
Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
Enzymes
2.2. Methods
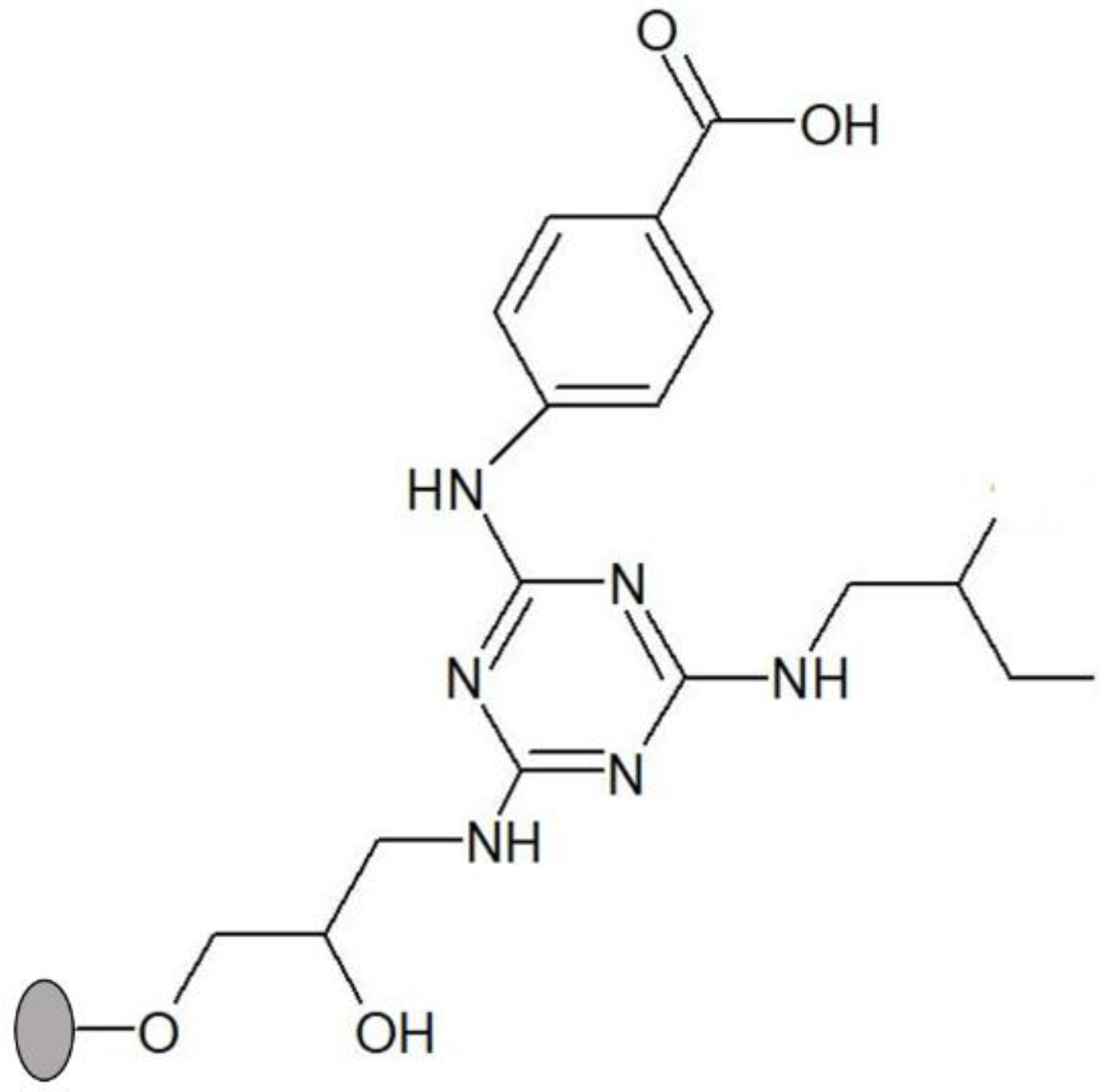
2.2.1. Solid-Phase Synthesis of Ligand 3′/11
Preparation of Dichlorotriazinyl Agarose
Nucleophilic Substitution of Chlorine Atoms with Selected Amines
2.2.2. Enzyme Adsorption to Solid-Phase Synthesized Ligands
2.2.3. Thermostability Assays with Free and Adsorbed Enzymes
2.2.4. Enzyme Activity Assays
Lipase Activity Assays
Invertase Activity Assays
2.2.5. Protein Determination by the BCA Assay
3. Results and Discussion
3.1. Characterization of Enzyme Preparations by SDS-PAGE
3.2. Enzymatic Activity Screening
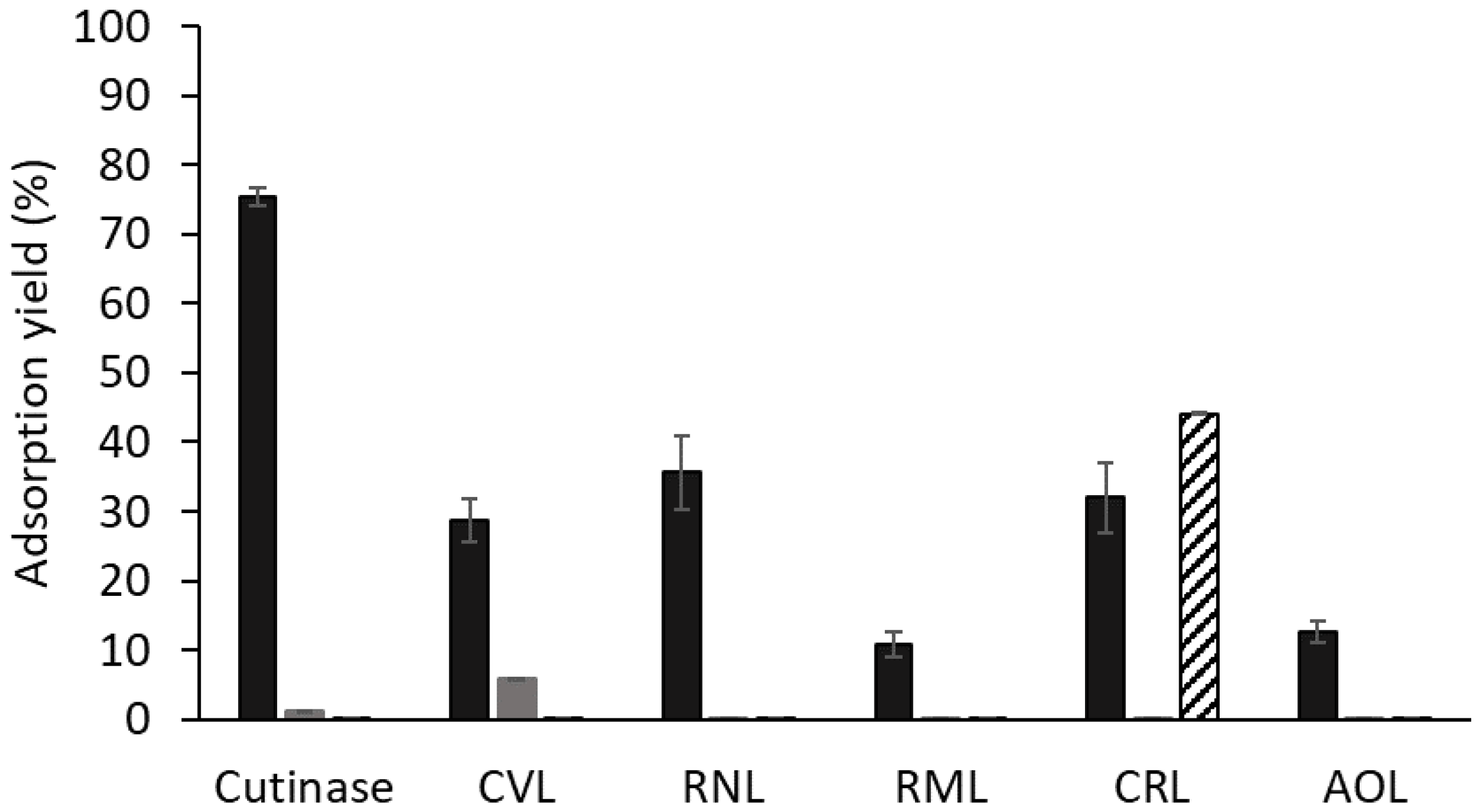
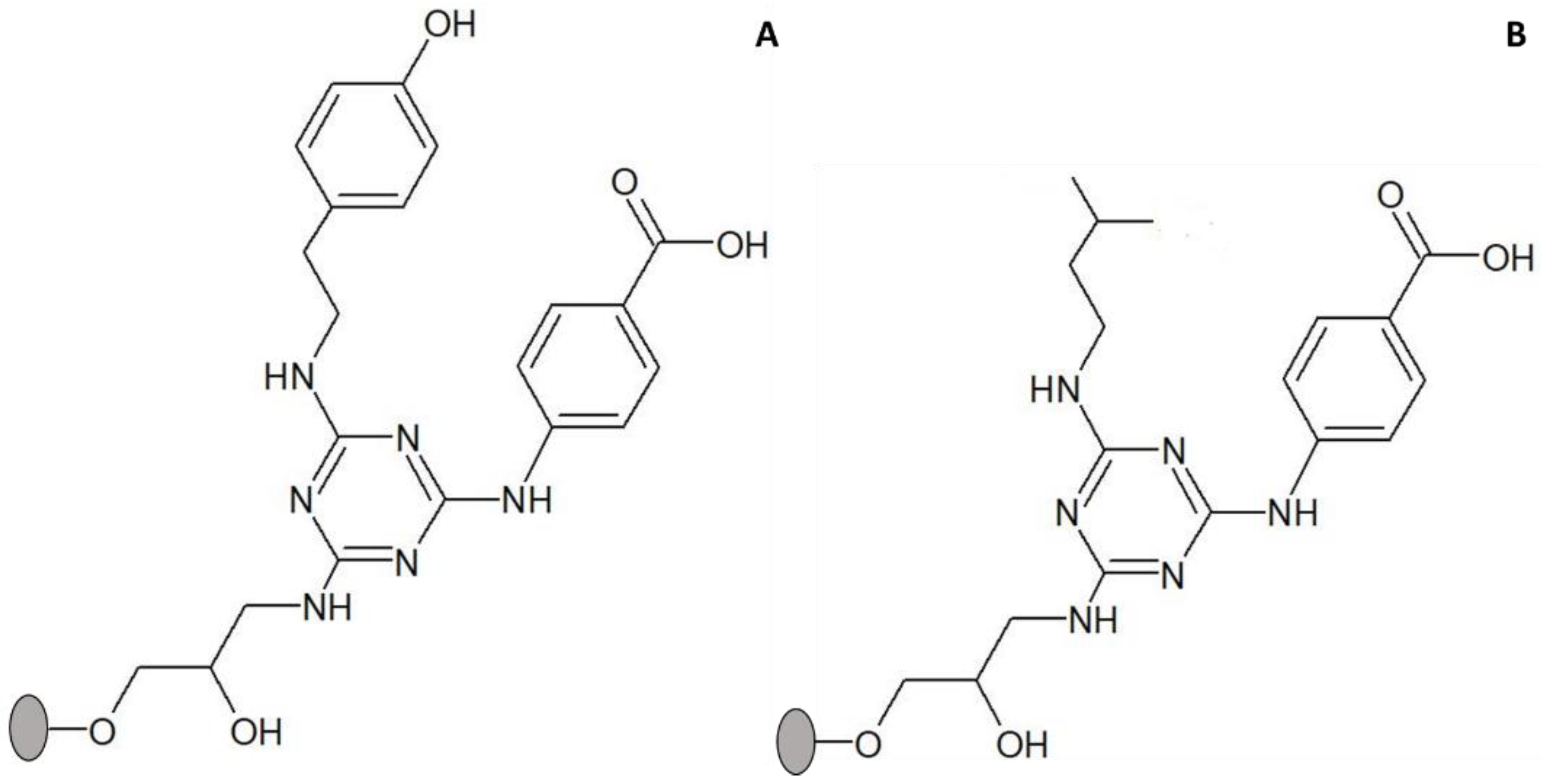
3.3. Assessment of Binding of Ligand 3′/11 to Different Lipases
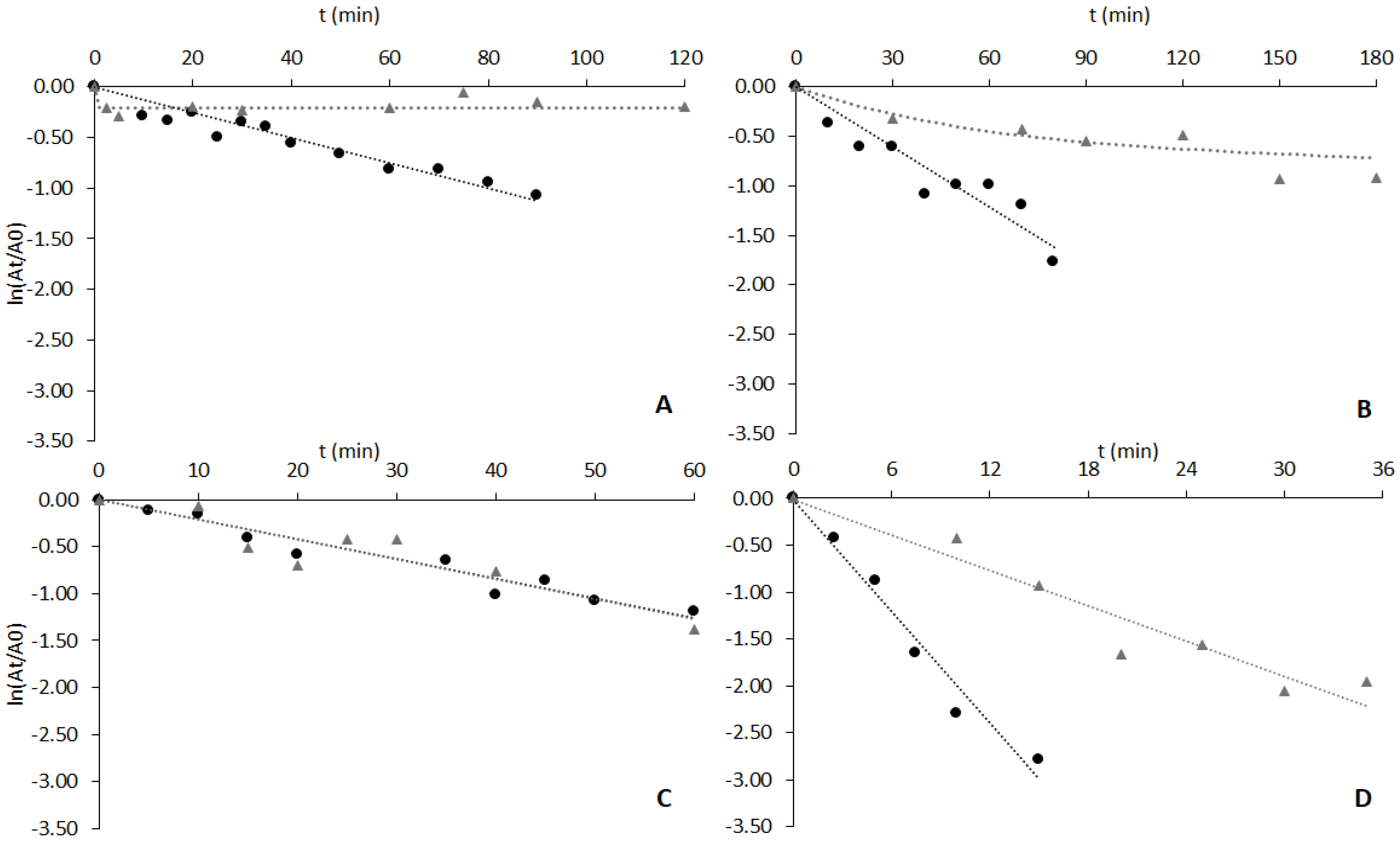
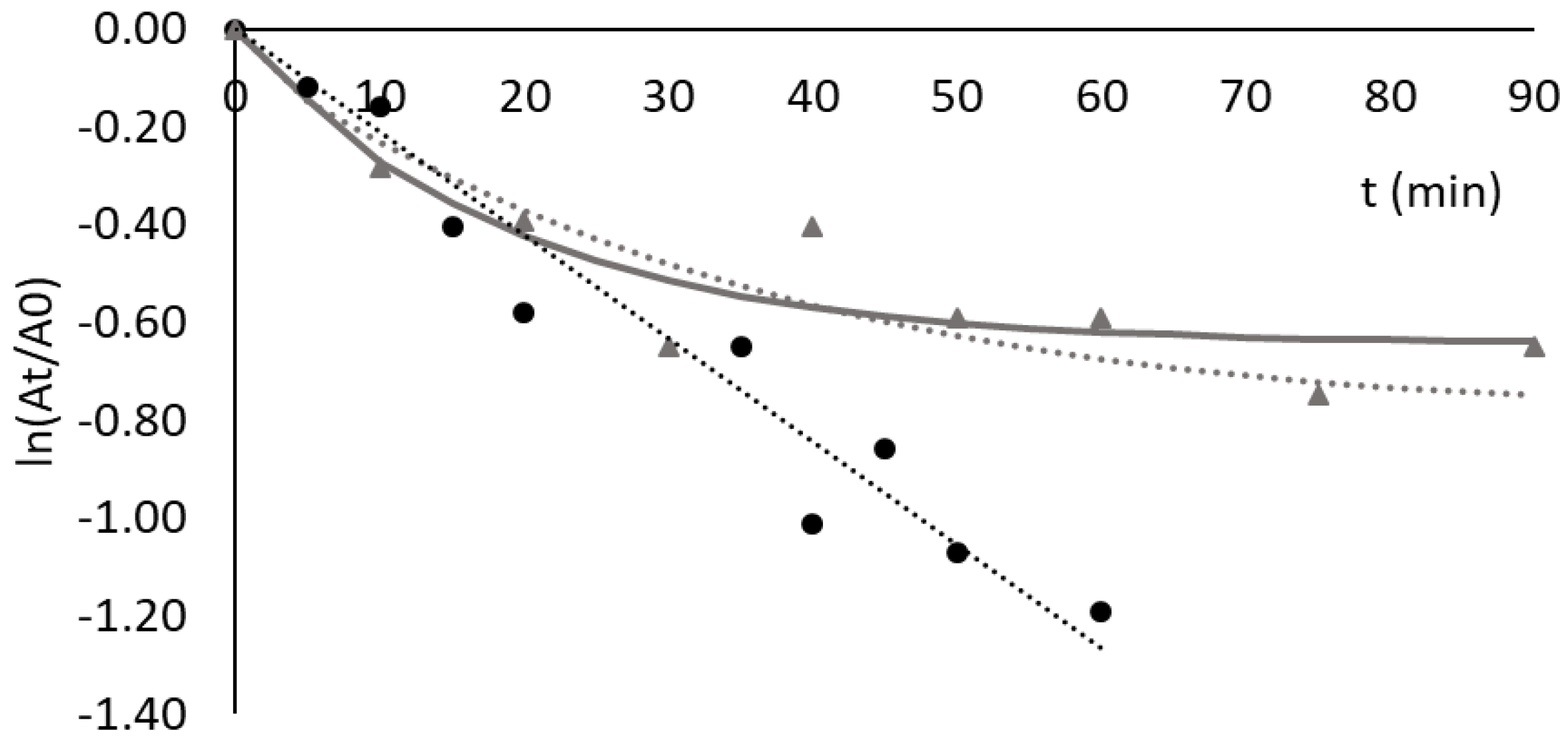
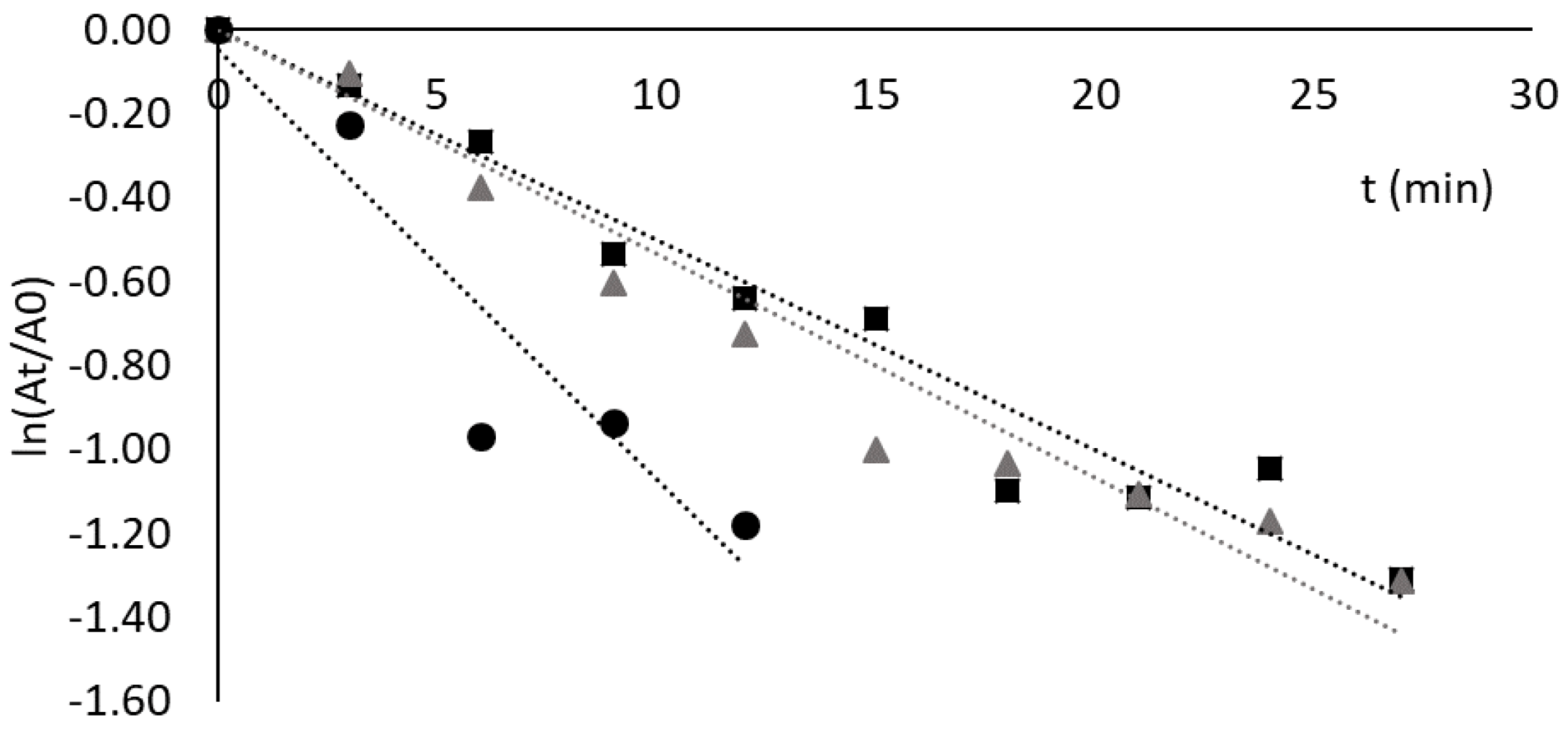
3.4. Thermal Stability of Lipases Free and Adsorbed to Ligand 3′/11
3.5. Adsorption and Thermostability Assays with CRL Adsorbed to Other Dipeptide-Mimic Ligands
3.6. Adsorption and Thermostability of Invertase Free and Adsorbed to Different Biomimetic Ligands
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Petersen, S.B.; Harald Jonson, P.; Fojan, P.; Petersen, E.I.; Neves Petersen, M.T.; Hansen, S.; Ishak, R.J.; Hough, E. Protein engineering the surface of enzymes. J. Biotechnol. 1998, 66, 11–26. [Google Scholar] [CrossRef]
- Jaenicke, R. Stability and stabilization of globular proteins in solution. J. Biotechnol. 2000, 79, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Melo, E.P.; Faria, T.Q.; Martins, L.O.; Gonçalves, A.M.; Cabral, J.M. Cutinase unfolding and stabilization by trehalose and mannosylglycerate. Proteins 2001, 42, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Pyne, P.; Samanta, N.; Ebbinghaus, S.; Mitra, R.K. Thermal stability modulation of the native and chemically-unfolded state of bovine serum albumin by amino acids. Phys. Chem. Chem. Phys. 2019, 22, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszyńska, D. Immobilization as a strategy for improving enzyme properties-application to oxidoreductases. Molecules 2014, 19, 8995–9018. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, L.; Pedrero, S.G.; Lopez-Carrobles, N.; Gorines, B.C.; Virgen-Ortíz, J.J.; Fernandez-Lafuente, R. Effect of protein load on stability of immobilized enzymes. Enzym. Microb. Technol. 2017, 98, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Boudrant, J.; Woodley, J.M.; Fernandez-Lafuente, R. Parameters necessary to define an immobilized enzyme preparation. Process Biochem. 2020, 90, 66–80. [Google Scholar] [CrossRef]
- Bilal, M.; Zhao, Y.; Rasheed, T.; Iqbal, H.M.N. Magnetic nanoparticles as versatile carriers for enzymes immobilization: A review. Int. J. Biol. Macromol. 2018, 120, 2530–2544. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.; Ripoll, M.; Jackson, E.; Grazú, V.; Betancor, L. Stabilization of b-Glucuronidase by Immobilization in Magnetic-Silica Hybrid Supports. Catalysts 2020, 10, 669. [Google Scholar] [CrossRef]
- Mansfeld, J.; Vriend, G.; Van den Burg, B.; Eijsink, V.G.H.; Ulbrich-Hofmann, R. Probing the unfolding region in a thermolysin-like protease by site-specific immobilization. Biochemistry 1999, 38, 8240–8245. [Google Scholar] [CrossRef]
- Mansfeld, J.; Ulbrich-Hofmann, R. Site-specific and random immobilization of thermolysin-like proteases reflected in the thermal inactivation kinetics. Biotechnol. Appl. Biochem. 2000, 32, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Nidetzky, B. Positively charged mini-protein zbasic2 as a highly efficient silica binding module: Opportunities for enzyme immobilization on unmodified silica supports. Langmuir 2012, 28, 10040–10049. [Google Scholar] [CrossRef] [PubMed]
- García-García, P.; Guisan, J.M.; Fernandez-Lorente, G. A mild intensity of the enzyme-support multi-point attachment promotes the optimal stabilization of mesophilic multimeric enzymes: Amine oxidase from Pisum sativum. J. Biotechnol. 2020, 318, 39–44. [Google Scholar] [CrossRef]
- Matos, M.J.B.; Pina, A.S.; Roque, A.C.A. Rational design of affinity ligands for bioseparation. J. Chromatogr. A 2020, 1619, 460871. [Google Scholar] [CrossRef]
- Ye, L.; Xu, A.; Cheng, C.; Zhang, L.; Huo, C.; Huang, F.; Xu, H.; Li, R. Design and synthesis of affinity ligands and relation of their structure with adsorption of proteins. J. Sep. Sci. 2011, 34, 3145–3150. [Google Scholar] [CrossRef]
- Kim, J.Y.H.; Lee, J.W.; Lee, W.S.; Ha, H.-H.; Vendrell, M.; Bork, J.T.; Lee, Y.; Chang, Y.-T. Combinatorial Solid-Phase Synthesis of 4,6-Diaryl and 4-Aryl, 6-Alkyl-1,3,5-triazines and Their Application to Efficient Biofuel Production. ACS Comb. Sci. 2012, 14, 395–398. [Google Scholar] [CrossRef]
- Ruiu, L.; Roque, A.C.A.; Taipa, M.A.; Lowe, C.R. De novo design, synthesis and screening of a combinatorial library of complementary ligands directed towards the surface of cutinase from Fusarium solani pisi. J. Mol. Recognit. 2006, 19, 372–378. [Google Scholar] [CrossRef]
- Sousa, I.T.; Ruiu, L.; Lowe, C.R.; Taipa, M.A. Synthetic affinity ligands as a novel tool to improve protein stability. J. Mol. Recognit. 2009, 22, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sousa, I.T.; Lourenço, N.M.T.; Afonso, C.A.M.; Taipa, M.A. Protein stabilization with a dipeptide-mimic triazine-scaffolded synthetic affinity ligand. J. Mol. Recognit. 2013, 26, 104–112. [Google Scholar] [CrossRef]
- Chen, S.; Su, L.; Chen, J.; Wu, J. Cutinase: Characteristics, preparation, and application. Biotechnol. Adv. 2013, 31, 1754–1767. [Google Scholar] [CrossRef]
- Roussel, A.; Amara, S.; Nyyssölä, A.; Mateos-Diaz, E.; Blangy, S.; Kontkanen, H.; Westerholm-Parvinen, A.; Carrière, F.; Cambillau, C. A cutinase from Trichoderma reesei with a lid-covered active site and kinetic properties of true lipases. J. Mol. Biol. 2014, 426, 3757–3772. [Google Scholar] [CrossRef]
- Creveld, L.D.; Amadei, A.; van Schaik, R.C.; Pepermans, H.A.M.; de Vlieg, J.; Berendsen, H.J.C. Identification of functional and unfolding motions of cutinase as obtained from molecular dynamics computer simulations. Proteins Struct. Funct. Bioinform. 1998, 33, 253–264. [Google Scholar] [CrossRef]
- Sousa, I.T.; Taipa, M.A. Biomimetic Affinity Ligands for Protein Purification. In Protein Downstream Processing: Design, Development and Application of High and Low-Resolution Methods; Labrou, N., Ed.; Methods in Molecular Biology Series; Springer Science: Berlin/Heidelberg, Germany, 2021; Volume 1129, Chapter 14; pp. 231–262. [Google Scholar] [CrossRef]
- Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules 2016, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Rodríguez, K.; Martínez, R. Integrating enzyme immobilization and protein engineering: An alternative path for the development of novel and improved industrial biocatalysts. Biotechnol. Adv. 2018, 36, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Sebastião, M.J.; Cabral, J.M.S.; Aires-Barros, M.R. Improved purification protocol of a Fusarium solani pisi recombinant cutinase by phase partitioning in aqueous two-phase systems of polyethylene glycol and phosphate. Enzym. Microb. Technol. 1996, 18, 251–260. [Google Scholar] [CrossRef]
- Henley, J.P.; Sadana, A. Categorization of enzyme deactivations using a series-type mechanism. Enzym. Microb. Technol. 1985, 7, 50–60. [Google Scholar] [CrossRef]
- Aymard, C.; Belarbi, A. Kinetics of thermal deactivation of enzymes: A simple three parameters phenomenological model can describe the decay of enzyme activity, irrespectively of the mechanism. Enzym. Microb. Technol. 2000, 27, 612–618. [Google Scholar] [CrossRef]
- Correa, S.; Puertas, S.; Gutiérrez, L.; Asín, L.; de la Fuente, J.M.; Grazú, V.; Betancor, L. Design of stable magnetic hybrid nanoparticles of Si-entrapped HRP. PLoS ONE 2019, 14, e0214004. [Google Scholar] [CrossRef]
- Gonçalves, A.M.; Serro, A.P.; Aires-Barros, M.R.; Cabral, J.M.S. Effects of ionic surfactants used in reversed micelles on cutinase activity and stability. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 2000, 1480, 92–106. [Google Scholar] [CrossRef]
- Vorderwülbecke, T.; Kieslich, K.; Erdmann, H. Comparison of lipases by different assays. Enzym. Microb. Technol. 1992, 14, 631–639. [Google Scholar] [CrossRef]
- Nunes, M.A.P.; Vila-Real, H.; Fernandes, P.C.B.; Ribeiro, M.H.L. Immobilization of Naringinase in PVA–Alginate Matrix Using an Innovative Technique. Appl. Biochem. Biotechnol. 2010, 160, 2129–2147. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S.; Czjzek, M.; Lamzin, V.; Nicolas, A.; Cambillau, C. Atomic resolution (1.0 A) crystal structure of Fusarium solani cutinase: Stereochemical analysis. J. Mol. Biol. 1997, 268, 779–799. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.B.; Fojan, P.; Petersen, E.I.; Petersen, M.T.N. The thermal stability of the Fusarium solani pisi cutinase as a function of pH. BioMed Res. Int. 2001, 1, 62–69. [Google Scholar] [CrossRef]
- Dutta, K.; Sen, S.; Veeranki, V.D. Production, characterization and applications of microbial cutinases. Process Biochem. 2009, 44, 127–134. [Google Scholar] [CrossRef]
- Martínez, A.; Maicas, S. Cutinases: Characteristics and Insights in Industrial Production. Catalysts 2021, 11, 1194. [Google Scholar] [CrossRef]
- Nyyssölä, A. Which properties of cutinases are important for applications? Appl. Microbiol. Biotechnol. 2015, 99, 4931–4942. [Google Scholar] [CrossRef] [PubMed]
- Nikolaivits, E.; Kanelli, M.; Dimarogona, M.; Topakas, E. A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases. Catalysts 2018, 8, 612. [Google Scholar] [CrossRef]
- Martinez, C.; De Geus, P.; Lauwereys, M.; Matthyssens, G.; Cambillau, C. Fusarium solani cutinase is a lipolytic enzyme with a catalytic serine accessible to solvent. Nature 1992, 356, 615–618. [Google Scholar] [CrossRef]
- Egmond, M.R.; de Vlieg, J. Fusarium solani pisi cutinase. Biochimie 2000, 82, 1015–1021. [Google Scholar] [CrossRef]
- Longhi, S.; Cambillau, C. Structure-activity of cutinase, a small lipolytic enzyme. Biochim. Biophys. Acta 1999, 1441, 185–196. [Google Scholar] [CrossRef]
- Lang, D.; Hofmann, B.; Haalck, L.; Hecht, H.-J.; Spener, F.; Schmid, R.D.; Schomburg, D. Crystal Structure of a Bacterial Lipase from Chromobacterium viscosum ATCC 6918 Refined at 1.6 Å Resolution. J. Mol. Biol. 1996, 259, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Toida, J.; Kondoh, K.; Fukuzawa, M.; Ohnishi, K.; Sekiguchi, J. Purification and Characterization of a Lipase from Aspergillus oryzae. Biosci. Biotechnol. Biochem. 1995, 59, 1199–1203. [Google Scholar] [CrossRef]
- Wu, X.Y.; JÄÄskelÄinen, S.; Linko, W.Y. Purification and partial characterization of Rhizomucor miehei lipase for ester synthesis. Appl. Biochem. Biotechnol. 1996, 59, 145–158. [Google Scholar] [CrossRef]
- Kohno, M.; Kugimiya, W.; Hashimoto, Y.; Morita, Y. Purification, characterization, and crystallization of two types of lipase from Rhizopus niveus. Biosci. Biotechnol. Biochem. 1994, 58, 1007–1012. [Google Scholar] [CrossRef]
- Benjamin, S.; Pandey, A. Candida rugosa lipases: Molecular biology and versatility in biotechnology. Yeast 1998, 14, 1069–1087. [Google Scholar] [CrossRef]
- Ghosh, P.K.; Saxena, R.K.; Gupta, R.; Yadav, R.P.; Davidson, S. Microbial lipases: Production and applications. Sci. Prog. 1996, 79 Pt 2, 119–157. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Hwang, S.; Chang, S.B.; Ku, J.; Chung, D.S. Acid dissociation constants of melamine derivatives from density functional theory calculations. J. Phys. Chem. A 2009, 113, 13036–13040. [Google Scholar] [CrossRef]
- Petersen, M.T.; Martel, P.; Petersen, E.I.; Drabløs, F.; Petersen, S.B. Surface and electrostatics of cutinases. Methods Enzym. 1997, 284, 130–154. [Google Scholar] [CrossRef]
- Palanisamy, U.D.; Winzor, D.J.; Lowe, C.R. Synthesis and evaluation of affinity adsorbents for glycoproteins: An artificial lectin. J. Chromatogr. B Biomed. Sci. Appl. 2000, 746, 265–281. [Google Scholar] [CrossRef]
- Scoble, J.A.; Scopes, R.K. Ligand structure of the divinylsulfone-based T-gel. J. Chromatogr. A 1997, 787, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Soares, C.M.F.; De Castro, H.F.; De Moraes, F.F.; Zanin, G.M. Characterization and utilization of Candida rugosa lipase immobilized on controlled pore silica. Appl. Biochem. Biotechnol. 1999, 79, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Enatsu, M.; Takee, R.; Kugimiya, W. Thermal stability of Rhizopus niveus lipase expressed in a kex2 mutant yeast. J. Biotechnol. 2000, 81, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef]
- Danisman, T.; Tan, S.; Kacar, Y.; Ergene, A. Covalent immobilization of invertase on microporous pHEMA–GMA membrane. Food Chem. 2004, 85, 461–466. [Google Scholar] [CrossRef]
- Andjelković, U.; Pićurić, S.; Vujčić, Z. Purification and characterisation of Saccharomyces cerevisiae external invertase isoforms. Food Chem. 2010, 120, 799–804. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Selected pH | Selected Temperature (°C) | Specific Activity (U/mg) |
|---|---|---|---|
| AOL | 8.0 | 37 | 3.4 ± 0.5 |
| CRL | 0.71 ± 0.07 | ||
| Cutinase * | 160 ± 70 | ||
| CVL | 30 | 98.5 ± 0.9 | |
| RML | 7.0 | 37 | 1.9 ± 0.5 |
| RNL | 8.0 | 0.0352 ± 0.0008 |
| Enzyme | ηprot (%) | ηact (%) |
|---|---|---|
| Cutinase | 75 ± 1 | 100 ± 0 |
| CVL | 29 ± 3 | 14.7 ± 0.6 |
| CRL | 36 ± 5 | 70.9 ± 0.6 |
| RNL | 32 ± 5 | ≈100 ± 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira-Faria, D.; Taipa, M.Â. Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment. Processes 2024, 12, 371. https://doi.org/10.3390/pr12020371
Ferreira-Faria D, Taipa MÂ. Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment. Processes. 2024; 12(2):371. https://doi.org/10.3390/pr12020371
Chicago/Turabian StyleFerreira-Faria, Diogo, and M. Ângela Taipa. 2024. "Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment" Processes 12, no. 2: 371. https://doi.org/10.3390/pr12020371
APA StyleFerreira-Faria, D., & Taipa, M. Â. (2024). Thermal Stabilization of Lipases Bound to Solid-Phase Triazine-Scaffolded Biomimetic Ligands: A Preliminary Assessment. Processes, 12(2), 371. https://doi.org/10.3390/pr12020371