Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification




Abstract
:1. Introduction
- Part 0:
- Sixt, M.; Strube, J. Systematic and model-assisted evaluation of solvent based- or pressurized hot water extraction for the extraction of Artemisinin from Artemisia annua L. Processes 2017, 5, 86, doi:10.3390/pr5040086 [3].
- Part I:
- Sixt, Schmidt et al. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part I: Conceptual process design and cost estimation. Processes 2018, 6, 161, doi:10.3390/pr6090161 [10].
- Part II:
- Schmidt, Sixt et al. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part II: Model-based design of agitated and packed columns for multistage extraction and scrubbing. Processes 2018, 6, 179, doi:10.3390/pr6100179 [11].
- Part III:
- Mestmäcker, Schmidt et al. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification (this article).
- Part IV:
- Huter, Schmidt et al. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part IV: Crystallization. Processes 2018, 6, 181, doi:10.3390/pr6100181 [12].
2. Materials and Methods
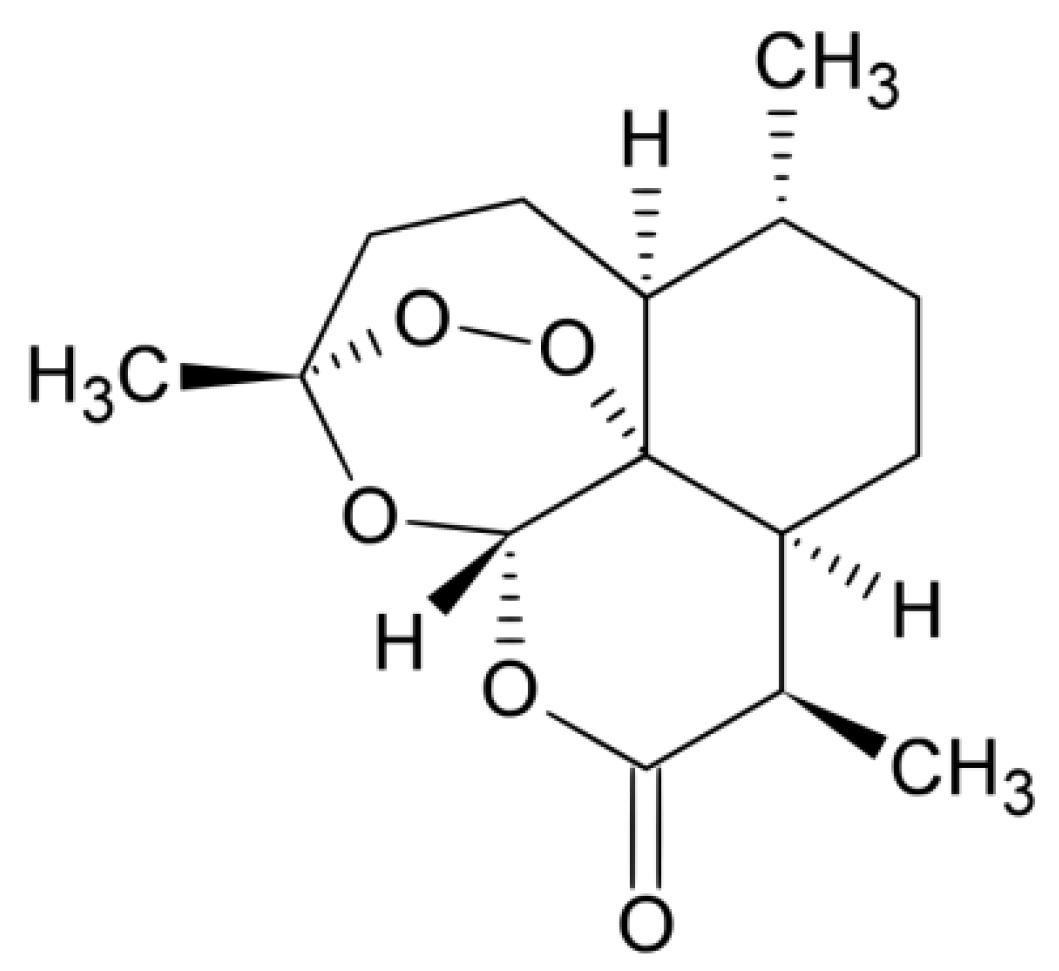
2.1. Chemicals
2.2. Feed Material for Chromatographic Measurements
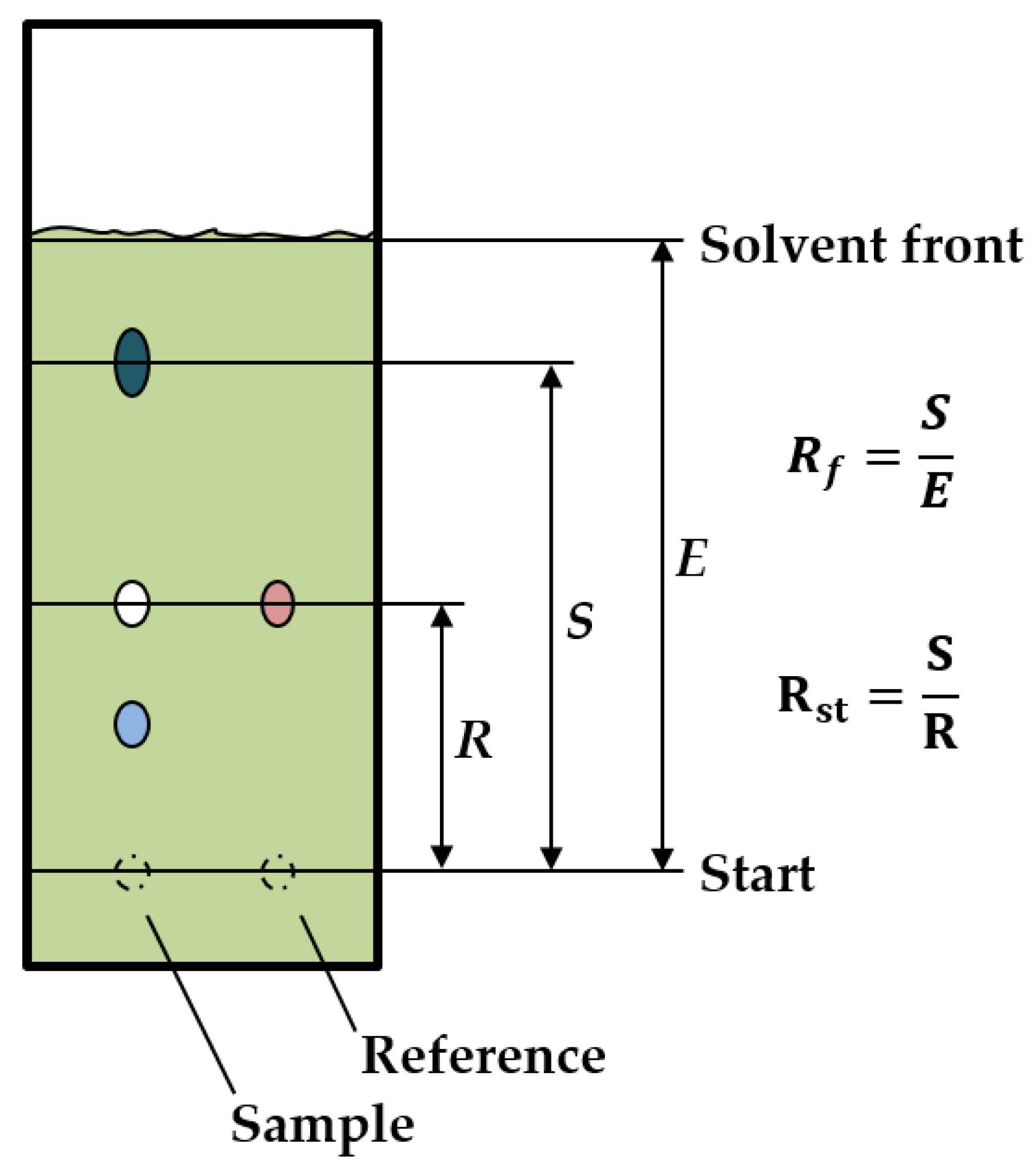
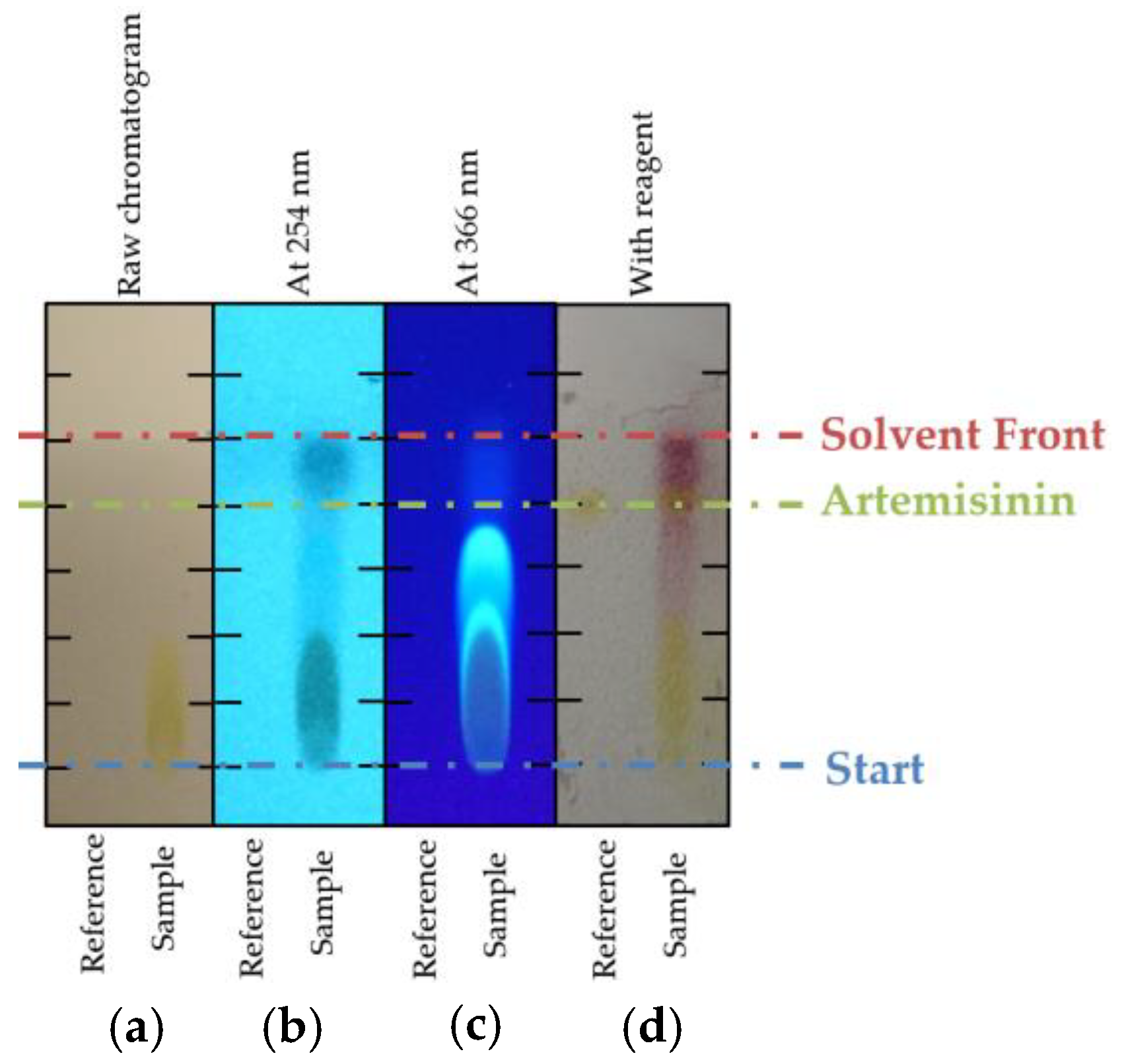
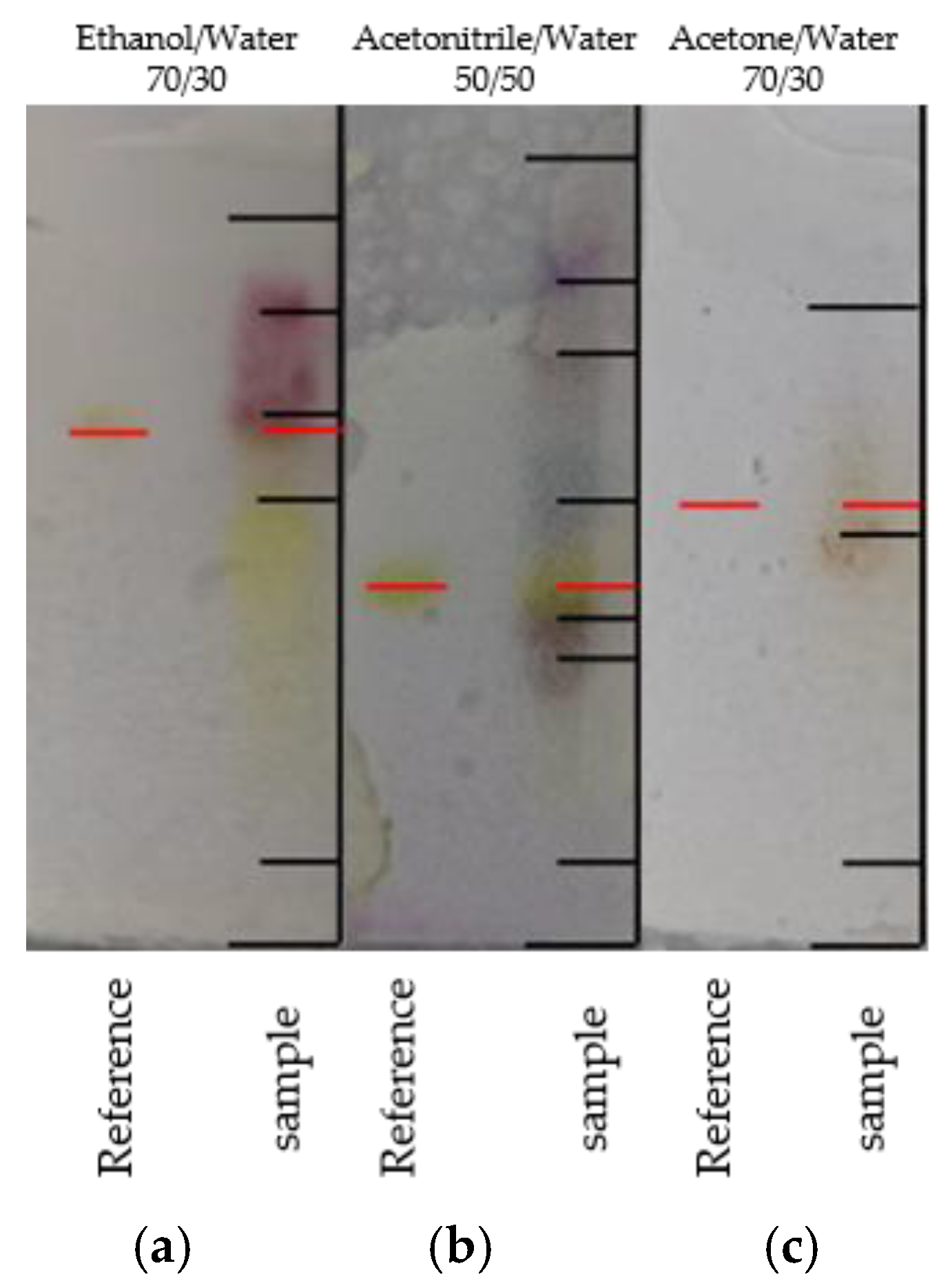
2.3. TLC Screening
2.4. HPLC Columns
2.5. Analytics
2.6. Devices and Instruments
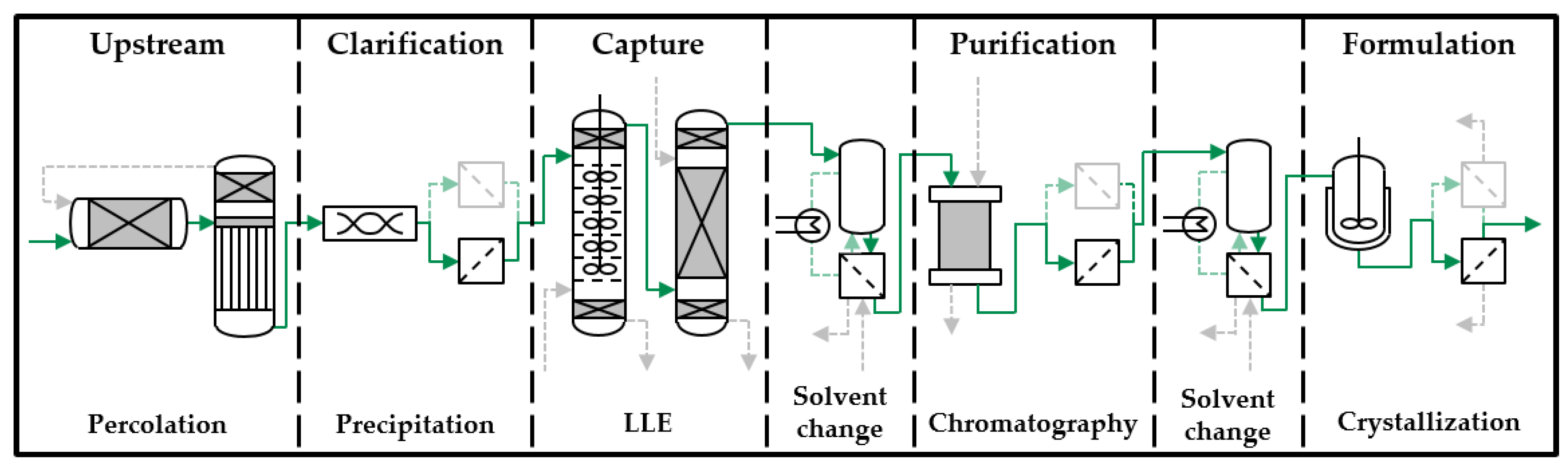
3. Results and Discussion
3.1. TLC Screening of Different Phases and Solvents
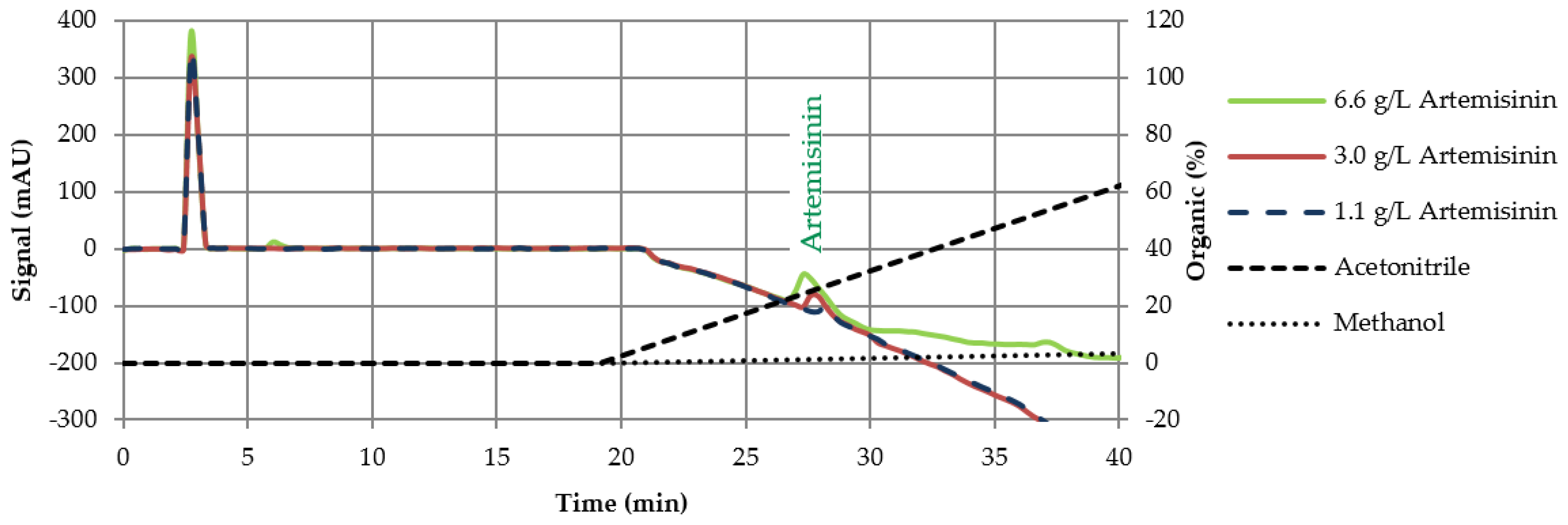
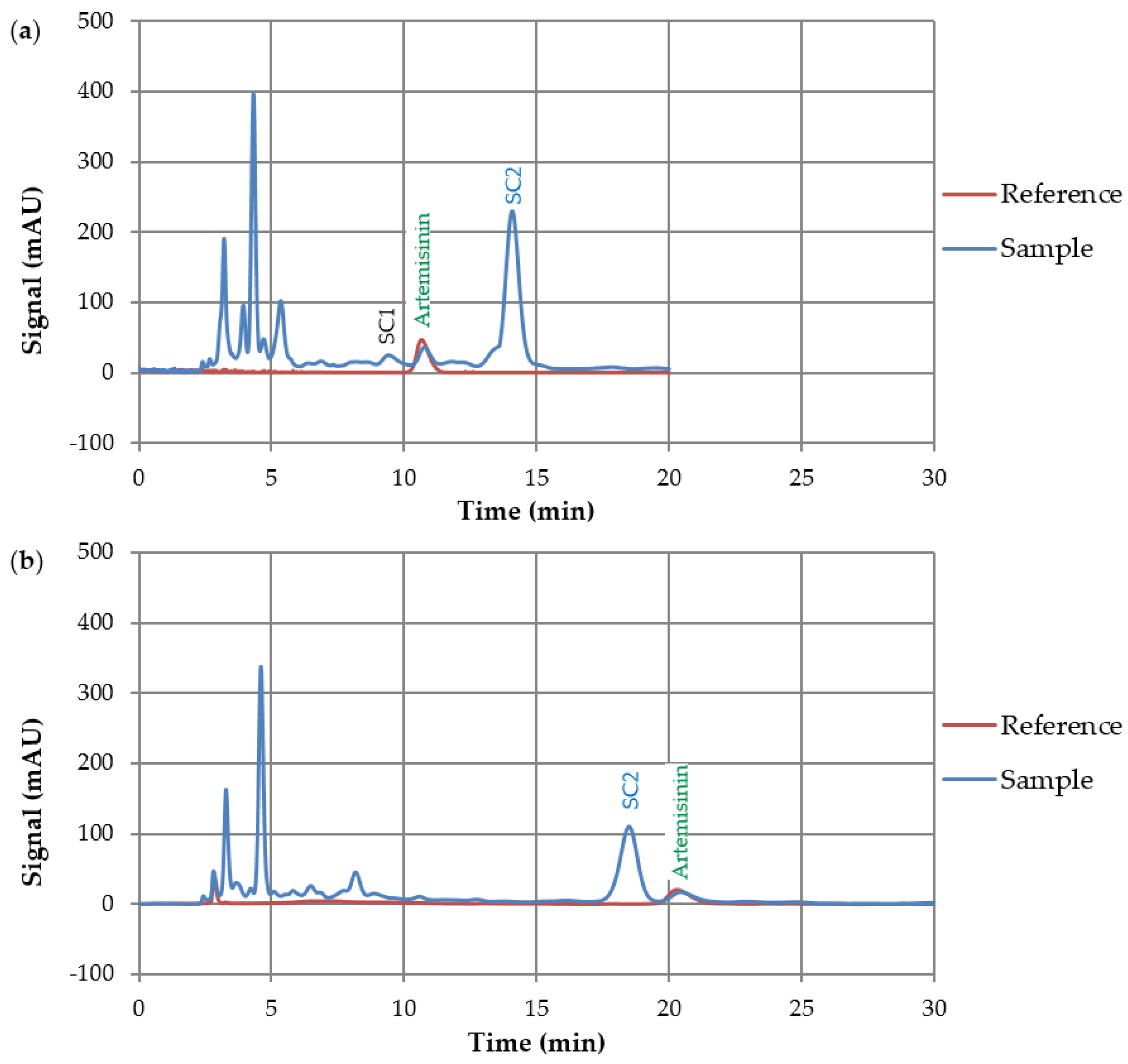
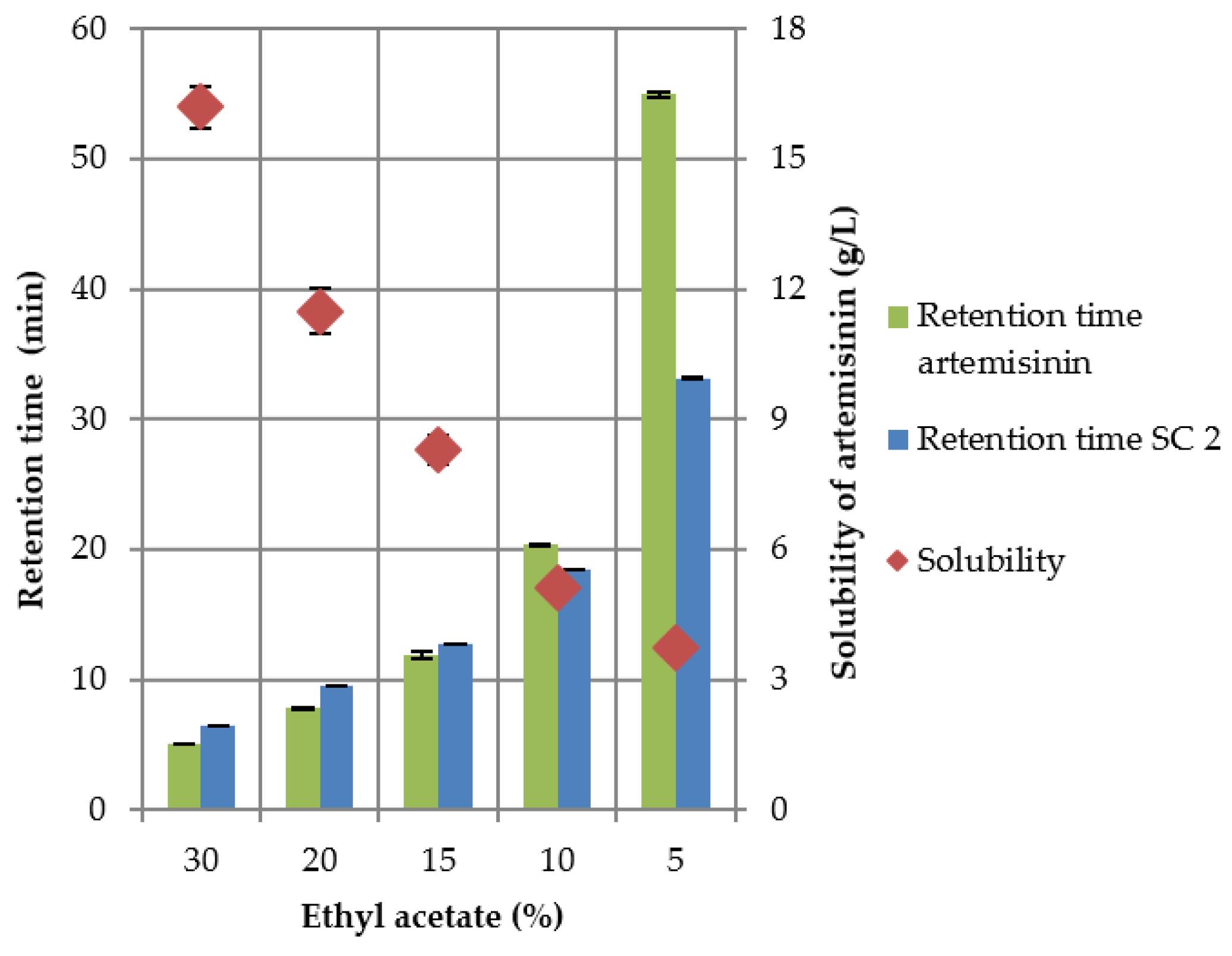
3.2. HPLC Screening
3.2.1. Column Experiments with Cyano Columns
3.2.2. Column Experiments with Normal Phase Columns
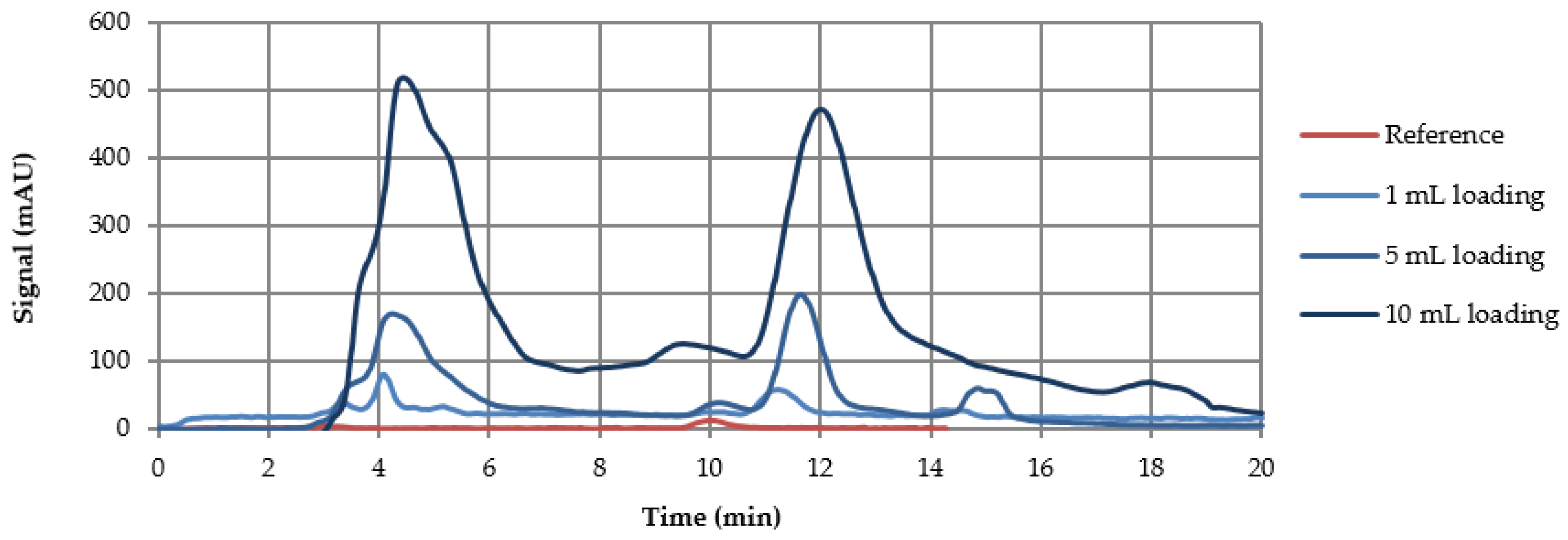
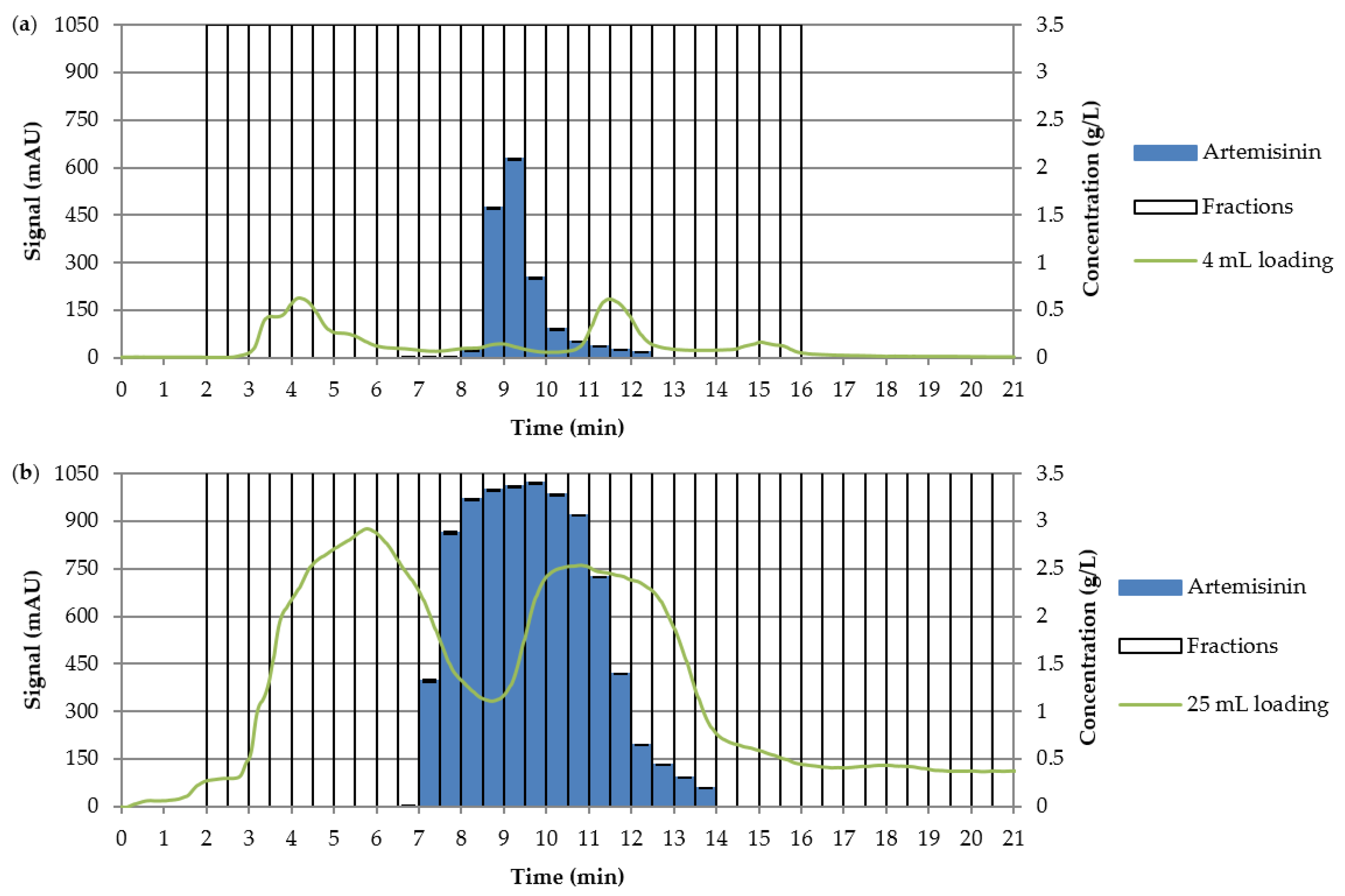
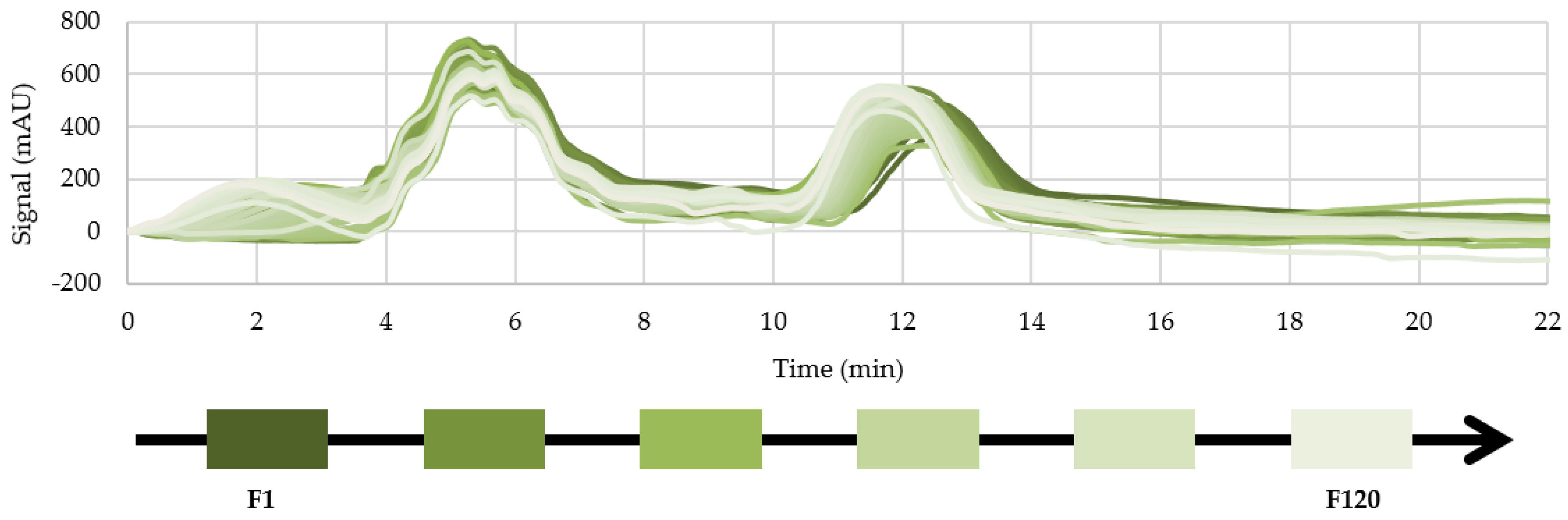
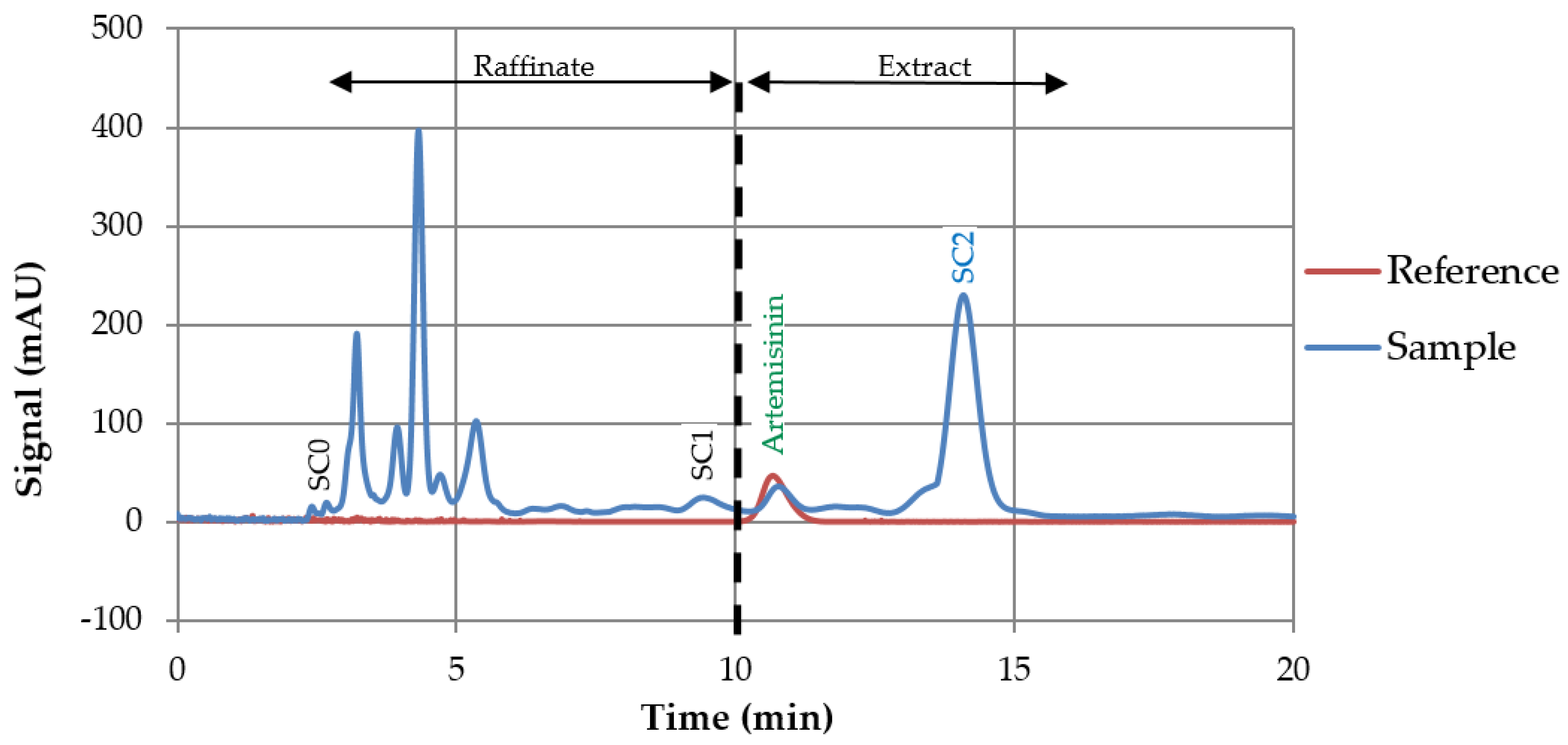
3.3. Preparative Experiments
3.3.1. Loading Experiments
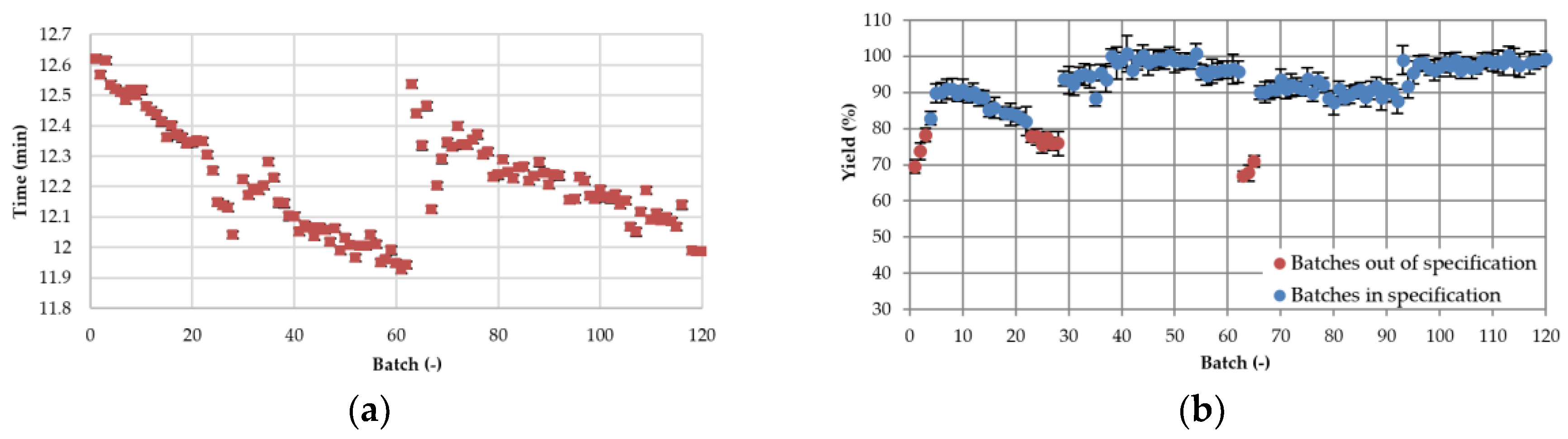
3.3.2. Process Stability
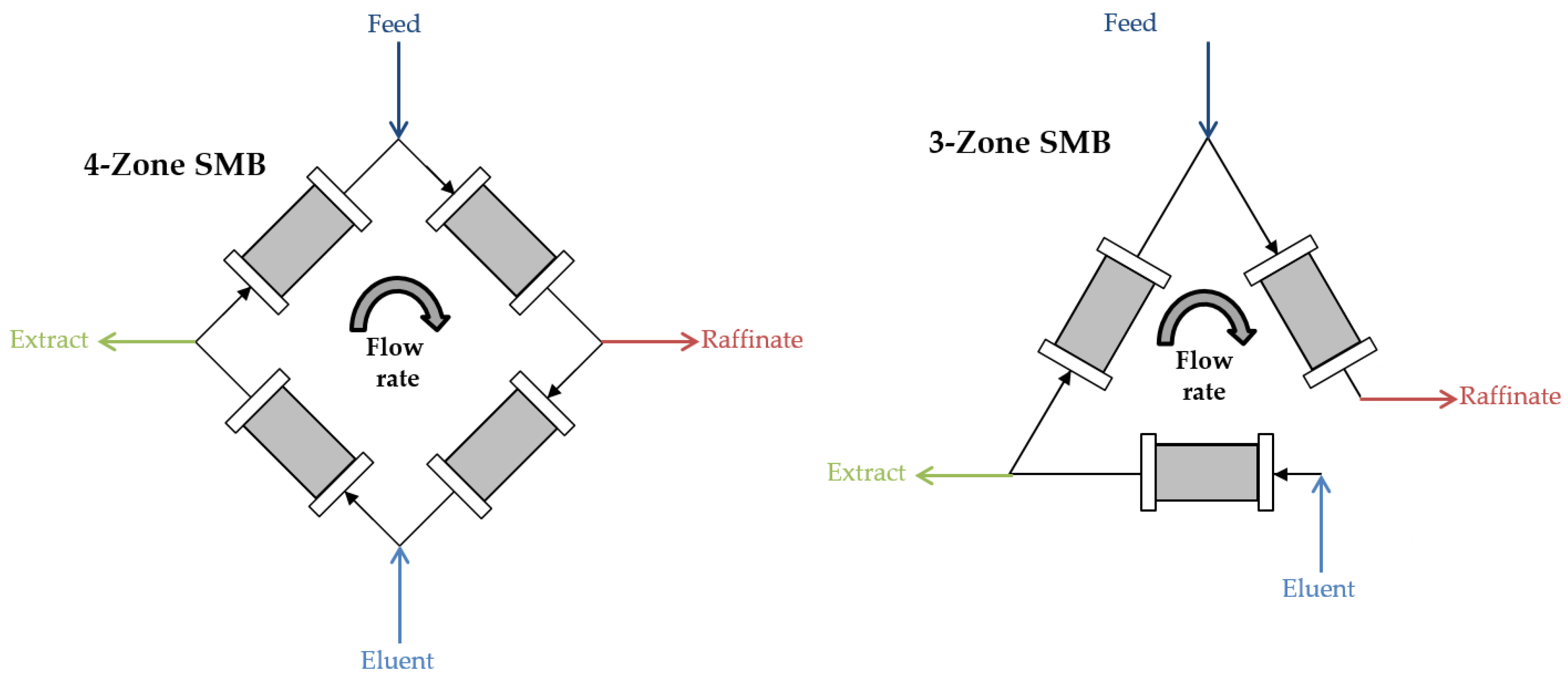
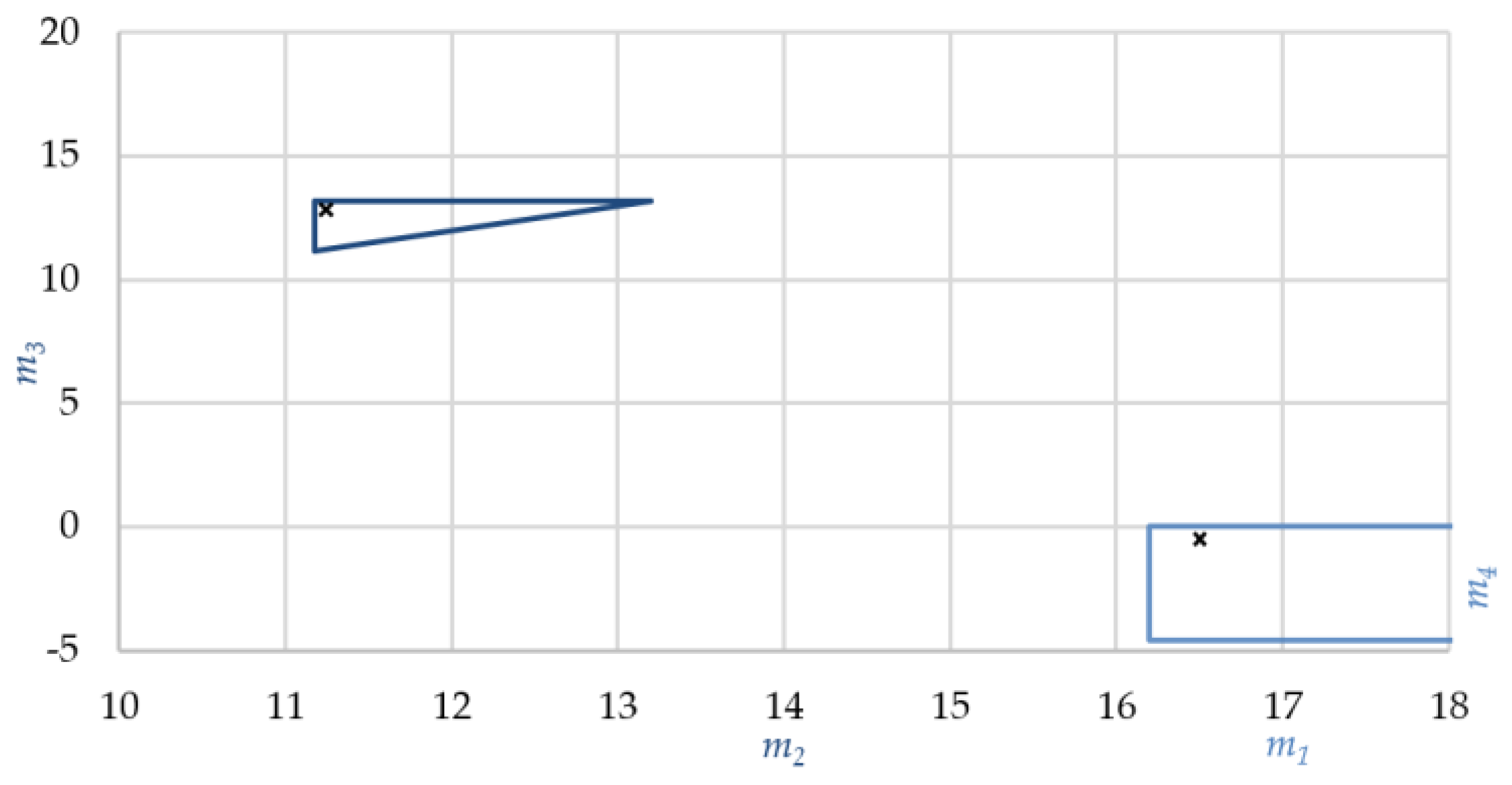
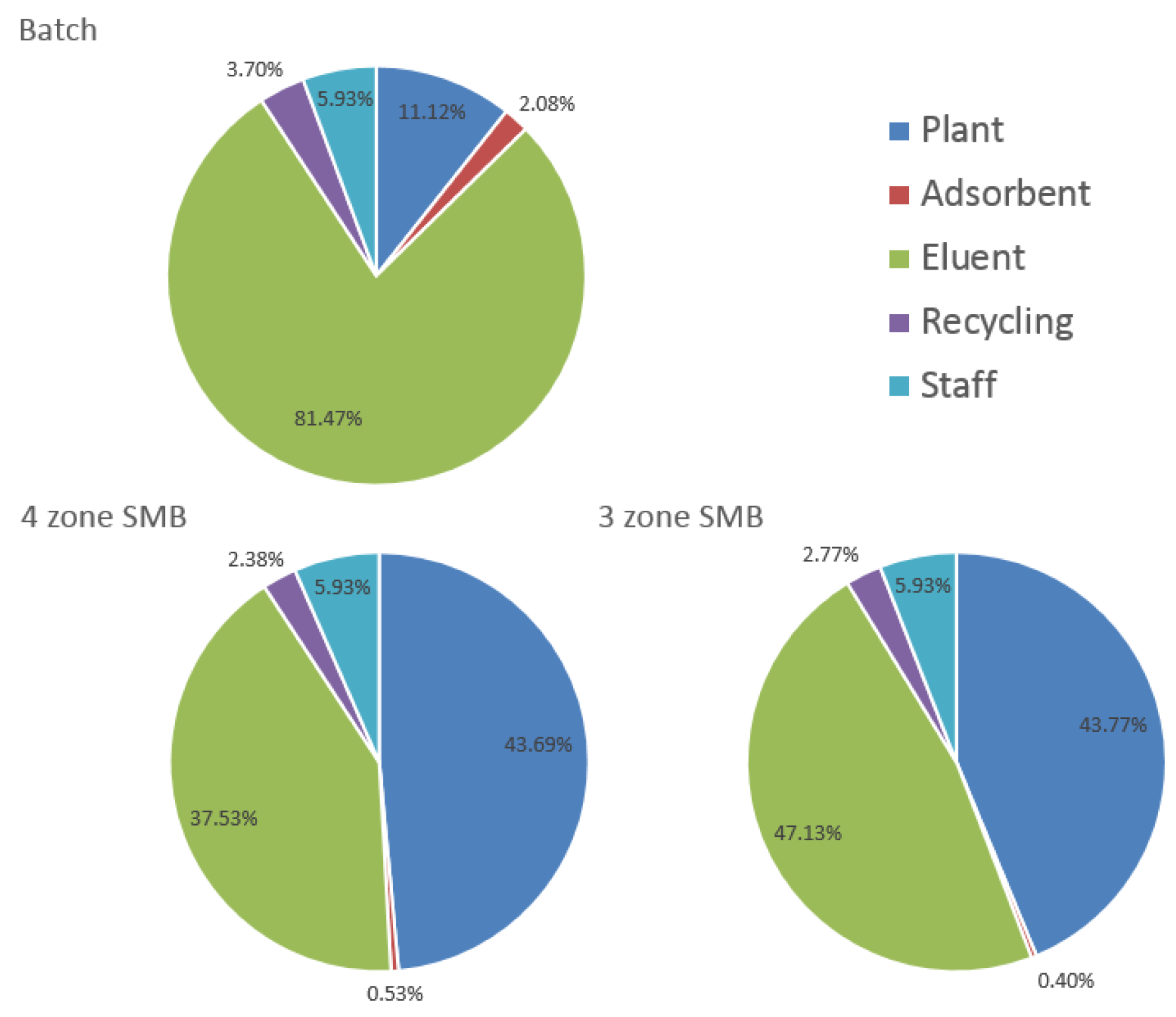
3.4. SMB Design
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Symbols and Abbreviations
| ε | Voidage |
| E | Migration distance of the eluent, cm |
| Capacity factor of component I | |
| mi | Volumetric phase ratio |
| R | Migration distance of the reference, cm |
| Rf | Retention factor |
| Rst | Retention factor for standard substance |
| S | Migration distance of the substance, cm |
| t0 | Dead time of the column, min |
| tR,I | Retention time of component I, min |
| tT | Cycle time, min |
| Volumetric flow in zone j, mL/min | |
| Vcolumn | Volume of the column, mL |
| CV | Column volume |
| ELSD | Evaporation light scattering detector |
| HPLC | High performance liquid chromatography |
| i.d. | Inner diameter |
| IPA | Isopropyl alcohol |
| LLE | Liquid-liquid extraction |
| n.a. | Not applicable |
| NMR | Nuclear magnetic resonance |
| NP | Normal phase |
| PHWE | Pressurized hot water extraction |
| RP | Reversed phase |
| SC | Side component |
| TLC | Thin-layer chromatography |
| UV | Ultra violet |
| VIS | Visible |
| SLE | Solid-liquid extraction |
| SMB | Simulated moving bed |
References
- Kassing, M.; Jenelten, U.; Schenk, J.; Hänsch, R.; Strube, J. Combination of rigorous and statistical modeling for process development of plant-based extractions based on mass balances and biological aspects. Chem. Eng. Technol. 2012, 35, 109–132. [Google Scholar] [CrossRef]
- Ditz, R.; Gerard, D.; Hagels, H.; Igl, N.; Schäffler, M.; Schulz, H.; Stürtz, M.; Tegtmeier, M.; Treutwein, J.; Strube, J.; et al. Phytoextracts-Proposal towards a New Comprehensive Research Focus; DECHEMA: Frankfurt, Germany, 2017. [Google Scholar]
- Sixt, M.; Strube, J. Systematic and model-assisted evaluation of solvent based- or pressurized hot water extraction for the extraction of Artemisinin from Artemisia annua L. Processes 2017, 5, 86. [Google Scholar] [CrossRef]
- Sixt, M.; Koudous, I.; Strube, J. Process design for integration of extraction, purification and formulation with alternative solvent concepts. C. R. Chim. 2016, 19, 733–748. [Google Scholar] [CrossRef]
- Koudous, I.; Both, S.; Gudi, G.; Schulz, H.; Strube, J. Process design based on physicochemical properties for the example of obtaining valuable products from plant-based extracts. C. R. Chim. 2014, 17, 218–231. [Google Scholar] [CrossRef]
- Koudous, I. Stoffdatenbasierte Verfahrensentwicklung zur Isolierung von Wertstoffen aus Pflanzenextrakten; Shaker: Aachen, Germany, 2017; ISBN 978-3-8440-5271-8. [Google Scholar]
- Both, S. Systematische Verfahrensentwicklung für pflanzlich basierte Produkte im regulatorischen Umfeld; Shaker: Aachen, Germany, 2015; ISBN 978-3-8440-3727-2. [Google Scholar]
- Chemat, F.; Strube, J. Green Extraction of Natural Products—Theory and Practice; Wiley VCH: Weinheim, Germany, 2015; ISBN 978-3-527-67682-8. [Google Scholar]
- Josch, J.P.; Both, S.; Strube, J. Characterization of feed properties for conceptual process design involving complex mixtures. Chem. Ing. Tech. 2012, 84, 918–931. [Google Scholar] [CrossRef]
- Sixt, M.; Schmidt, A.; Mestmäcker, F.; Huter, M.J.; Uhlenbrock, L.; Strube, J. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part I: Conceptual process design and cost estimation. Processes 2018, 6, 161. [Google Scholar] [CrossRef]
- Schmidt, A.; Sixt, M.; Huter, M.; Mestmäcker, F.; Strube, J. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part II: Model-based design of agitated and packed columns for multistage extraction and scrubbing. Processes 2018, 6, 179. [Google Scholar] [CrossRef]
- Huter, M.; Schmidt, A.; Mestmäcker, F.; Sixt, M.; Strube, J. Systematic and model-assisted process design for the extraction and purification of Artemisinin from Artemisia annua L.—Part IV: Crystallization. Processes 2018, 6, 181. [Google Scholar] [CrossRef]
- Poole, C. Instrumental Thin-Layer Chromatography; Elsevier Science: Burlington, MA, USA, 2014; ISBN 978-0-12-417223-4. [Google Scholar]
- Zlatkis, A.; Kaiser, R.E. HPTLC-High Performance Thin-Layer Chromatography; Elsevier Scientific Pub. Co.: New York, NY, USA, 1977. [Google Scholar]
- Horváth, Z.; Horosanskaia, E.; Lee, J.W.; Lorenz, H.; Gilmore, K.; Seeberger, P.H.; Seidel-Morgenstern, A. Recovery of Artemisinin from a complex reaction mixture using continuous chromatography and crystallization. Org. Process Res. Dev. 2015, 19, 624–634. [Google Scholar] [CrossRef]
- O’Brien, A.G.; Horváth, Z.; Lévesque, F.; Lee, J.W.; Seidel-Morgenstern, A.; Seeberger, P.H. Continuous synthesis and purification by direct coupling of a flow reactor with simulated moving-bed chromatography. Angew. Chem. Int. Ed. 2012, 51, 7028–7030. [Google Scholar] [CrossRef] [PubMed]
- Malwade, C.R.; Qu, H.; Rong, B.-G.; Christensen, L.P. Conceptual process synthesis for recovery of natural products from plants: A case study of Artemisinin from Artemisia annua. Ind. Eng. Chem. Res. 2013, 52, 7157–7169. [Google Scholar] [CrossRef]
- Qu, H.; Christensen, K.B.; Fretté, X.C.; Tian, F.; Rantanen, J.; Christensen, L.P. Chromatography-crystallization hybrid process for Artemisinin purification from Artemisia annua. Chem. Eng. Technol. 2010, 33, 791–796. [Google Scholar] [CrossRef]
- Malwade, C.R.; Buchholz, H.; Rong, B.-G.; Qu, H.; Christensen, L.P.; Lorenz, H.; Seidel-Morgenstern, A. Crystallization of Artemisinin from chromatography fractions of Artemisia annua extract. Org. Process Res. Dev. 2016, 20, 646–652. [Google Scholar] [CrossRef] [Green Version]
- Ndocko Ndocko, E.; Bäcker, W.; Strube, J. Process design method for manufacturing of natural compounds and related molecules. Sep. Sci. Technol. 2008, 43, 642–670. [Google Scholar] [CrossRef]
- Lapkin, A.A.; Plucinski, P.K.; Cutler, M. Comparative assessment of technologies for extraction of artemisinin. J. Nat. Prod. 2006, 69, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- ElSohly, H.N.; Croom, E.M.; El-Feraly, F.S.; El-Sherei, M.M. A large-scale extraction technique of Artemisinin from Artemisia annua. J. Nat. Prod. 1990, 53, 1560–1564. [Google Scholar] [CrossRef]
- Juza, M. Development of a high-performance liquid chromatographic simulated moving bed separation from an industrial perspective. J. Chromatogr. A 1999, 865, 35–49. [Google Scholar] [CrossRef]
- Juza, M.; Mazzotti, M.; Morbidelli, M. Simulated moving-bed chromatography and its application to chirotechnology. Trends Biotechnol. 2000, 18, 108–118. [Google Scholar] [CrossRef]
- Mazzotti, M. Equilibrium theory based design of simulated moving bed processes for a generalized Langmuir isotherm. J. Chromatogr. A 2006, 1126, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.E. Simulated Moving Bed Technology—Principles, Design and Process Applications; Butterworth-Heinemann: Oxford, UK, 2015; ISBN 978-0-12-802024-1. [Google Scholar]
- Storti, G.; Masi, M.; Carrà, S.; Morbidelli, M. Optimal design of multicomponent countercurrent adsorption separation processes involving nonlinear equilibria. Chem. Eng. Sci. 1989, 44, 1329–1345. [Google Scholar] [CrossRef]
- Zobel, S.; Helling, C.; Ditz, R.; Strube, J. Design and operation of continuous countercurrent chromatography in biotechnological production. Ind. Eng. Chem. Res. 2014, 53, 9169–9185. [Google Scholar] [CrossRef]
- Mazzotti, M.; Storti, G.; Morbidelli, M. Optimal operation of simulated moving bed units for nonlinear chromatographic separations. J. Chromatogr. A 1997, 769, 3–24. [Google Scholar] [CrossRef]
- Jermann, S.; Mazzotti, M. Three column intermittent simulated moving bed chromatography: 1. Process description and comparative assessment. J. Chromatogr. A 2014, 1361, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Jermann, S.; Alberti, A.; Mazzotti, M. Three-column intermittent simulated moving bed chromatography: 2. Experimental implementation for the separation of Tröger’s Base. J. Chromatogr. A 2014, 1364, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Jermann, S.; Meijssen, M.; Mazzotti, M. Three column intermittent simulated moving bed chromatography: 3. Cascade operation for center-cut separations. J. Chromatogr. A 2015, 1378, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Jork, H.; Funk, W.; Fischer, W.; Wimmer, H. Dünnschicht-Chromatographie. Reagenzien und Nachweismethoden, 1st ed.; Wiley-VCH: Weinheim, Germany, 1990. [Google Scholar]
- Lapkin, A.A.; Walker, A.; Sullivan, N.; Khambay, B.; Mlambo, B.; Chemat, S. Development of HPLC analytical protocols for quantification of artemisinin in biomass and extracts. J. Pharm. Biomed. Anal. 2009, 49, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; lü, H.; Pang, F. Solubility of Artemisinin in seven different pure solvents from (283.15 to 323.15) K. J. Chem. Eng. Data 2009, 54, 762–764. [Google Scholar] [CrossRef]
- Nti-Gyabaah, J.; Gbewonyo, K.; Chiew, Y.C. Solubility of Artemisinin in different single and binary solvent mixtures between (284.15 and 323.15) K and NRTL interaction parameters. J. Chem. Eng. Data 2010, 55, 3356–3363. [Google Scholar] [CrossRef]
- Lapkin, A.A.; Peters, M.; Greiner, L.; Chemat, S.; Leonhard, K.; Liauw, M.A.; Leitner, W. Screening of new solvents for artemisinin extraction process using ab initio methodology. Green Chem. 2010, 12, 241–251. [Google Scholar] [CrossRef]
- Wang, L.-H.; Song, Y.-T.; Chen, Y.; Cheng, Y.-Y. Solubility of Artemisinin in Ethanol + Water from (278.2 to 343.2) K. J. Chem. Eng. Data 2007, 52, 757–758. [Google Scholar] [CrossRef]
- Haynes, R.K.; Fugmann, B.; Stetter, J.; Rieckmann, K.; Heilmann, H.-D.; Chan, H.-W.; Cheung, M.-K.; Lam, W.-L.; Wong, H.-N.; Croft, S.L.; et al. Artemisone—A Highly Active Antimalarial Drug of the Artemisinin Class. Angew. Chem. 2006, 118, 2136–2142. [Google Scholar] [CrossRef]
- Strube, J.; Altenhöner, U.; Meurer, M.; Schmidt-Traub, H. Optimierung kontinuierlicher Simulated-Moving-Bed-Chromatographie-Prozesse durch dynamische Simulation. Chem. Ing. Tech. 1997, 69, 328–331. [Google Scholar] [CrossRef]
- Subramanian, G. Biopharmaceutical Production Technology, 1st ed.; Wiley-VCH: Weinheim, Germany, 2012; ISBN 978-3-527-33029-4. [Google Scholar]
- Seidel-Morgenstern, A.; Keßler, L.C.; Kaspereit, M. New Developments in Simulated Moving Bed Chromatography. Chem. Eng. Technol. 2008, 31, 826–837. [Google Scholar] [CrossRef]
- Strube, J. Technische Chromatographie. Auslegung, Optimierung, Betrieb und Wirtschaftlichkeit; Shaker: Aachen, Germany, 2000. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plate | Solvents |
|---|---|
| Nano-Sil CN/UV | Acetone, acetonitrile, butyl acetate, ethanol, ethyl acetate, isopropyl alcohol, methanol, methyl-tert-butyl ether, n-heptane, n-hexane, tetrahydrofurane, toluene, water |
| Silica gel 60 RP-18 F254S | Acetone, acetonitrile, butyl acetate, ethanol, ethyl acetate, n-heptane, n-hexane, water |
| Silica gel 60 F254S | Acetone, acetonitrile, butyl acetate, ethanol, ethyl acetate, isopropyl alcohol, methanol, n-hexane |
| Column | Solvents |
|---|---|
| LiChrospher 300 CN | Acetone, acetonitrile, ethanol, methanol, water, phosphate buffer |
| PharmaPrep P100 RP-18e | Acetone, acetonitrile, butyl acetate, ethanol, n-hexane, water |
| LiChrospher Si60 | Butyl acetate, ethyl acetate, ethanol, isopropyl alcohol, n-hexane, n-heptane, toluene |
| Solvent | Temperature (K) | Solubility (g/L) | Reference |
|---|---|---|---|
| Acetone | Ambient temperature a | 242.2 | [18] |
| 298.15 | 32.1 | [35] | |
| 298.15 | 31.6 | [36] | |
| Acetonitrile | Ambient temperature a | 267.7 | [18] |
| 293.15 | 157.7 | [37] | |
| 298.15 | 2.8 | [35] | |
| 298.15 | 9.7 | [36] | |
| Ethanol | Ambient temperature a | 31.6 | [18] |
| 303.15 | 14.0 | [38] | |
| 298.15 | 22.3 | [36] | |
| Ethyl acetate | Ambient temperature a | 153.2 | [18] |
| 298.15 | 42.3 | [35] | |
| 298.15 | 39.4 | [36] | |
| Hexane | Ambient temperature a | 2.5 | [18] |
| 293.15 | 1.8 | [37] | |
| 298.15 | 0.28 | [36] | |
| Heptane | 298.15 | 0.25 | [36] |
| Isopropyl alcohol | Ambient temperature a | 20.1 | [18] |
| Methanol | Ambient temperature a | 46.8 | [18] |
| 298.15 | 9.9 | [35] | |
| 298.15 | 9.7 | [36] | |
| Toluene | 293.15 | 90.7 | [37] |
| 298.15 | 141.6 | [35] | |
| 298.15 | 90.7 | [36] | |
| Water (pH 7.2) | n.a. | 0.063 | [39] |
| Eluent Consumption | k-Value for Artemisinin |
|---|---|
| n-heptane/butyl acetate (95/5 vol %) | <0.01 |
| n-heptane/ethanol (95/5 vol %) | n.a. |
| n-heptane/IPA (10 CV gradient to 30 vol % IPA) | 4.76 |
| n-hexane | 9.04 |
| n-hexane/butyl acetate (70/30 vol %) | <0.01 |
| n-hexane/butyl acetate (95/5 vol %) | n.a. |
| n-hexane/ethyl acetate (70/30 vol %) | 0.81 |
| n-hexane/ethyl acetate (80/20 vol %) | 1.77 |
| n-hexane/ethyl acetate (85/15 vol %) | 3.28 |
| n-hexane/ethyl acetate (90/10 vol %) | 6.28 |
| n-hexane/ethyl acetate (95/5 vol %) | 18.62 |
| n-hexane/ethyl acetate (Gradient 5 vol % to 15 vol % ethyl acetate) | n.a. |
| Loading (mL) | Yield (%) | Purity (%) | Productivity (g·Lads−1·d−1) |
|---|---|---|---|
| 4 | 97.8 | >99 | 44.9 |
| 10 | 96.6 | 97.9 | 110.8 |
| 25 | 93.8 | 97.7 | 161.4 |
| Parameter | Batch Process | 4-Zone SMB | 3-Zone SMB |
|---|---|---|---|
| Yield (%) | 93.9 | 100 | 100 |
| Purity (%) | >99 | >99 | >99 |
| Productivity (g·Lads−1·d−1) | 107.4 | 190.6 | 254.1 |
| Eluent consumption (Ldes·gartemisinin−1) | 4.2 | 2.7 | 3.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mestmäcker, F.; Schmidt, A.; Huter, M.; Sixt, M.; Strube, J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification. Processes 2018, 6, 180. https://doi.org/10.3390/pr6100180
Mestmäcker F, Schmidt A, Huter M, Sixt M, Strube J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification. Processes. 2018; 6(10):180. https://doi.org/10.3390/pr6100180
Chicago/Turabian StyleMestmäcker, Fabian, Axel Schmidt, Maximilian Huter, Maximilian Sixt, and Jochen Strube. 2018. "Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification" Processes 6, no. 10: 180. https://doi.org/10.3390/pr6100180
APA StyleMestmäcker, F., Schmidt, A., Huter, M., Sixt, M., & Strube, J. (2018). Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part III: Chromatographic Purification. Processes, 6(10), 180. https://doi.org/10.3390/pr6100180