erosion. Adv. Drug Deliv. Rev. 2002, 54, 911–931. [Google Scholar] [CrossRef]
- Sevim, K.; Pan, J. A model for hydrolytic degradation and erosion of biodegradable polymers. Acta Biomater. 2018, 66, 192–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lao, L.L.; Peppas, N.A.; Boey, F.Y.C.; Venkatraman, S.S. Modeling of drug release from bulk-degrading polymers. Int. J. Pharm. 2011, 418, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Faisant, N.; Akiki, J.; Richard, J.; Benoit, J.P. Effect of the size of biodegradable microparticles on drug release: Experiment and theory. J. Control. Release 2004, 96, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Batycky, R.P.; Hanes, J.; Langer, R.; Edwards, D.A. A theoretical model of erosion and macromolecular drug release from biodegrading microspheres. J. Pharm. Sci. 1997, 86, 1464–1477. [Google Scholar] [CrossRef]
- Guo, Q.; Knight, P.T.; Mather, P.T. Tailored drug release from biodegradable stent coatings based on hybrid polyurethanes. J. Control. Release 2009, 137, 224–233. [Google Scholar] [CrossRef]
- Siegel, R.A.; Rathbone, M.J. Overview of controlled release mechanisms. Fundam. Appl. Control. Release Drug Deliv. 2012, 2, 19–43. [Google Scholar]
- Bajpai, A.K.; Shukla, S.K.; Bhanu, S.; Kankane, S. Responsive polymers in controlled drug delivery. Prog. Polym. Sci. 2008, 33, 1088–1118. [Google Scholar] [CrossRef]
- Hotha, K.K.; Roychowdhury, S.; Subramanian, V. Drug-excipient interactions: Case studies and overview of drug degradation pathways. Am. J. Anal. Chem. 2016, 7, 107–140. [Google Scholar] [CrossRef] [Green Version]
- von Ahn, A.; Dallegrave, A.; dos Santos, J.H.Z. Evaluation of the Cefalexin Drug Degradation Profile in Pharmaceutical Capsule Forms Based on Forced Degradation Studies. Chromatographia 2022, 85, 263–279. [Google Scholar] [CrossRef]
- Prabhu, S.; Hossainy, S. Modeling of degradation and drug release from a biodegradable stent coating. J. Biomed. Mater. Res. Part A 2007, 80, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, S.; Cameron, R.E. The effect of initial polymer morphology on the degradation and drug release from polyglycolide. Biomaterials 2002, 23, 2401–2409. [Google Scholar] [CrossRef]
- Di Colo, G. Controlled drug release from implantable matrices based on hydrophobia polymers. Biomaterials 1992, 13, 850–856. [Google Scholar] [CrossRef]
- Weaver, J.C.; Vaughan, T.E.; Chizmadzhev, Y. Theory of electrical creation of aqueous pathways across skin transport barriers. Adv. Drug Deliv. Rev. 1999, 35, 21–39. [Google Scholar] [CrossRef]
- Wischke, C.; Behl, M.; Lendlein, A. Drug-releasing shape-memory polymers–the role of morphology, processing effects, and matrix degradation. Expert Opin. Drug Deliv. 2013, 10, 1193–1205. [Google Scholar] [CrossRef]
- Visan, A.I.; Popescu-Pelin, G.; Socol, G. Degradation Behavior of Polymers Used as Coating Materials for Drug Delivery—A Basic Review. Polymers 2021, 13, 1272. [Google Scholar] [CrossRef]
- Proikakis, C.S.; Mamouzelos, N.J.; Tarantili, P.A.; Andreopoulos, A.G. Swelling and hydrolytic degradation of poly (D, L-lactic acid) in aqueous solutions. Polym. Degrad. Stab. 2006, 91, 614–619. [Google Scholar] [CrossRef]
- Kaunisto, E.; Marucci, M.; Borgquist, P.; Axelsson, A. Mechanistic modelling of drug release from polymer-coated and swelling and dissolving polymer matrix systems. Int. J. Pharm. 2011, 418, 54–77. [Google Scholar] [CrossRef]
- Rizwan, M.I.; Damodharan, N. Mathematical modelling of dissolution kinetics in dosage forms. Res. J. Pharm. Technol. 2020, 13, 1339–1345. [Google Scholar]
- Rossi, F.; Castiglione, F.; Ferro, M.; Moioli, M.; Mele, A.; Masi, M. The role of drug–drug interactions in hydrogel delivery systems: Experimental and model study. ChemPhysChem 2016, 17, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Iftime, M.M.; Dobreci, D.L.; Irimiciuc, S.A.; Agop, M.; Petrescu, T.; Doroftei, B. A theoretical mathematical model for assessing diclofenac release from chitosan-based formulations. Drug Deliv. 2020, 27, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Clercq, S.; Temelli, F.; Badens, E. In-Depth Study of Cyclodextrin Complexation with Carotenoids toward the Formation of Enhanced Delivery Systems. Mol. Pharm. 2021, 18, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Comin, L.M.; Temelli, F.; Saldaña, M.D. Barley β-glucan aerogels as a carrier for flax oil via supercritical CO2. J. Food Eng. 2012, 111, 625–631. [Google Scholar] [CrossRef]
- Zhao, L.; Temelli, F. Preparation of liposomes using supercritical carbon dioxide via depressurization of the supercritical phase. J. Food Eng. 2015, 158, 104–112. [Google Scholar] [CrossRef]
- Zhao, L.; Temelli, F.; Curtis, J.M.; Chen, L. Encapsulation of lutein in liposomes using supercritical carbon dioxide. Food Res. Int. 2017, 100, 168–179. [Google Scholar] [CrossRef]
- Li, X.; Sun, H.; Li, H.; Hu, C.; Luo, Y.; Shi, X.; Pich, A. Multi-Responsive Biodegradable Cationic Nanogels for Highly Efficient Treatment of Tumors. Adv. Funct. Mater. 2021, 31, 2100227. [Google Scholar] [CrossRef]
- Liang, K.; Li, Z.; Luo, Y.; Zhang, Q.; Yin, F.; Xu, L.; Chwen, H.; Wang, H. Intelligent Nanocomposites with Intrinsic Blood–Brain-Barrier Crossing Ability Designed for Highly Specific MR Imaging and Sonodynamic Therapy of Glioblastoma. Small 2020, 16, 1906985. [Google Scholar] [CrossRef]
- Trucillo, P.; Martino, M.; Reverchon, E. Supercritical Assisted Production of Lutein-Loaded Liposomes and Modelling of Drug Release. Processes 2021, 9, 1162. [Google Scholar] [CrossRef]
- Sharmeen, S.; Rahman, A.M.; Lubna, M.M.; Salem, K.S.; Islam, R.; Khan, M.A. Polyethylene glycol functionalized carbon nanotubes/gelatin-chitosan nanocomposite: An approach for significant drug release. Bioact. Mater. 2018, 3, 236–244. [Google Scholar] [CrossRef]
- Salgado, M.; Santos, F.; Rodríguez-Rojo, S.; Reis, R.L.; Duarte, A.R.C.; Cocero, M.J. Development of barley and yeast β-glucan aerogels for drug delivery by supercritical fluids. J. CO2 Util. 2017, 22, 262–269. [Google Scholar] [CrossRef]
- Haghiralsadat, F.; Amoabediny, G.; Helder, M.N.; Naderinezhad, S.; Sheikhha, M.H.; Forouzanfar, T.; Zandieh-Doulabi, B. A comprehensive mathematical model of drug release kinetics from nano-liposomes, derived from optimization studies of cationic PEGylated liposomal doxorubicin formulations for drug-gene delivery. Artif. Cells Nanomed. Biotechnol. 2018, 46, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drakoulas, G.; Kokkinos, C.; Fotiadis, D.; Kokkinos, S.; Loukas, K.; Moulas, A.N.; Semertzioglou, A. Coupled FEA Model with Continuum Damage Mechanics for the Degradation of Polymer-based coatings on Drug-Eluting Stents. In Proceedings of the 43rd Annual International Conference of the IEEE Engineering in Medicine & Biology Society, Virtual, 30 October–5 November 2021; pp. 4319–4323. [Google Scholar]
- Yang, Z.; Luo, Y.; Hu, Y.; Liang, K.; He, G.; Chen, Q.; Wang, Q.; Chen, H. Photothermo-Promoted Nanocatalysis Combined with H2S-Mediated Respiration Inhibition for Efficient Cancer Therapy. Adv. Funct. Mater. 2021, 31, 2007991. [Google Scholar] [CrossRef]
- Li, J.; Yu, X.; Jiang, Y.; He, S.; Zhang, Y.; Luo, Y.; Pu, K. Second near-infrared photothermal semiconducting polymer nanoadjuvant for enhanced cancer immunotherapy. Adv. Mater. 2021, 33, 2003458. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yu, T.; Li, X.; Lei, Y.; Li, J.; Wang, X.; Peng, P.; Ni, D.; Wang, X.; Luo, Y. Second near-infrared photothermal-amplified immunotherapy using photoactivatable composite nanostimulators. J. Nanobiotechnol. 2021, 19, 433. [Google Scholar] [CrossRef]
- Zhang, C.; He, S.; Zeng, Z.; Cheng, P.; Pu, K. Smart Nano-PROTACs Reprogram Tumor Microenvironment for Activatable Photo-metabolic Cancer Immunotherapy. Angew. Chem. 2022, 61, e202114957. [Google Scholar]
- Sun, X.; Agate, S.; Salem, K.S.; Lucia, L.; Pal, L. Hydrogel-based sensor networks: Compositions, properties, and applications—A review. ACS Appl. Bio. Mater. 2000, 4, 140–162. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- Elmas, A.; Akyüz, G.; Bergal, A.; Andaç, M.; Andac, O. Mathematical modelling of drug release. Res. Eng. Struct. Mater. 2000, 6, 63–86. [Google Scholar] [CrossRef]
- Quiño, J.; Franke, K.; Ivanova, M.; Kareth, S.; Petermann, M.; Braeuer, A.S. In situ measurement of drug transport in porous silica gel. Microporous Mesoporous Mater. 2018, 260, 17–23. [Google Scholar] [CrossRef]
- Badens, E.; Masmoudi, Y.; Mouahid, A.; Crampon, C. Current situation and perspectives in drug formulation by using supercritical fluid technology. J. Supercrit. Fluids 2018, 134, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Bouledjouidja, A.; Masmoudi, Y.; Sergent, M.; Trivedi, V.; Meniai, A.; Badens, E. Drug loading of foldable commercial intraocular lenses using supercritical impregnation. Int. J. Pharm. 2016, 500, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Metwally, A.A.; Hathout, R.M. Computer-assisted drug formulation design: Novel approach in drug delivery. Mol. Pharm. 2015, 12, 2800–2810. [Google Scholar] [CrossRef] [PubMed]
- Mizera, M.; Muratov, E.N.; Alves, V.M.; Tropsha, A.; Cielecka-Piontek, J. Computer-aided discovery of new solubility-enhancing drug delivery system. Biomolecules 2020, 10, 913. [Google Scholar] [CrossRef]
- Piotto, S.; Di Biasi, L.; Fino, R.; Parisi, R.; Sessa, L. Yada: A novel tool for molecular docking calculations. J. Comput.-Aided Mol. Des. 2016, 30, 753–759. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors and Scientists | Nr. of Full Text Mentions | Use Frequency, % |
|---|---|---|
| Crank and Baker | 2748 | 5 |
| Peppas | 16,117 | 27 |
| Higuchi | 19,243 | 33 |
| Hixson-Crowell | 2094 | 3 |
| Peppas and Sahlin | 640 | 1 |
| Bake & Lonsdale | 1716 | 3 |
| Hopfenberg | 403 | 1 |
| Corrigan | 10,297 | 18 |
| Weibull | 4238 | 7 |
| Sivak | 1047 | 2 |
| Phenomena | Definition | Ref. |
|---|---|---|
| Degradation | Scission of a polymer chain in oligomers or monomers | [110] |
| Bio-degradation | Degradation due to the action of a biological system | [111] |
| Erosion | Mass loss phenomenon from the polymeric interphase | [112] |
| Bio-erosion | Erosion due to the action of a biological system | [113] |
| Phenomena | Reference |
|---|---|
| External water invades the inner core of the matrix | [114,115] |
| Drug degradation | [116,117] |
| Polymer degradation | [118,119] |
| Creation of aqueous pores | [120,121] |
| Degradative phenomena of interference drug/polymer | [122,123] |
| Polymer swelling | [124,125] |
| Drug/polymer adsorption | [126,127] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trucillo, P. Drug Carriers: A Review on the Most Used Mathematical Models for Drug Release. Processes 2022, 10, 1094. https://doi.org/10.3390/pr10061094
Trucillo P. Drug Carriers: A Review on the Most Used Mathematical Models for Drug Release. Processes. 2022; 10(6):1094. https://doi.org/10.3390/pr10061094
Chicago/Turabian StyleTrucillo, Paolo. 2022. "Drug Carriers: A Review on the Most Used Mathematical Models for Drug Release" Processes 10, no. 6: 1094. https://doi.org/10.3390/pr10061094
APA StyleTrucillo, P. (2022). Drug Carriers: A Review on the Most Used Mathematical Models for Drug Release. Processes, 10(6), 1094. https://doi.org/10.3390/pr10061094