1. Introduction
The energy-efficient membrane gas separation technology can play significant roles in performance enhancement and process intensification for the advanced fossil energy systems with CO
2 capture capacity. For the emerging coal- and natural gas-based IGCC power production with CO
2 capture and sequestration (CCS), cost effective air separation unit (ASU) for the gasifier and H
2/CO
2 separation units for pre-combustion CO
2 capture are critical to commercial success. However, both the O
2 and CO
2 gas separations currently remain challenging that hinders the realization of the IGCC-CCS technology. Traditional industrial O
2 separation by cryogenic distillation or pressure swing adsorption (PSA) is highly energy intensive and involves large capital investment. Membrane separation has shown promises as energy-efficient alternative for O
2 separation in the past few decades [
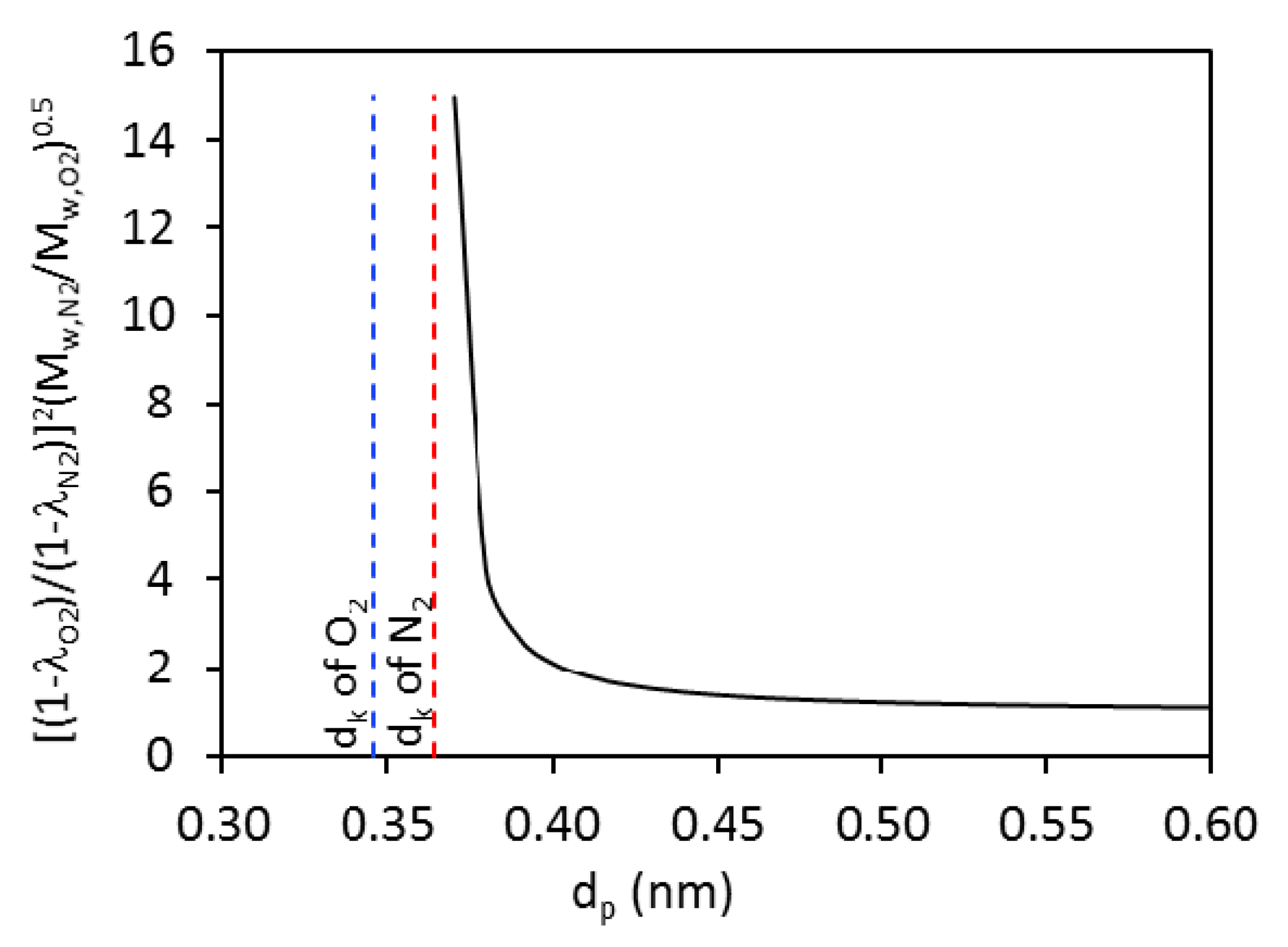
1]. However, because of the very close kinetic diameters (
dk) of the O
2 (
dk = 0.346 nm) and N
2 (
dk = 0.364 nm) molecules and similarly weak adsorbing behaviors of both gases in common membrane materials, low temperature O
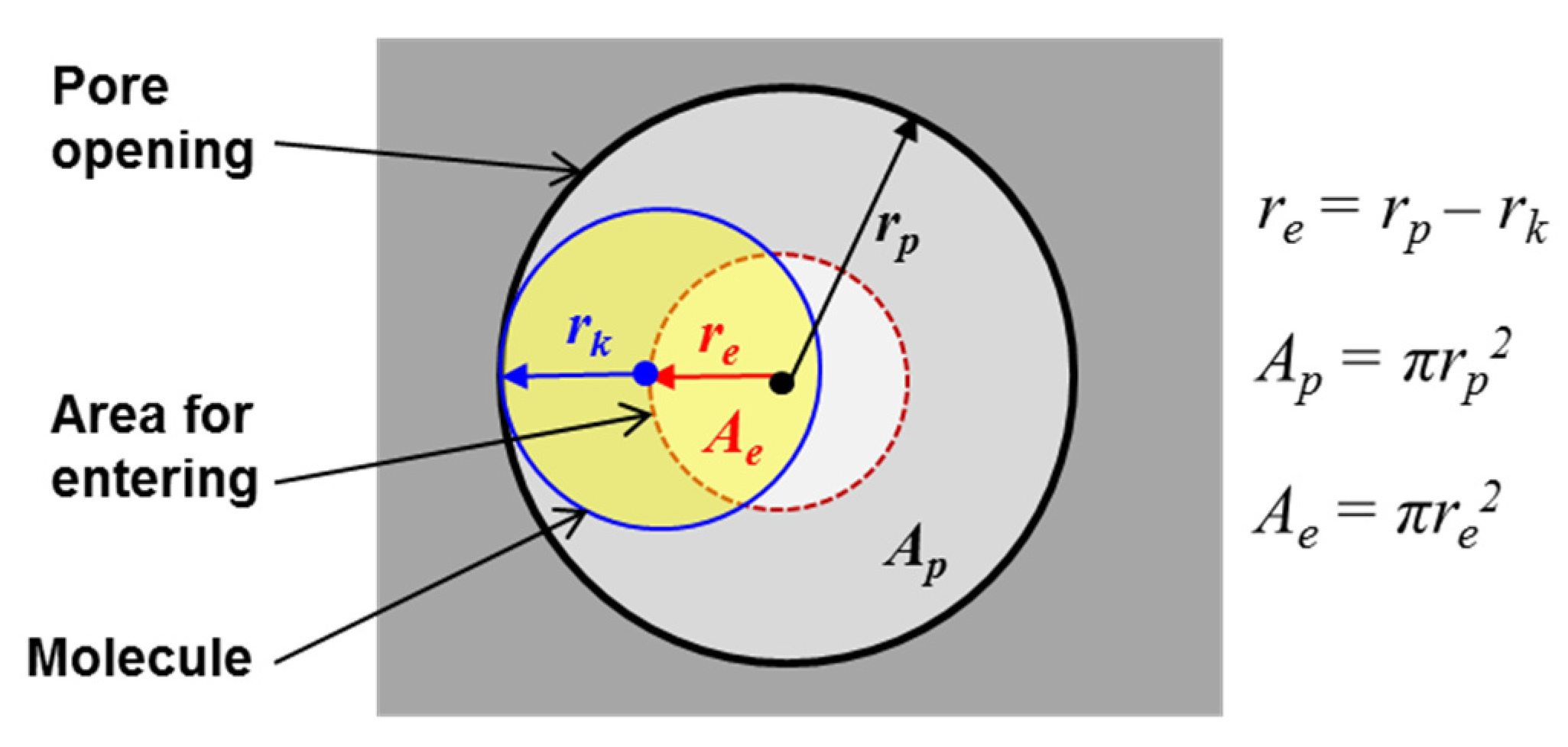
2 separation membranes reported in the literature have been of relatively poor performance with limited selectivity and low permeance. The microporous zeolite membranes, because of their extraordinary stability and molecular sieving effects enabled by their uniform sub-nanometer pore sizes, are promising for separating many small molecule gases. The kinetic molecular theory of gas transport in zeolitic pores suggests that, in order to achieve O
2/N
2 selectivity by non-adsorptive diffusion mechanism, the zeolite pore size must situate in between or very near the kinetic diameters of O
2 and N
2 [
2,
3]. Therefore, in order to achieve size-selectivity between O
2 and N
2 molecules, the zeolite membranes must possess pore diameter (
dp) very close to the kinetic size of N
2, i.e., <0.4 nm.
Membranes of zeolites with pore openings defined by 8-membered rings are promising candidates for O
2 separation because their
dp are around 0.4 nm. The highly siliceous DDR zeolite (
dp ~ 0.4 nm) membrane has been reported to exhibit O
2/N
2 permselectivity around 2 but the O
2 permeance was only in the order of 10
−9–10
−8 mol/m
2·s·Pa at 298 K [
4,
5,
6]. Since O
2 and N
2 are both weakly adsorbing with similarly small adsorption amounts in the DDR zeolite [
5], the very low O
2 permeance in the DDR zeolite membrane is a result of the large resistance for O
2 transport in the critically sized zeolitic pores over the entire membrane thickness. The membrane of NaA zeolite (
dp ~ 0.41 nm) was once reported to have surprisingly high O
2/N
2 separation factor of ~7 and O
2 permeance of 2.6 × 10
−7 mol/m
2·s·Pa at room temperature [
7]. This performance though was observed under vacuum pressure and its reproducibility is yet to be confirmed. In general, for zeolite membranes of very large aluminum contents, such as the A-type membranes, the gas permeation properties are often severely affected by the presence of trace water vapor due to its extremely ionic and hydrophilic surface [
8]. In addition, zeolite membranes of such high-aluminum contents are known to experience gradual degradation when subjected to dehydration in vacuum over extended time [
9].
Zeolites with pore openings formed by 10-membered rings have effective
dp of around 0.6 nm, which are much less resistive to small gas transport but become incapable of achieving size-selectivity or molecular sieving effect between the closely sized molecules like O
2, N
2, H
2 and CO
2. However, such relatively large pore size allows for depositing molecular modifiers to the internal pore wall to fine tune the effective channel openings. In the literature, highly siliceous MFI-type zeolite membranes, which have nearly cylindrical zeolitic channels formed by 10-membered rings with a diameter of ~0.56 nm, were successfully modified by depositing mono-silica species on their internal pore surfaces. The effective
dp of the internally modified zeolitic channels was estimated to be around 0.36 nm based on the observation of obvious permeance cut-off effects between H
2 and N
2 or CO
2 [
3,
10,
11]. The modification of the MFI-type zeolite membranes by deposition of mono-silica on the internal pore wall using methyldiethoxysilane (MDES) as precursor was first reported by Masuda et al. [
10]. The MDES molecule has a linear geometry with nominal dimension of 0.41 nm × 0.91 nm, which can penetrate into the pristine MFI zeolitic channels of 0.56 nm in diameter. The modification was conducted by pre-loading MDES in the zeolitic channels at low temperature followed by calcination to thermally decompose the MDES molecules and deposit mono-silica inside the zeolite channels [
10]. After modification, the H
2/N
2 separation factor increased from 1.5~4.5 to 90~140 at 383 K but the H
2 permeance decreased by an order of magnitude to only 2 × 10
−8 mol/m
2·s·Pa. The drastic loss of H
2 permeance was attributed to the deposition of mono-silica over the entire channel length that caused enormous resistance to molecular diffusion. An on-stream catalytic cracking deposition (CCD) method was developed in our lab to avoid excessive silica deposition over the channel length [
12]. In the on-stream CCD modification process, the MDES precursor is carried in a feed stream during membrane gas permeation operation at around 450 °C where the CCD process can occur at catalytically active sites such as the alumina tetrahedron sites in channel surface without inducing non-catalytic MDES thermal decomposition [
3,
13,
14]. Thus, in principle, after CCD deposition of silica at a single site, this mono-silica deposit creates a narrow “gate” to prevent MDES molecules from entering the rest of channel length. Consequently, the deposition of mono-silica is effectively limited in a small segment of the zeolitic channel while large part of the channel remains in its original size (~0.56 nm) for fast diffusion [
3,
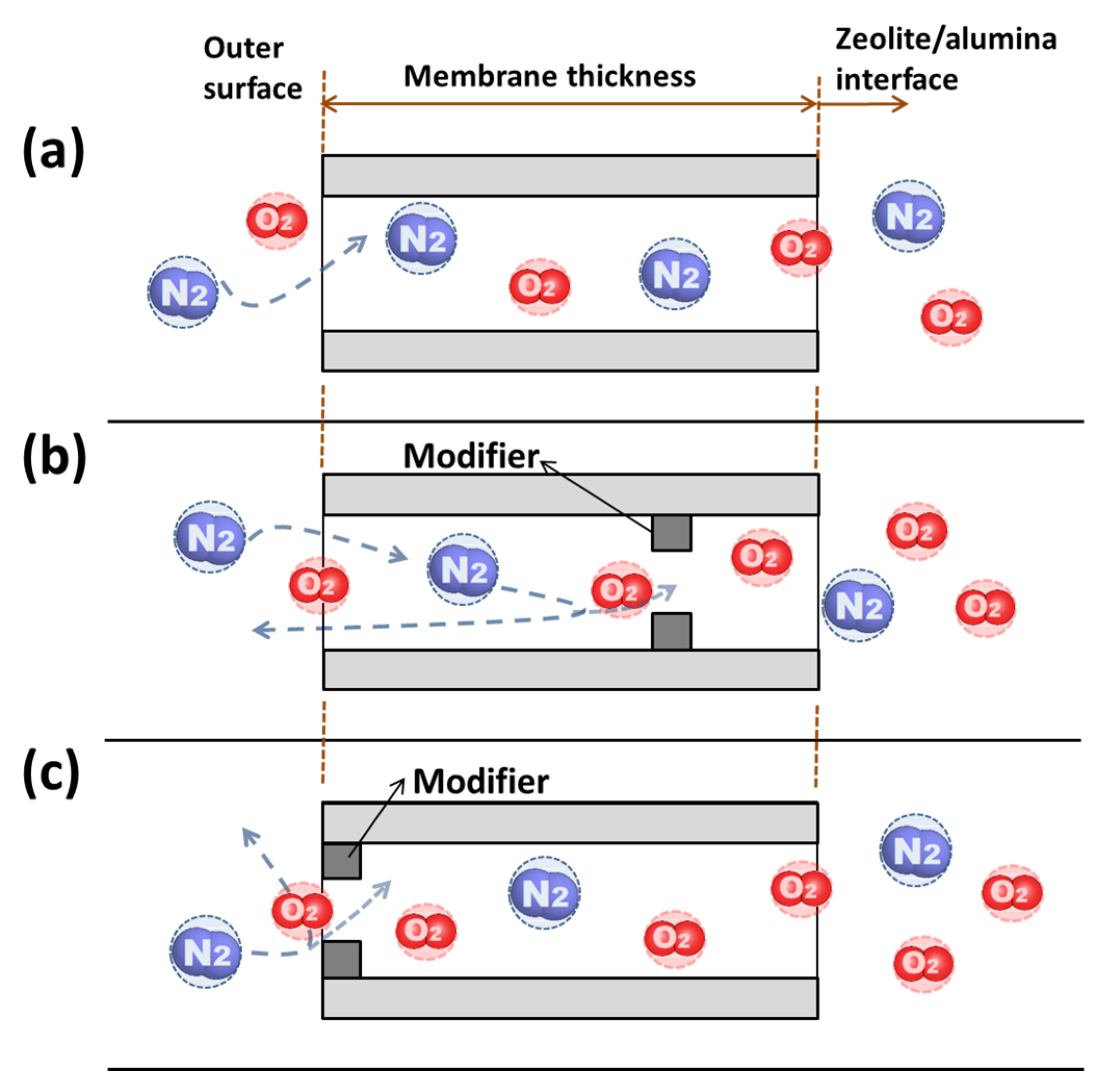
15]. The on-stream CCD method alleviated the mass transport resistance increase while achieving selectivity improvement. However, the supported zeolite membrane had largely random distribution of catalytic sites and the locations of CCD depositions were consequentially uncontrolled along the membrane thickness, which is a multiple of individual crystal size in the film. It is conceivable that when the modified site, which could be thought of as a “gate,” locates deep down the channel length, the less permeable molecules will have to back diffuse from the gated point to the feed stream during gas mixture permeation. Such a back-diffusion process would inevitably hinder the permeation of the more permeable component.
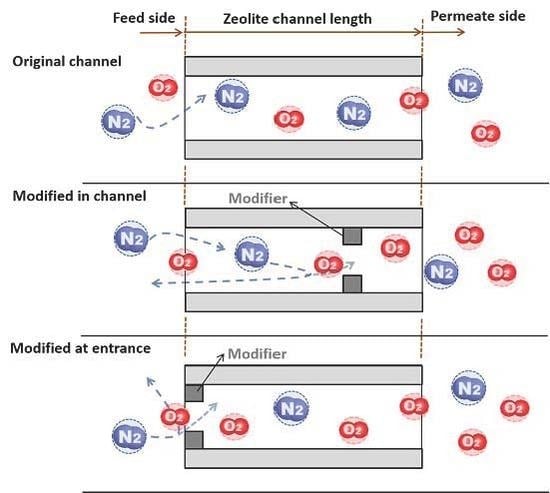
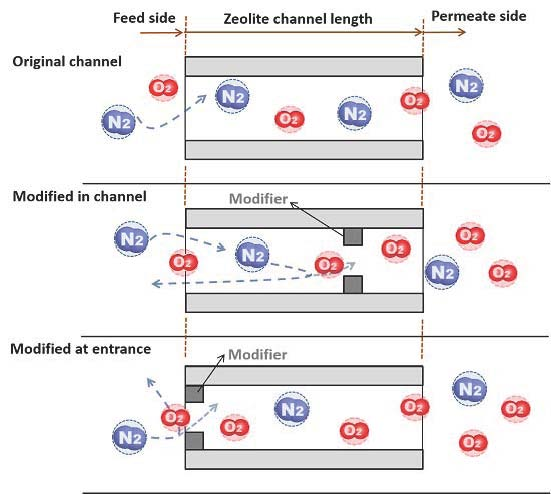
Here we report the synthesis and on-stream CCD modification of an MFI-type zeolite membrane, which has a pure-silica (i.e., silicalite) base layer with an outer surface of aluminum-containing framework (i.e., ZSM-5), in attempt to effectively control the pore modification within a small depth near the membrane outer surface. The hypothesis is that such a surface “gated” channel structure can improve the gas permeance by avoiding the back diffusion of the less permeable gases without sacrificing permeation selectivity. The modified MFI-type zeolite membranes are examined by permeation for a number of small molecule gases including O2 and N2.
2. Experimental
2.1. Materials and Chemicals
The porous α-alumina disc substrates were made of α-alumina powders with an average diameter of 0.46 µm (SG16, Alcoa, TN, USA). The alumina disc was 2-mm thick and 2.6-cm in diameter with porosity and average pore diameter (dp) of 27–30% and 0.1 µm, respectively. The edge of the disc was sealed by glass and the active membrane area was ~2.5 cm2 after excluding the glass sealed edge area. The surface of the disc used for membrane coating was polished by 800 mesh sandpaper and washed and dried in an oven at 313 K for overnight before membrane synthesis. The following chemicals and materials were used in the present work: sodium hydroxide (99.99% trace metal basis, pellet, Sigma-Aldrich, USA), fumed silica (0.007 µm, Aldrich, USA), tetrapropylammonium hydroxide (TPAOH, 1 M, Aldrich, USA), sodium aluminate (anhydrous, Al content as Al2O3: 50–56 wt. %, Riedel-de Haën, Germany), HNO3 solution (1.0 M, Fluka, USA) and methyldiethoxysilane (MDES, 96%, Aldrich, USA). All chemicals were used as received. Gases used for membrane permeation tests included H2 (99.999%), He (99.999%), N2 (99.999%), O2 (99.6%), CO2 (99.99%), CH4 (99.999%), i-C4H10 (99.5%) and SF6 (99%). All gases were obtained from Wright Brothers Inc. (Cincinnati, OH, USA) and used as received.
2.2. Synthesis of MFI Zeolite Membranes
Two kinds of MFI-type zeolite membranes were synthesized by the in situ hydrothermal crystallization method under different conditions. The first was a silicalite membrane without an aluminum-containing zeolite surface and the second was a silicalite membrane with an aluminum-containing MFI-zeolite (ZSM-5) outer surface obtained through a two-stage synthesis procedure.
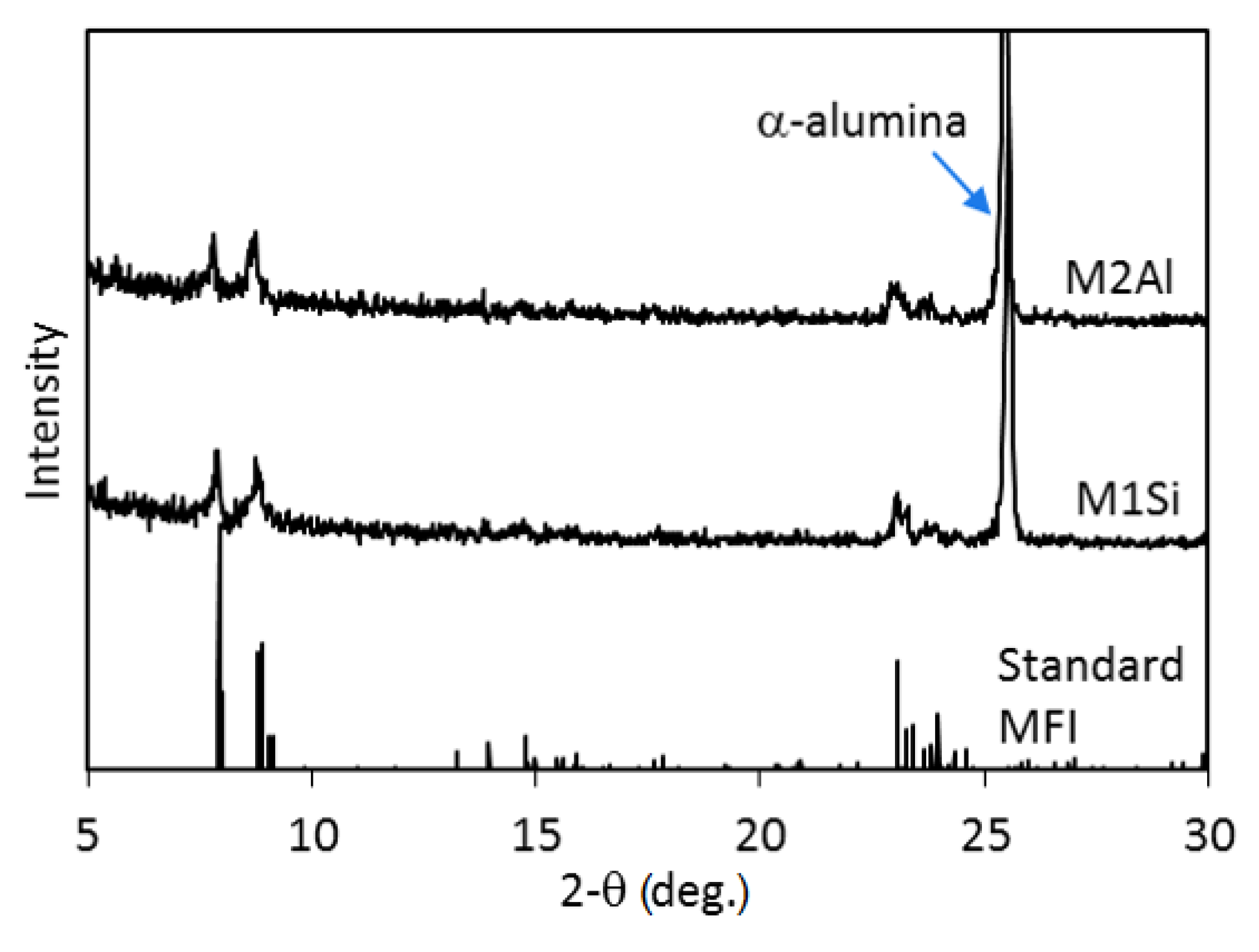
The silicalite membrane without a ZSM-5 surface was obtained from an alumina-free synthesis precursor, which was prepared by mixing 0.35 g NaOH, 5 g SiO2 and 25 mL 1 M TPAOH solution under vigorous stirring at 353 K. The TPAOH was used as the structure directing agent (SDA). The synthesis solution was aged for four hours at room temperature and then transferred into the synthesis autoclave where the disc substrate was placed horizontally at the Teflon-lined bottom with the polished side facing upward. About 25 mL of the synthesis solution was poured into the autoclave alone the autoclave wall to immerse the alumina disc. The hydrothermal synthesis was conducted at 453 K for 5 h and then the disc membrane was recovered and washed with DI water until the water pH was close to 7. The cleaned membrane disc was dried in an oven at 313 K and then calcined in air at 773 K for 6 h to remove the SDA from the zeolitic pores. This membrane is denoted as M1Si hereafter.
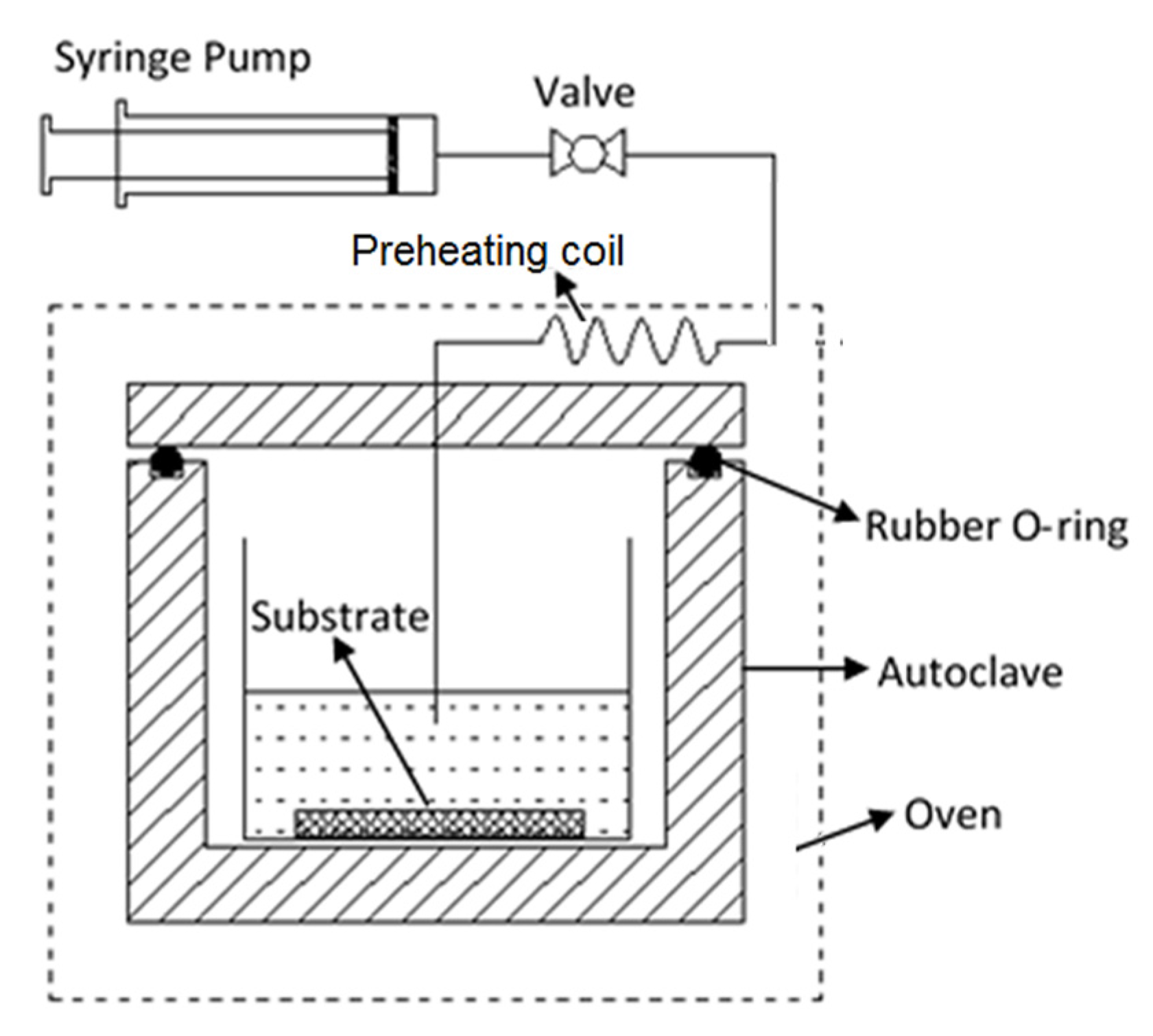
The membrane with a silicalite base layer and a ZSM-5 outer surface was obtained by a two-stage synthesis procedure where the liquid phase composition was varied for the second stage. A specially designed synthesis autoclave, as schematically shown in
Figure 1, was employed for the continuous two-stage synthesis. The zeolite precursor solution and hydrothermal reaction conditions for the first stage of synthesis were identical to those used for synthesizing the M1Si. However, the hydrothermal treatment was not terminated in 5 h; and instead, 1.2 mL of sodium aluminate solution (21 wt. % NaAlO
2) was injected into the liquid phase by a high-pressure precision syringe pump (KD Scientific, KDS-410, Holliston, MA, USA) and the hydrothermal treatment continued for another hour at 453 K. The reaction was then terminated and the membrane was recovered. The thus synthesized membrane is denoted as M2Al hereafter. Membrane M2Al underwent the same washing, drying and calcining processes as used for preparation of M1Si.
To confirm the formation of a ZSM-5 thin skin on the outer surface of the silicalite crystals by the above described synthesis process, silicalite crystals were synthesized with and without the second stage treatment in NaAlO2 containing solution following the exact procedures used for the syntheses of the two kinds of membranes. The pure-silica MFI-type zeolite (i.e., silicalite) crystals were synthesized using the same aluminum-free precursor and reaction temperature (453 K) and duration (5 h). The thus obtained silicalite particles are denoted as P1Si. In synthesis of the silicalite crystals with a ZSM-5 outer surface, 1.2 mL 21wt. % NaAlO2 solution was introduced into the above silicalite synthesis solution (~25 mL) after 5 h of reaction and the hydrothermal treatment continued for another hour. The thus obtained zeolite particles, which have a ZSM-5 outer surface, are denoted as P2Al.
Both zeolite particulate samples were extensively washed to ensure that any aluminum ions adsorbed on the external surface were completely removed. The washing process including the following consecutive steps: (1) repeated process of zeolite particle dispersion in DI water followed by filtration and rinsing until the filtered water reached pH of ~7; (2) surface cleaning by treating the particles in a 0.1 M NaOH solution under stirring and subsequent ultrasonication for 1 h; (3) washing the NaOH solution-treated zeolite particles by DI water until pH of the washing water reached pH of ~7; (4) treating the cleaned particles in 0.1 M HNO3 solution under stirring and subsequent ultrasonication for 1 h; and (5) final cleaning by rinsing the zeolite particles with DI water until the filtered water reached pH of ~7. This rigorous surface cleaning process was repeated twice to ensure the complete removal of any aluminum ions and silicate species adsorbed to the external surface of the zeolites. The zeolite particles were then dried and calcined at 450 °C for SDA removal.
2.3. Material Characterizations
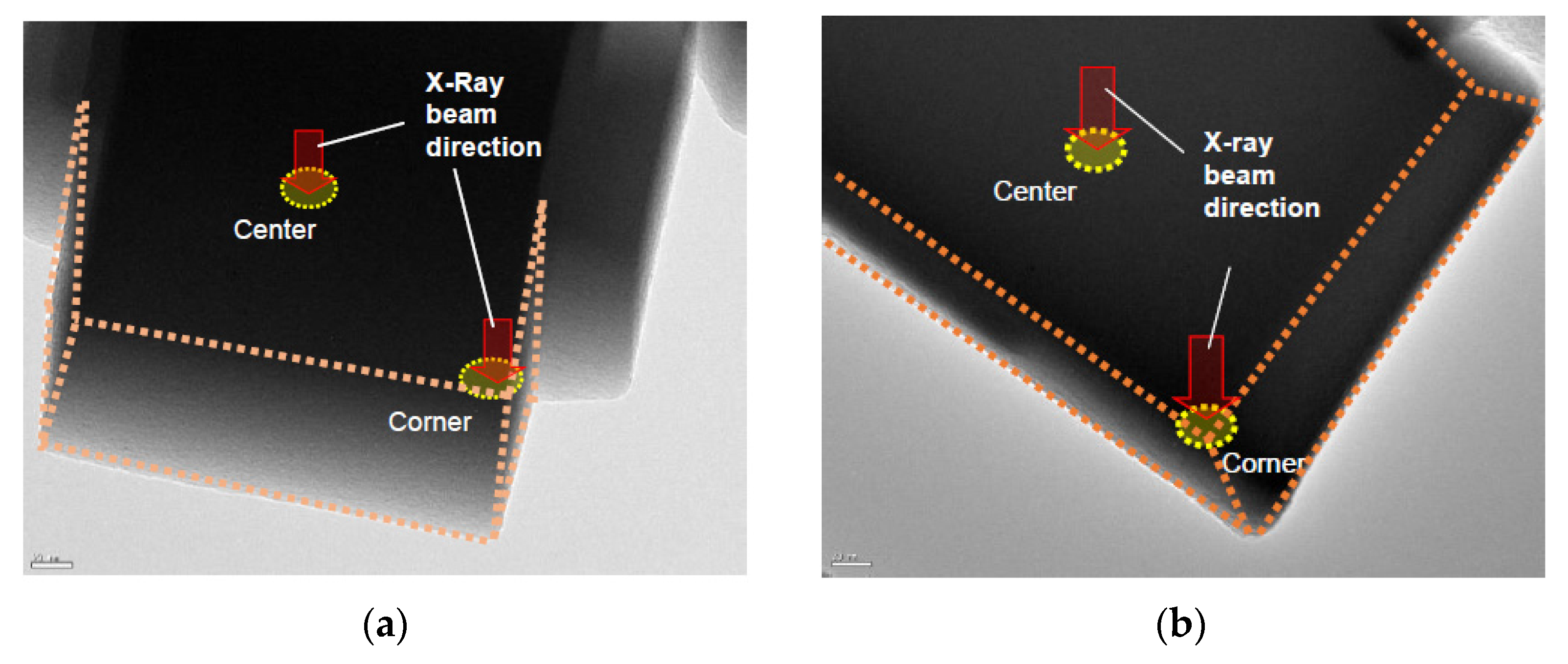
The zeolite membranes were examined by X-ray diffraction (XRD, PANalytical, X’Pert Pro MPD, The Netherlands) to confirm the crystal phase and purity. Scanning electron microscopy (SEM, FEI, XL30, Philips, USA) was used to observe the membrane morphology and determine the individual crystallite size and membrane thickness. The energy dispersive X-ray spectroscopy (EDS, EDAX, PV9761/70, NJ, USA) was employed to estimate the elemental compositions at different locations of the zeolite crystals in combination with the transmission electron microscopy (TEM; FEI CM20, Philips, USA).
2.4. Membrane Modification
Figure 2 shows the schematic diagram of the experimental system for CCD modification of the membranes that was essentially the same as that described in our previous publication [
12]. This apparatus was also used for membrane gas mixture separation measurements. The disc-shaped zeolite membrane was mounted in a stainless-steel permeation cell using soft graphite gasket seals (Mercer Gasket & Shim, NJ, USA). The membrane permeation cell was placed in a temperature-programmable furnace.
The CCD modification started with permeation of an equimolar H2/CO2 mixture gas fed at a total flow rate of 40 cm3 (STP)/min when the permeate side was swept by a helium stream at a flow rate of 30 cm3 (STP)/min. The partial pressure of MDES in the H2/CO2 carrier gas was determined to be ~4.3 kPa from the room temperature saturator. The temperature for CCD modification was 723 K at which MDES decomposition occurs catalytically at [AlO2−] sites but not on pure silicalite surface, which is noncatalytic. During the gas permeation, the membrane cell was heated up to 723 K at a heating rate of 0.5 K/min and kept at 723 K throughout the modification process. The H2/CO2 mixture feed was then switched to bubble through the MDES liquid column to carry MDES vapor before entering the membrane cell. The permeate stream was continuously analyzed by an online gas chromatographer (GC, Agilent 6890N, USA) equipped with a Carboxen 1000 packed column (Supelco, USA) and a thermal conductivity detector (TCD). The H2/CO2 separation through the zeolite membrane was continuously monitored during the entire CCD modification process. The feed gas flow was switched back to dry gas feeding route to stop the MDES supply when the online monitored H2/CO2 separation factor and gas permeance were stabilized for 2 h.
2.5. Gas Permeation
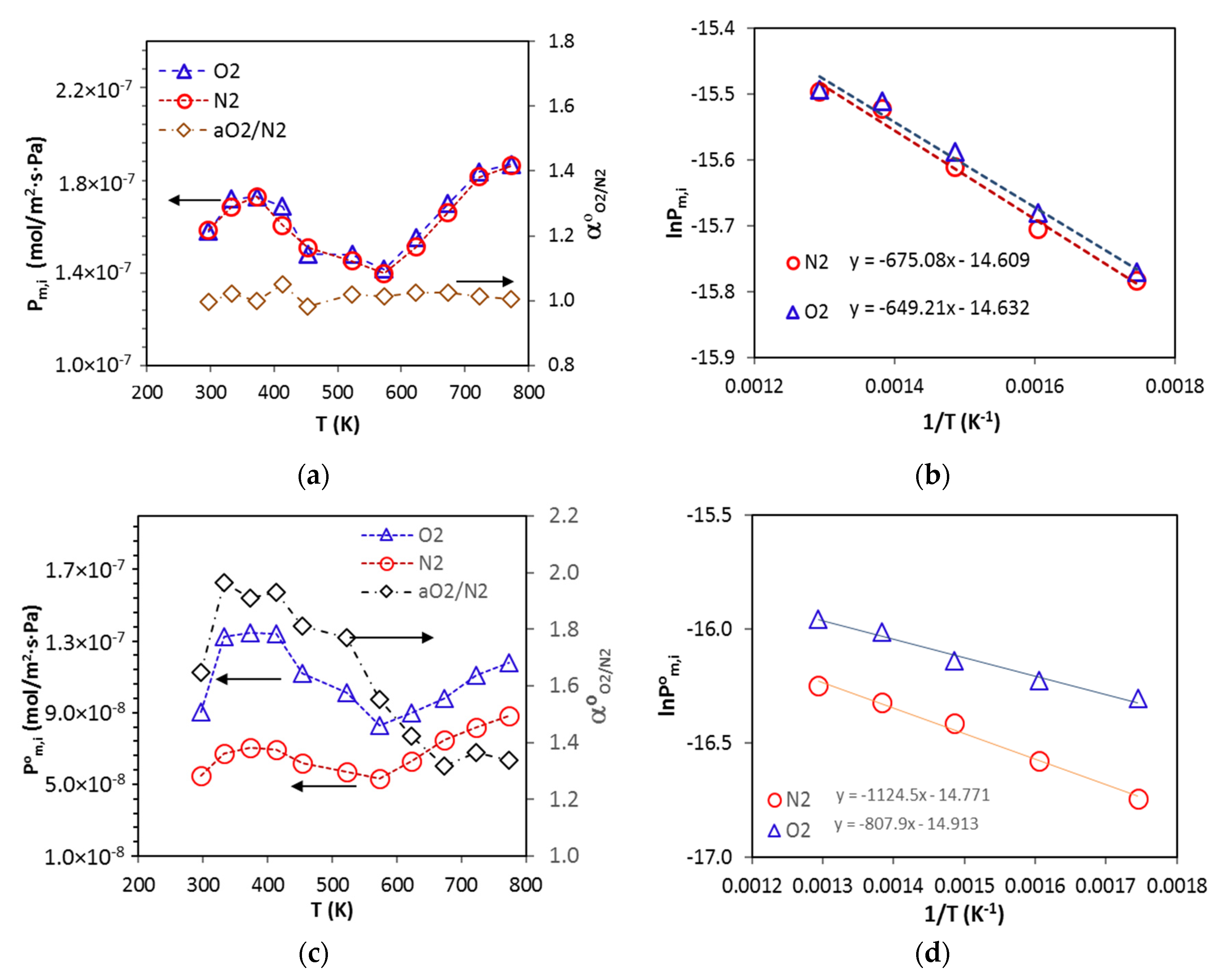
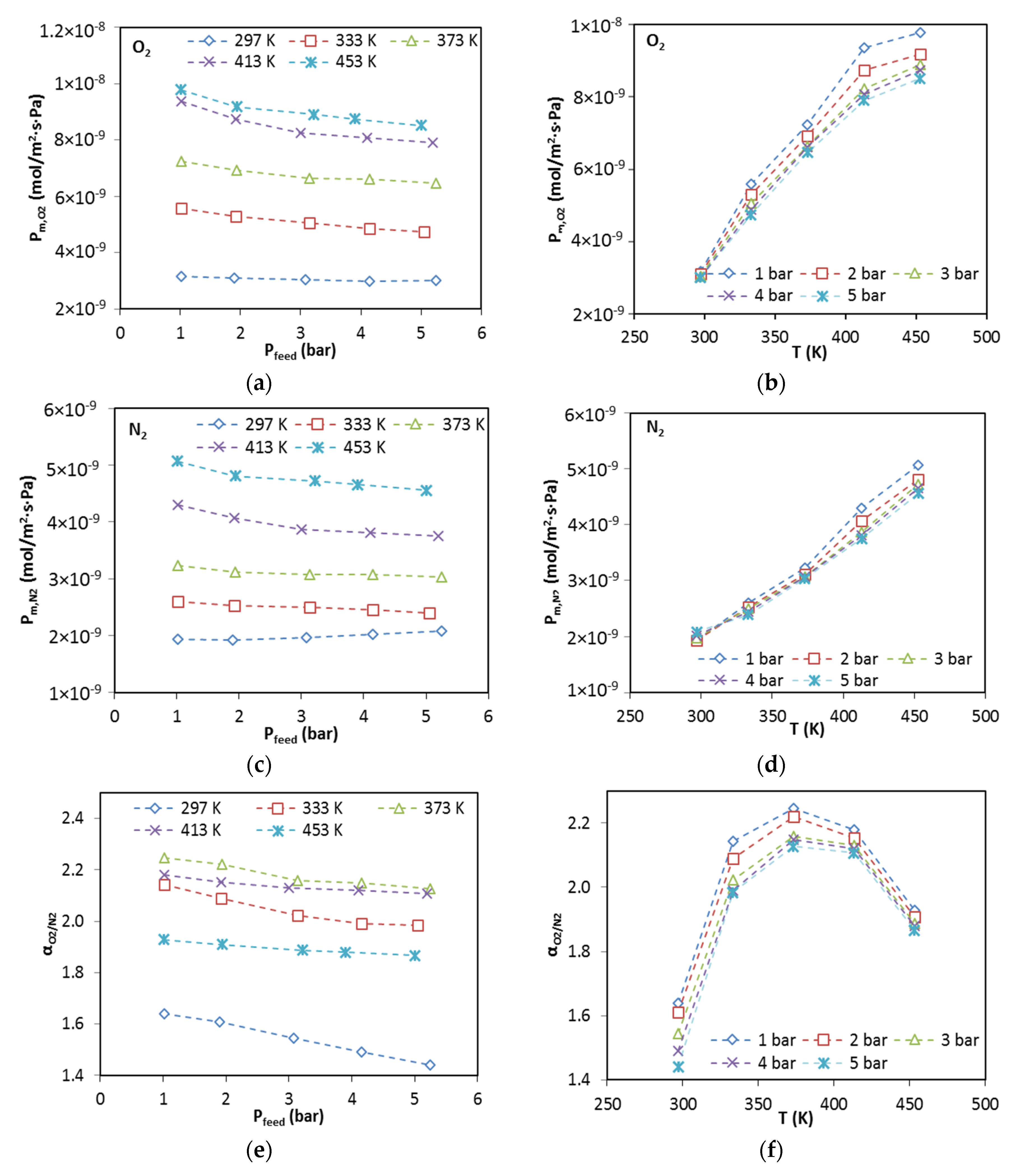
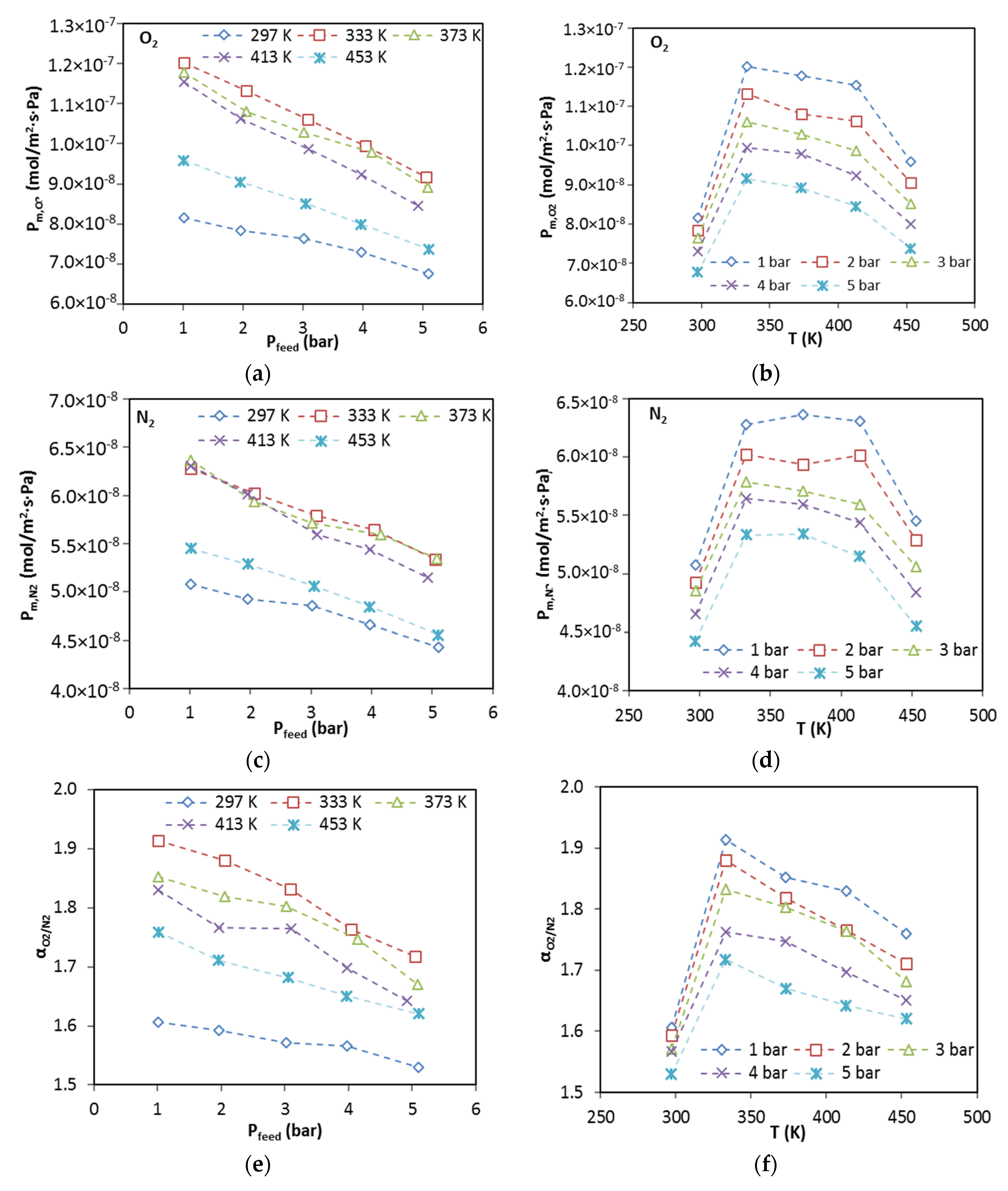
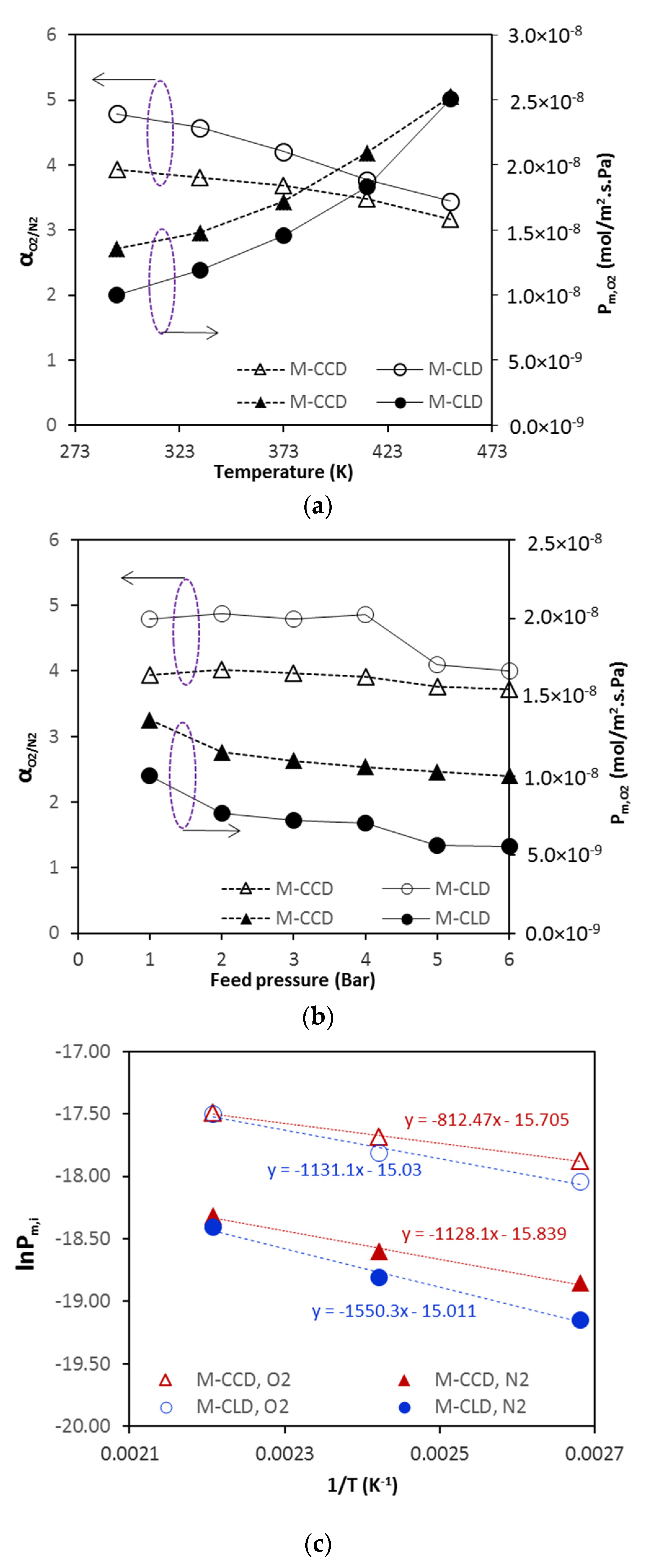
The membrane was degassed in the permeation cell by vacuuming at 453 K for 12 h prior to testing permeation for each gas. The permeance of pure gases was measured by the transient permeation method in a temperature range from 297 to 773 K with feed pressure of 2 bar using an experimental setup reported a previous publication [
16]. The separation of gas mixtures was performed by the conventional steady-state permeation setup (
Figure 2) at a feed flow rate of 40 cm
3 (STP)/min and a helium sweep flow rate of 30 cm
3 (STP)/min. The permeance for gas
i (
Pm,i), permselectivity (i.e., ideal selectivity) of gas
i over gas
j (
) and separation factor of gas
i over gas
j in mixture (
) are defined as follows:
where
Q (mol) is the moles of gas
i permeated through the membrane over a time period of
t (s); Δ
Pi is the partial pressure difference (i.e., Δ
Pi =
Pi,f −
Pi,p, where
Pi,f and
Pi,p are the partial pressures of gas component
i in the feed and permeate sides, respectively);
and
are pure gas permeance of gas
i and gas
j, respectively; and
x and
y are molar compositions of the feed and permeate gas mixtures, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













