Accelerating Biologics Manufacturing by Modeling: Process Integration of Precipitation in mAb Downstream Processing



Abstract
:1. Introduction
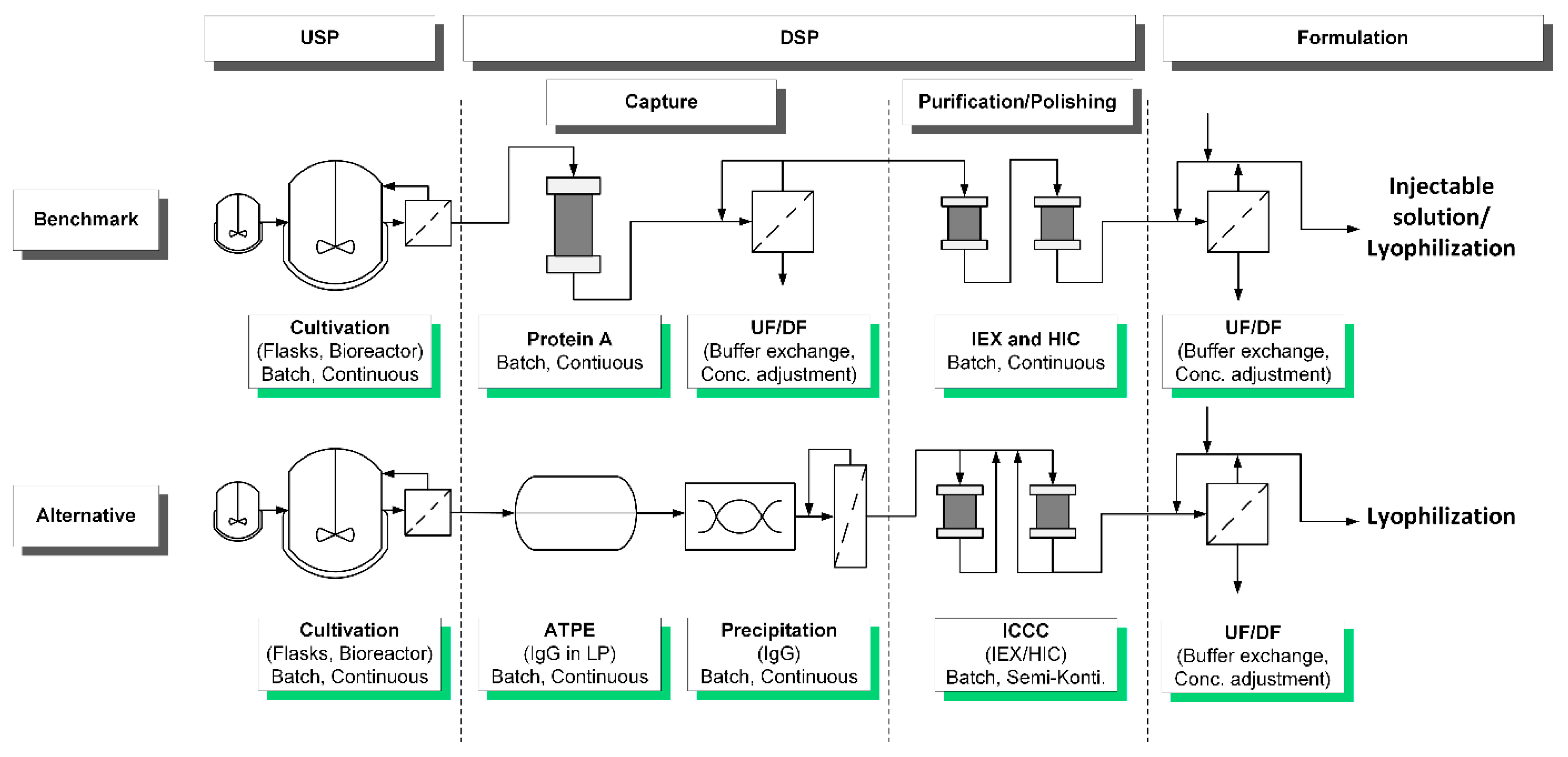
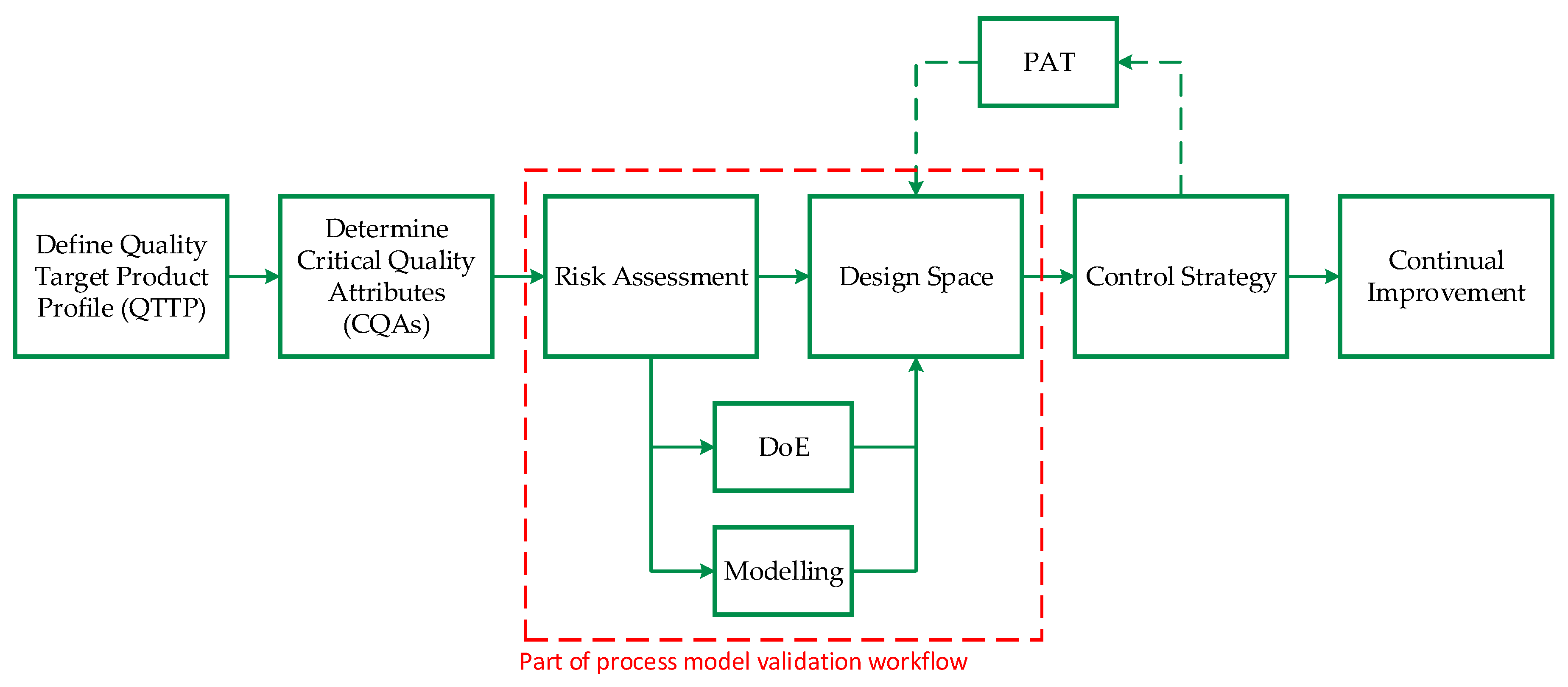
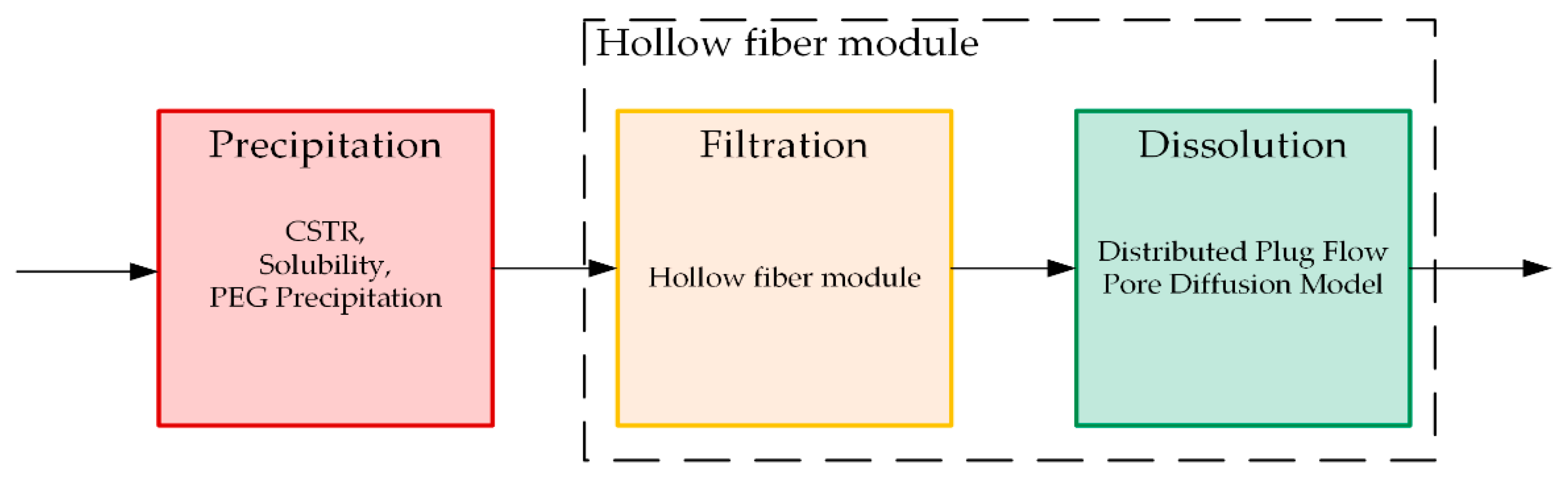
2. Process Integration Applying Quality by Design
- Precipitation of IgG;
- Loading of hollow fiber membrane;
- Precipitate wash;
- Dissolution of the target;
- Filtration (intermediate product).
3. Material and Methods
3.1. Cultivation
3.2. Aqueous Two Phase Extraction (ATPE)
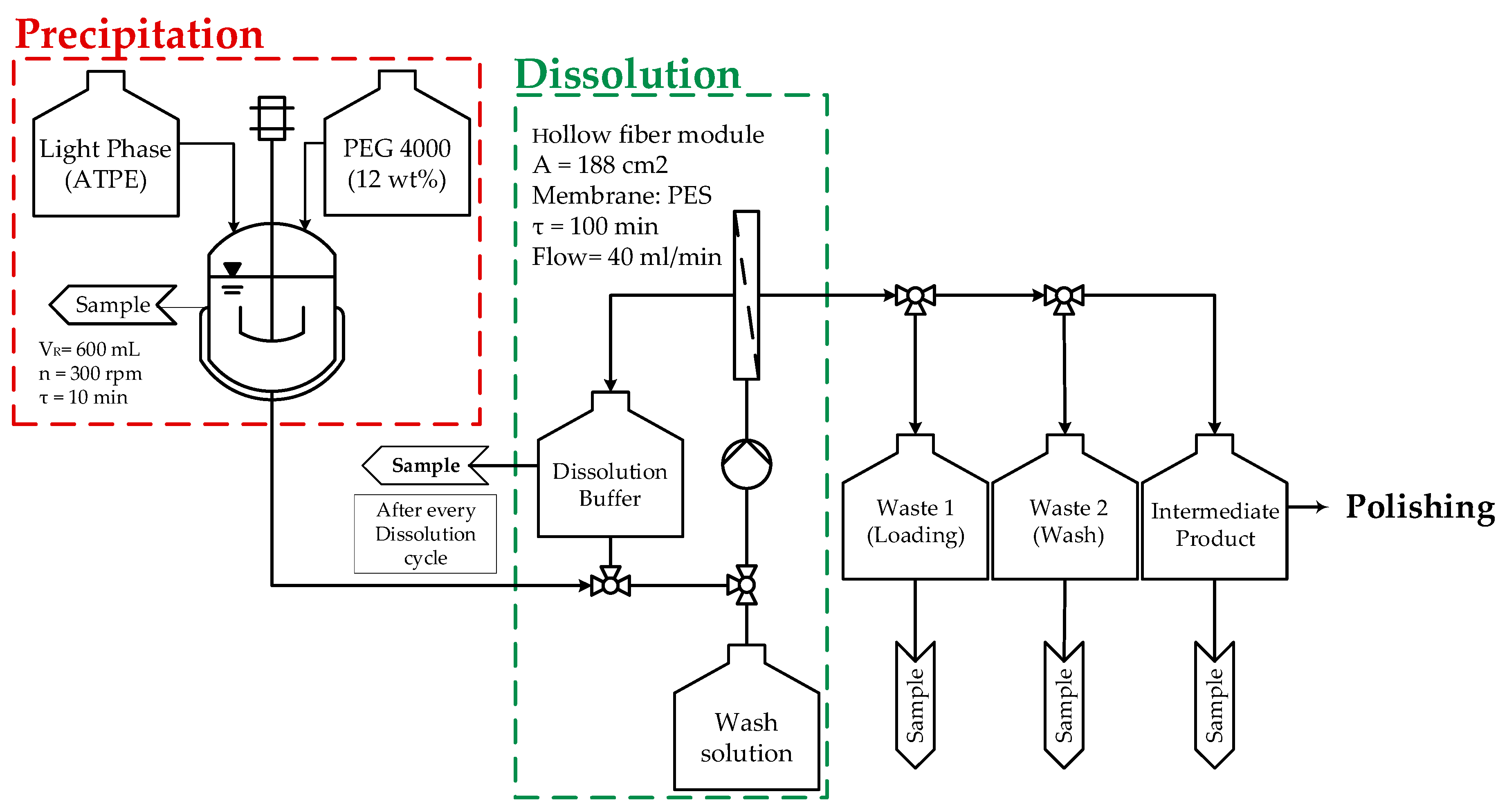
3.3. Precipitation
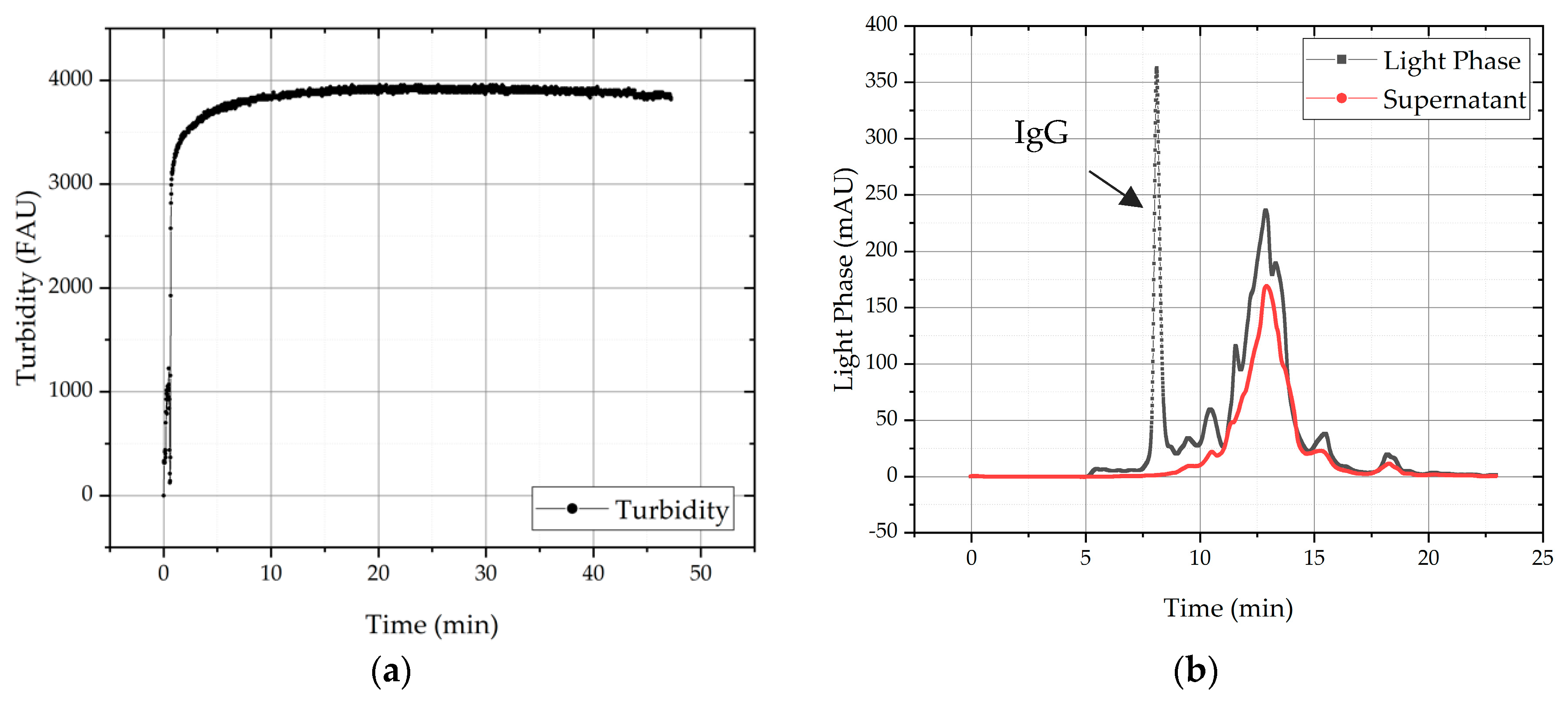
3.4. Solid–Liquid Separation and Precipitate Wash
3.5. Dissolution
3.6. Screening of Washing Solutions
3.7. Analytics
4. Results
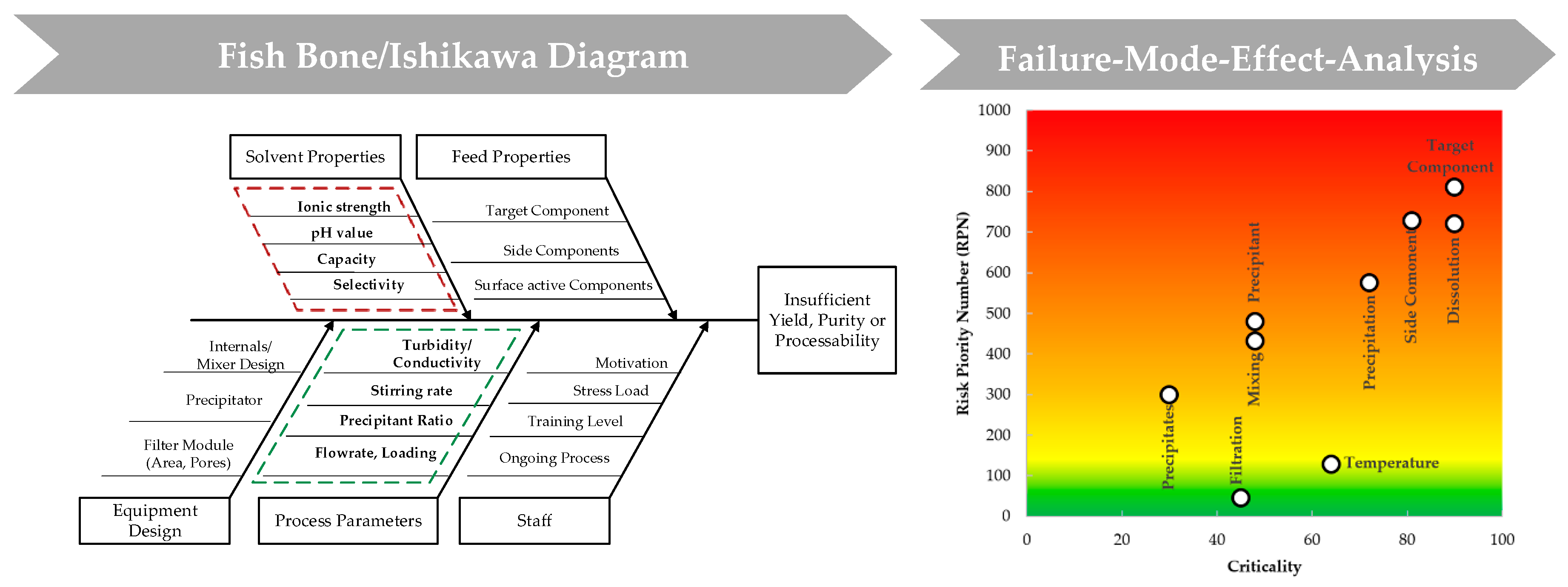
4.1. Results Risk Assessment: Precipitation
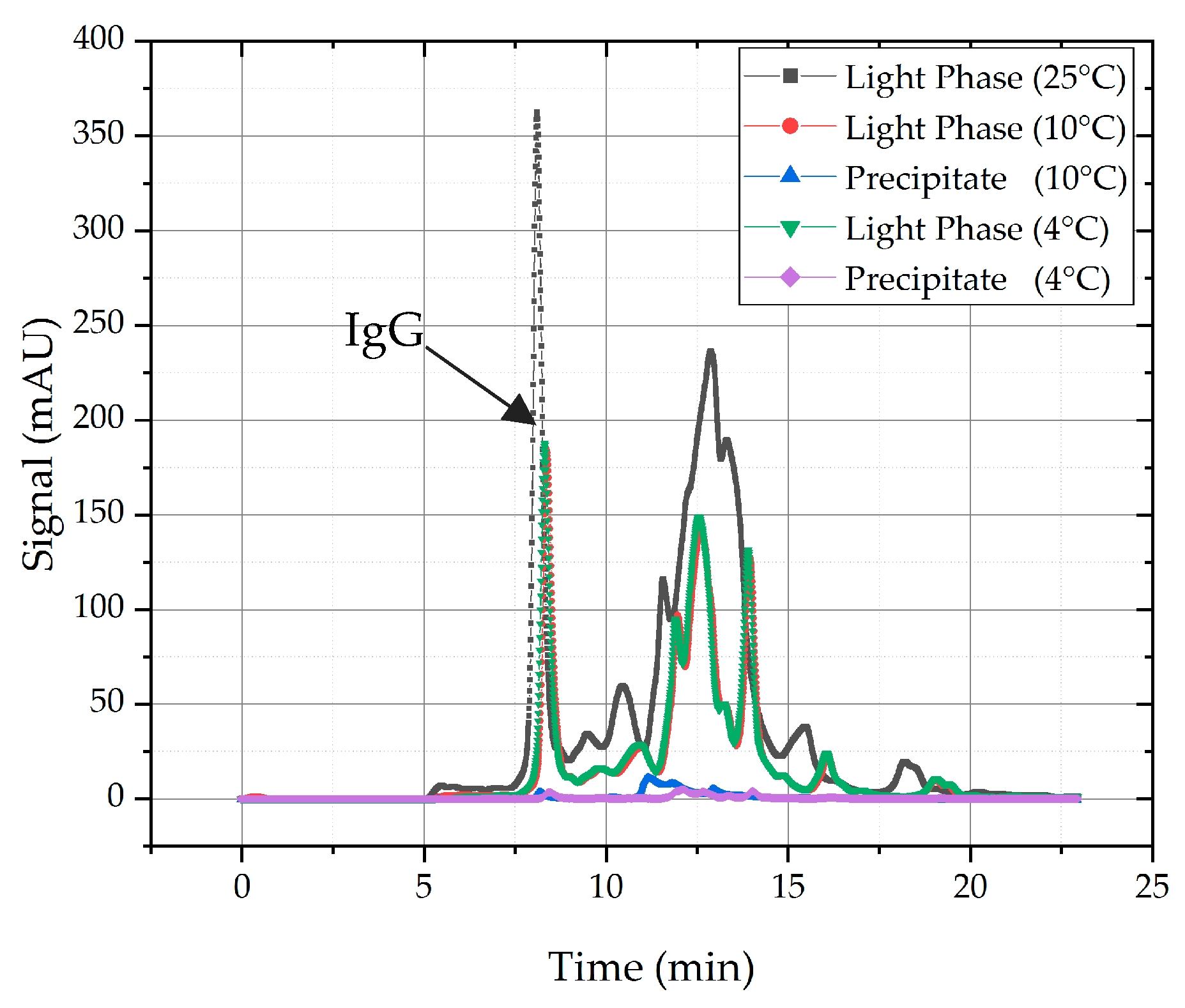
4.1.1. Temperature
4.1.2. Mixing Time
4.1.3. Precipitant
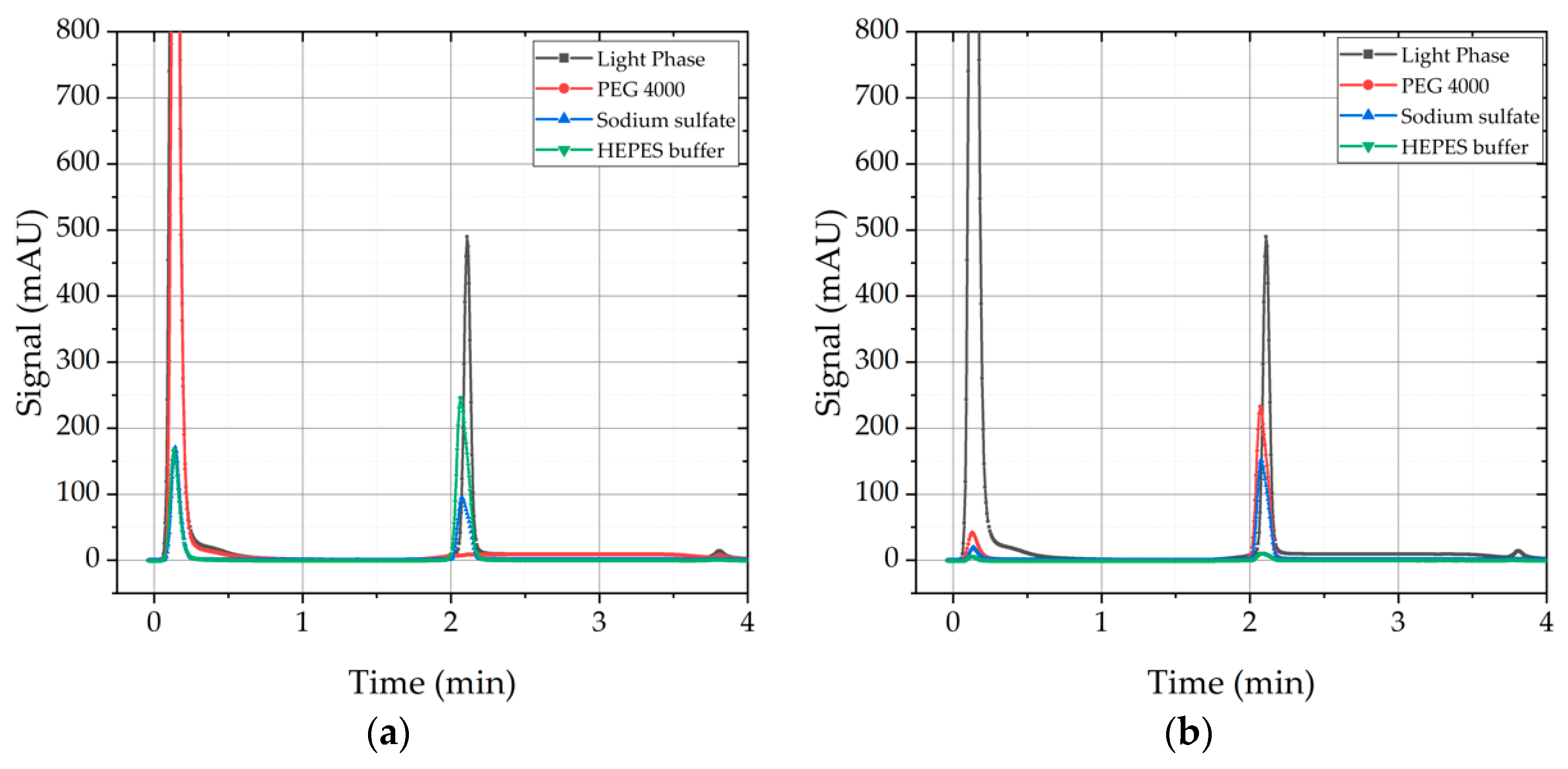
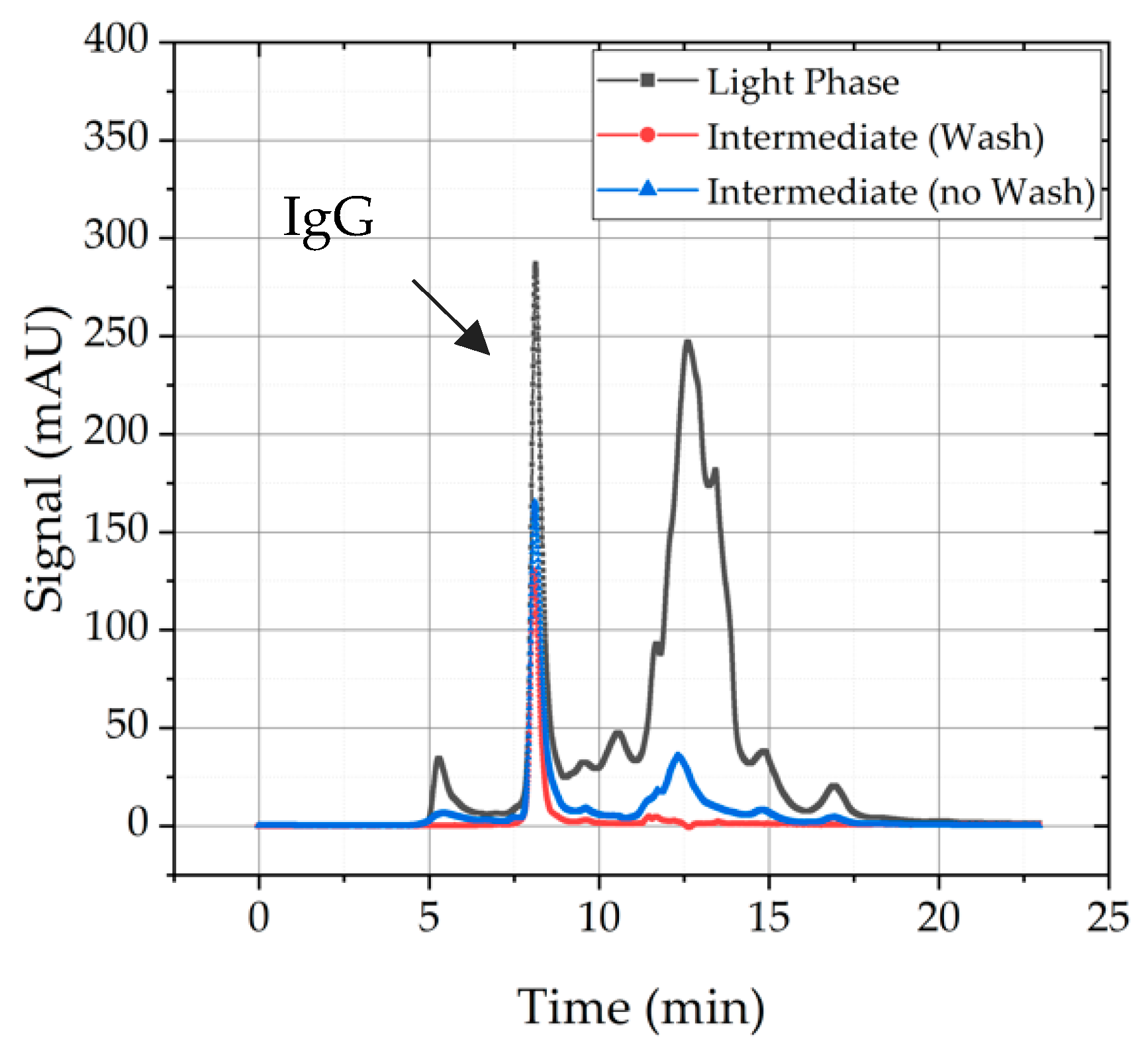
4.1.4. Precipitate Wash
4.2. Results Risk Assessment: Dissolution
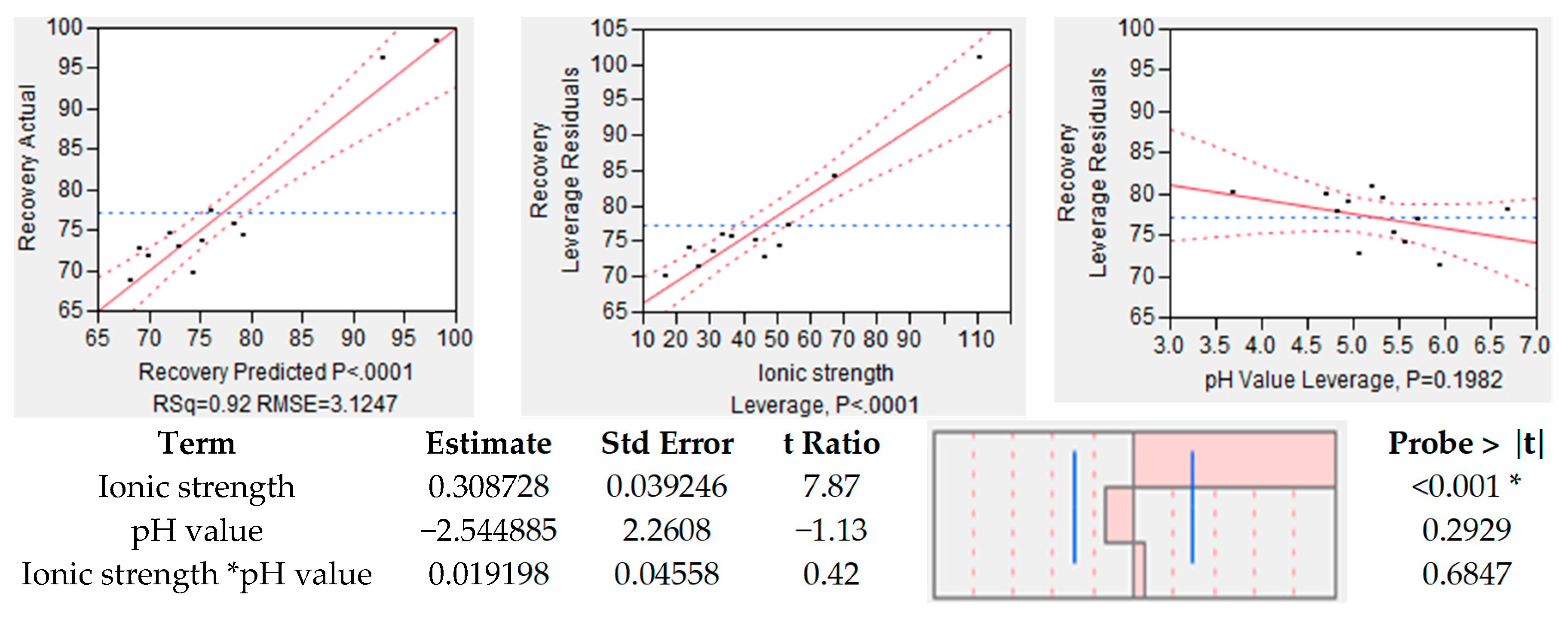
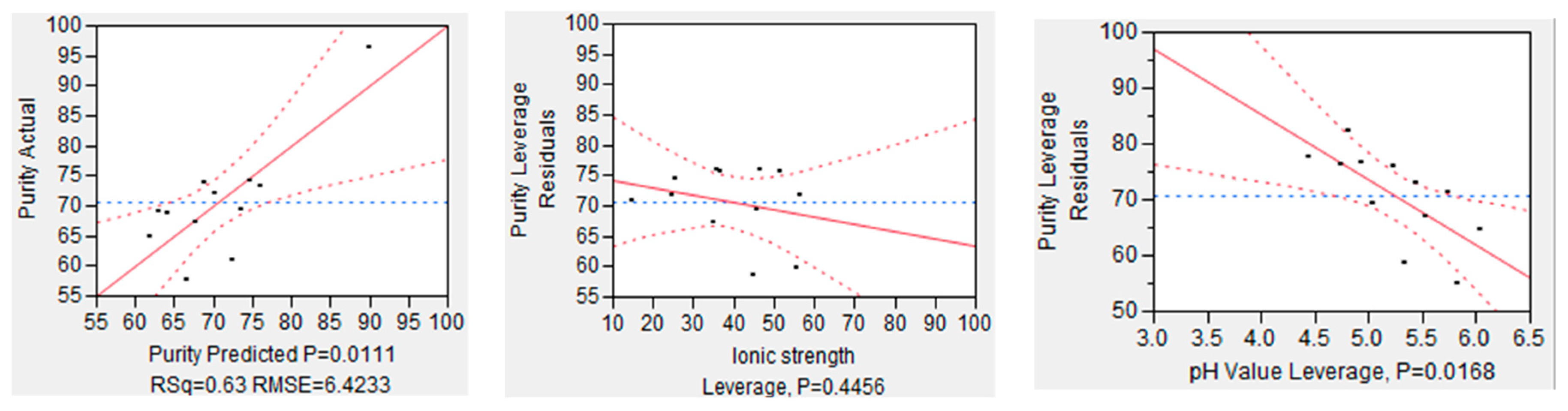
4.2.1. Ionic Strength and pH-Value
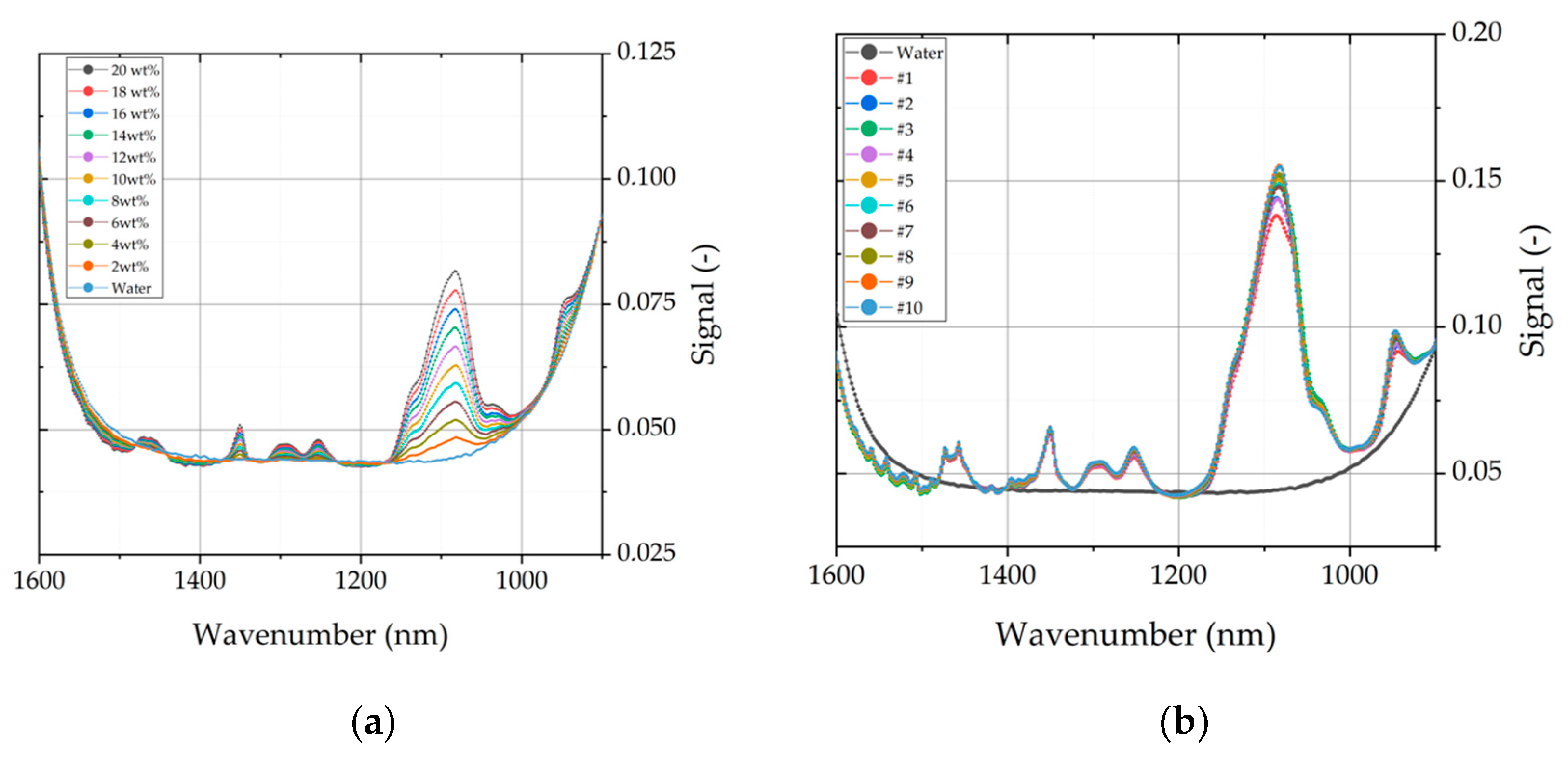
4.2.2. Determination of PEG Content
4.2.3. Dissolution Cycles and Recovery
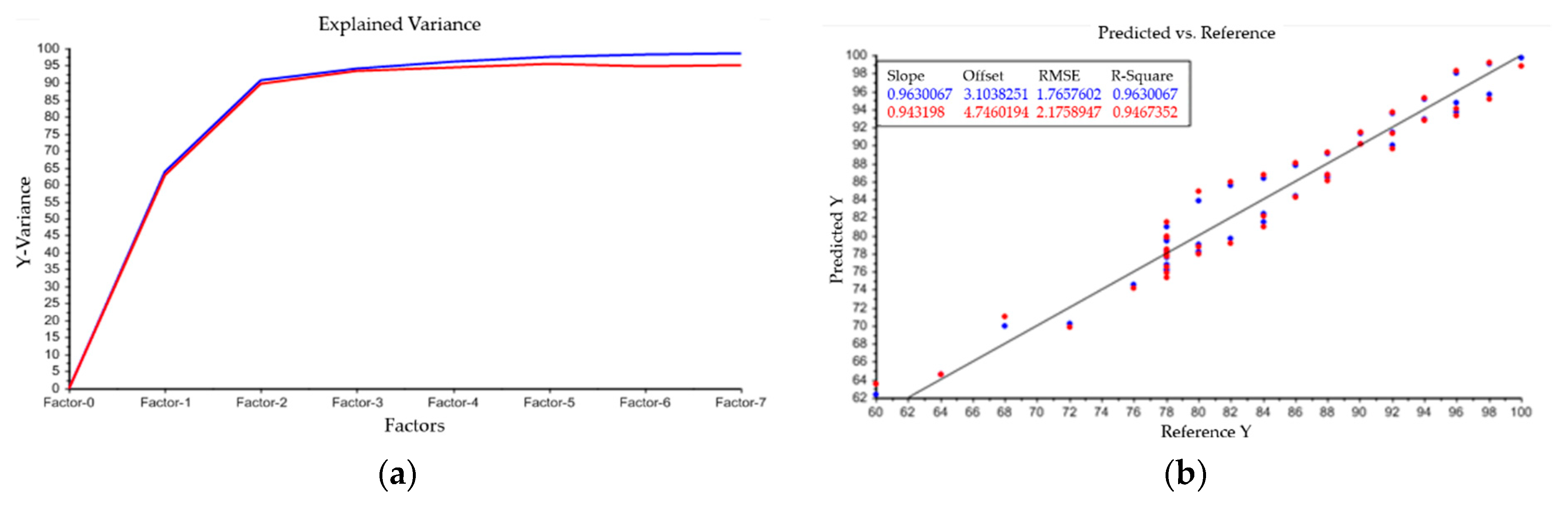
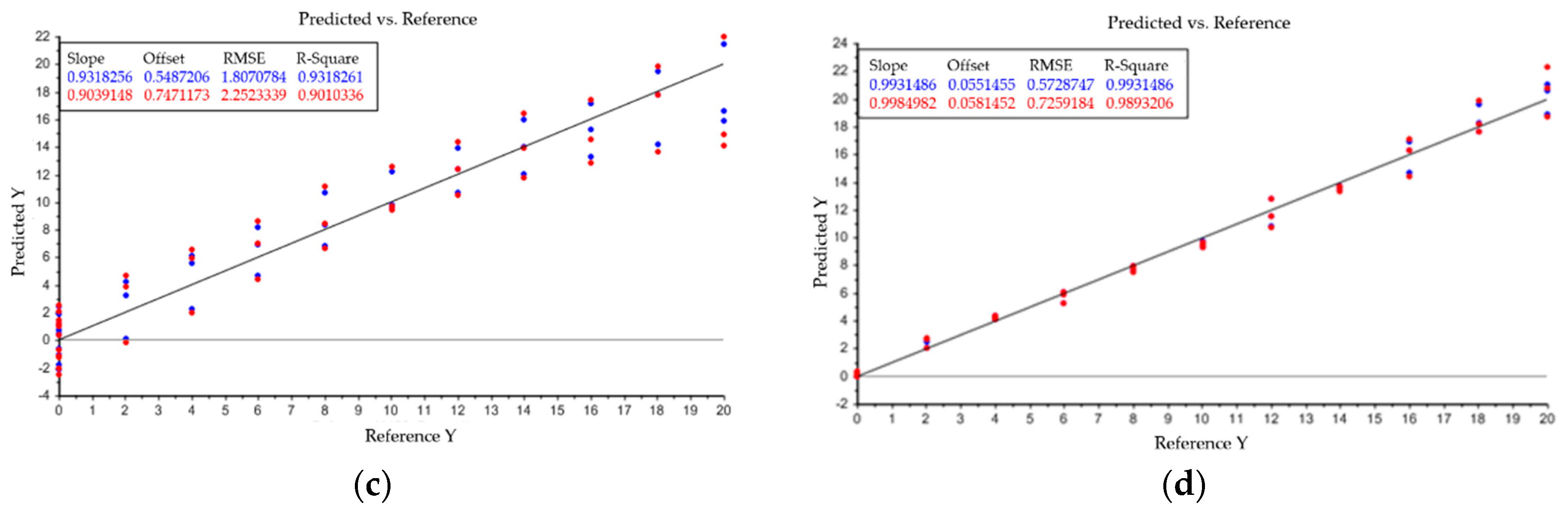
5. Modeling of Precipitation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

| Severity (S) | Occurrence (O) | Detection (D) | Criticality (SxO) | RPN (SxOxD) | Comment | |
|---|---|---|---|---|---|---|
| Target Component | 10 | 9 | 9 | 90 | 810 | Is expected to be in good control, once characterized in feed. However, detection is offline and cannot be monitored during process. Must be controlled to avoid yield loss. Parameters: recovery and purity |
| Side Component | 9 | 9 | 9 | 81 | 729 | Is expected to be in good control, once characterized in feed. However, detection is offline and cannot be monitored during process. Must be controlled to avoid ineffectiveness and underperformance of unit. Parameters: purity |
| Precipitation | 9 | 6 | 9 | 54 | 486 | Scope of precipitation => complete/incomplete. Must be controlled for effective precipitation and to avoid yield loss. Precipitation of target component demands that biological activity is retained. |
| Precipitant | 8 | 6 | 10 | 48 | 480 | Suitable precipitant for selective precipitation. Might be harmful for following purification step. Precipitant and protein ratio is essential for complete precipitation. Difficulties by detecting correct ratio. |
| Precipitate/Particles | 6 | 5 | 10 | 30 | 300 | Shape of precipitates is not crucial for the product after the unit, because it is a temporary state. It might influence dissolution and be harmful to filters. Detection of shape during process is difficult. |
| Temperature | 8 | 8 | 2 | 64 | 128 | Is expected to be in good control, once temperature range is characterized. Temperature shifts lead to crystallization/precipitation and can effect purity of the target. Temperature can be detected easily. |
| Mixing | 8 | 6 | 9 | 48 | 432 | Mixing is essential for precipitation. Complete and selective precipitation occurs due to the correct ratio of precipitant and protein. Mixing state is difficult to detect and influences scope of precipitation. It has to be considered particularly in large scale. Parameters: stirring rate, mixing time, vessel volume. |
| Filtration | 9 | 5 | 1 | 45 | 45 | Filtration is a function of flux, mAb concentration and pressure. In small scale less important. Has to be considered in Up-Scale. |
| Dissolution | 10 | 9 | 8 | 90 | 720 | Dissolution is essential for recovery of product and leads to high yield loss and ineffectiveness of the unit. Parameters: selectivity, capacity, dissolution buffer ratio and dwell time. |
| Process Step | Flow Chart |
|---|---|
| Precipitation: Precipitant: PEG 4000 (12 wt%) VR = 600 mL τ = 10 min n = 300 rpm |  |
| Loading: Hollow fiber module is used for solid-liquid separation Pore size: 0.2 µm A = 470 cm2 P = 250 mbar Membrane: Polyether sulfone (PES) |  |
| Wash: PEG 4000 (12 wt%) is selected as wash solution This amount of PEG 4000 ensures no dissolution of mAb Reduction of surficial HCPs on precipitates |  |
| Dissolution: Dissolution ratio can be adjusted by added volume of dissolution buffer Dissolution ration of 1:1 is desired Buffer volume is recycled 10 times to enlarge dwell time but small dissolution ratio |  |
| Filtration/Intermediate Product Filtration of recycled dissolution buffer through filter module |  |
| Process Parameters | Process Parameter Range | Unit | Purity IgG | Yield IgG | Rationale |
|---|---|---|---|---|---|
| Precipitation | |||||
| Temperature | 4 to 25 | °C | No | No | No impact in this range, thus not include in this study. Precipitation occurs more rapidly by low temperatures but it does not affect purity and yield of mAb. |
| Mixing Time | 1 to 60 | min | Low | Low | Low effect of Precipitation duration; precipitation occurs immediately. |
| Stirring rate | 200 to 400 | rpm | No | No | Less important in small scale, precipitation occurs directly after addition of PEG. Should be taken into account for large scale operation. |
| Precipitant | 1450–12000 | MW | High | High | Screening of different PEGs. Due to prior knowledge PEG 1450 was set as starting point. |
| Precipitant ratio | 10 to 16 | wt% PEG | High | High | No mAb was found in supernatant when precipitant ratio was higher than 12 wt%; below this concentration complete precipitation could not be guaranteed/ensured for PEG 4000. |
| Precipitate Wash | Yes/no | - | Medium | Low | Wash solutions: Ammonium sulfate, Sodium sulfate, PEG 4000, HEPES buffer. Improved Purity due to wash, but low effect on recovery of mAb from precipitate. |
| Dissolution | |||||
| Ionic Strength | 0 to 150 | mM | High | High | Exploitation of salting-in effect. Up to 100 mM addition of salt has a stabilizing effect on proteins in solution, various salts have different effectiveness. |
| pH-Value | 3 to 6 | - | High | High | low pH-Values lead to better dissolution of mAb; simultaneously dissolution of side components might occur. |
| Dissolution volume ratio | 1–16 | - | Medium | Medium | Dissolution volume ratio has a medium impact on dissolution. Yield of dissolution IgG is higher when concentration gradient is greater. |
| Dissolution cycles | 1–10 | - | Medium | Medium | Capacity and concentration gradient is important for dissolution. |


References
- BioPharm. The Renaissance of Protein Precipitation. BioPharm Int. 2006, 19, 8. [Google Scholar]
- EvaluatePharma. World Preview 2017: Outlook to 2022. 2017. Available online: www.evaluate.com/PharmaWorldPreview2017 (accessed on 4 September 2019).
- Dos Santos, R.; Carvalho, A.L.; Roque, A.C.A. Renaissance of protein crystallization and precipitation in biopharmaceuticals purification. Biotechnol. Adv. 2017, 35, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Arunkumar, A.; Chollangi, S.; Tan, Z.G.; Borys, M.; Li, Z.J. Clarification technologies for monoclonal antibody manufacturing processes: Current state and future perspectives. Biotechnol. Bioeng. 2016, 113, 698–716. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Hubbuch, J. Downstream process development strategies for effective bioprocesses: Trends, progress, and combinatorial approaches. Eng. Life Sci. 2017, 17, 1142–1158. [Google Scholar] [CrossRef]
- Cho, B.S.; Kim, J.O.; Ha, D.H.; Yi, Y.W. Exosomes derived from human adipose tissue-derived mesenchymal stem cells alleviate atopic dermatitis. Stem Cell Res. Ther. 2018, 9, 187. [Google Scholar] [CrossRef] [Green Version]
- Fuenmayor, J.; Gòdia, F.; Cervera, L. Production of virus-like particles for vaccines. New Biotechnol. 2017, 39, 174–180. [Google Scholar] [CrossRef]
- Gagnon, P. Technology trends in antibody purification. J. Chromatogr. A 2012, 1221, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Spitali, M.; Norrant, E.L.; Bracewell, D.G. Precipitation as an Enabling Technology for the Intensification of Biopharmaceutical Manufacture. Trends Biotechnol. 2019, 37, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerschmidt, N.; Tscheliessnig, A.; Sommer, R.; Helk, B.; Jungbauer, A. Economics of recombinant antibody production processes at various scales: Industry-standard compared to continuous precipitation. Biotechnol. J. 2014, 9, 766–775. [Google Scholar] [CrossRef]
- Burgstaller, D.; Jungbauer, A.; Satzer, P. Continuous integrated antibody precipitation with two-stage tangential flow microfiltration enables constant mass flow. Biotechnol. Bioeng. 2019, 116, 1053–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Gu, Q.; Coffman, J.L.; Przybycien, T.; Zydney, A.L. Continuous precipitation for monoclonal antibody capture using countercurrent washing by microfiltration. Biotechnol. Prog. 2019, e2886. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, N.; Hobiger, S.; Jungbauer, A. Continuous polyethylene glycol precipitation of recombinant antibodies: Sequential precipitation and resolubilization. Process Biochem. 2016, 51, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Swartz, A.R.; Xu, X.; Traylor, S.J.; Li, Z.J.; Chen, W. One-step affinity capture and precipitation for improved purification of an industrial monoclonal antibody using Z-ELP functionalized nanocages. Biotechnol. Bioeng. 2018, 115, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Gisela, S.M. The Affinity Precipitat Ion for the Isolat Ion of Biomolecules; EPFL: Lausanne, Switzerland, 2007. [Google Scholar]
- Hilbrig, F.; Freitag, R. Protein purification by affinity precipitation. J. Chromatogr. B 2003, 790, 79–90. [Google Scholar] [CrossRef]
- Scopes, R.K. Protein Purification. Principles and Practice, 3rd ed.; Springer: New York, NY, USA, 1994; ISBN 0387940723. [Google Scholar]
- Chmiel, H. Bioprozesstechnik; 3rd neu bearb. Aufl.; Spektrum Akademischer Verlag: Heidelberg, Germany, 2011. [Google Scholar]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Forrer, K.; Helk, B.; Trout, B.L. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol. J. 2011, 6, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Glycosylation of recombinant antibody therapeutics. Biotechnol. Prog. 2005, 21, 11–16. [Google Scholar] [CrossRef]
- Raju, T.S. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr. Opin. Immunol. 2008, 20, 471–478. [Google Scholar] [CrossRef]
- Hodoniczky, J.; Zheng, Y.Z.; James, D.C. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol. Prog. 2005, 21, 1644–1652. [Google Scholar] [CrossRef]
- Abès, R.; Teillaud, J.-L. Impact of Glycosylation on Effector Functions of Therapeutic IgG. Pharmaceuticals 2010, 3, 146–157. [Google Scholar] [CrossRef]
- Wright, A.; Morrison, S.L. Effect of glycosylation on Effect og glycosylation on antibody function: Implications for genetic engineering. TIBTECH 1997, 15, 26–32. [Google Scholar] [CrossRef]
- del Val, I.J.; Kontoravdi, C.; Nagy, J.M. Towards the implementation of quality by design to the production of therapeutic monoclonal antibodies with desired glycosylation patterns. Biotechnol. Prog. 2010, 26, 1505–1527. [Google Scholar] [CrossRef] [PubMed]
- Atha, D.H.; Ingham, K.C. Mechanism of Precipiation of Proteins by polyethylene Glycols. J. Biochem. Chem. 1981, 256, 12108–12117. [Google Scholar]
- Thompson, R.W.; Latypov, R.F.; Wang, Y.; Lomakin, A.; Meyer, J.A.; Vunnum, S.; Benedek, G.B. Evaluation of effects of pH and ionic strength on colloidal stability of IgG solutions by PEG-induced liquid-liquid phase separation. J. Chem. Phys. 2016, 145, 185101. [Google Scholar] [CrossRef] [PubMed]
- Oelmeier, S.A.; Ladd-Effio, C.; Hubbuch, J. Alternative separation steps for monoclonal antibody purification: Combination of centrifugal partitioning chromatography and precipitation. J. Chromatogr. A 2013, 1319, 118–126. [Google Scholar] [CrossRef]
- Brodsky, Y.; Zhang, C.; Yigzaw, Y.; Vedantham, G. Caprylic acid precipitation method for impurity reduction: An alternative to conventional chromatography for monoclonal antibody purification. Biotechnol. Bioeng. 2012, 109, 2589–2598. [Google Scholar] [CrossRef]
- Christen, P.; Jaussi, R.; Benoit, R. Biochemie und Molekularbiologie; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Großhans, S.; Wang, G.; Fischer, C.; Hubbuch, J. An integrated precipitation and ion-exchange chromatography process for antibody manufacturing: Process development strategy and continuous chromatography exploration. J. Chromatogr. A 2018, 1533, 66–76. [Google Scholar] [CrossRef]
- Sim, S.-L.; He, T.; Tscheliessnig, A.; Mueller, M.; Tan, R.B.H.; Jungbauer, A. Protein precipitation by polyethylene glycol: A generalized model based on hydrodynamic radius. J. Biotechnol. 2012, 157, 315–319. [Google Scholar] [CrossRef]
- Giese, G.; Myrold, A.; Gorrell, J.; Persson, J. Purification of antibodies by precipitating impurities using Polyethylene Glycol to enable a two chromatography step process. J. Chromatogr. B 2013, 938, 14–21. [Google Scholar] [CrossRef]
- Zhou, Y. Mathematical Modelling of Protein Precipitation Based on the Phase Equilibrium for an Antibody Fragment from E. coli Lysis. J. Bioprocess. Biotech. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.C.; Hämmerling, F.; Kittelmann, J.; Dürr, C.; Görlich, F.; Hubbuch, J. Influence of structure properties on protein-protein interactions-QSAR modeling of changes in diffusion coefficients. Biotechnol. Bioeng. 2017, 114, 821–831. [Google Scholar] [CrossRef]
- Hämmerling, F.; Ladd Effio, C.; Andris, S.; Kittelmann, J.; Hubbuch, J. Investigation and prediction of protein precipitation by polyethylene glycol using quantitative structure-activity relationship models. J. Biotechnol. 2017, 241, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.-L.; He, T.; Tscheliessnig, A.; Mueller, M.; Tan, R.B.H.; Jungbauer, A. Branched polyethylene glycol for protein precipitation. Biotechnol. Bioeng. 2012, 109, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Asakura, S.; Oosawa, F. Interaction between particles suspended in solutions of macromolecules. J. Polym. Sci. 1958, 33, 183–192. [Google Scholar] [CrossRef]
- Iverius, P.H.; Laurent, T.C. Precipitation of some plasma proteins by the addition of dextran or polyethylene glycol. Biochim. Biophys. Acta (BBA) Protein Struct. 1967, 133, 371–373. [Google Scholar] [CrossRef]
- Cohn, E.J.; Strong, L.E. Preparation and properties of serum and plasma proteins; a system for the separation into fractions of the protein and lipoprotein components of biological tissues and fluids. J. Am. Chem. Soc. 1946, 68, 459–475. [Google Scholar] [CrossRef]
- Glynn, J. Process-Scale Precipitation of Impurities in mammalian Cell Culture Broth. In Process Scale Purification of Antibodies; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 309–324. [Google Scholar]
- Wang, G. Advancing Downstream Process Development—Mechanistic Modeling and Artificial Intelligence. Available online: https://d-nb.info/1163320358/34 (accessed on 7 January 2020).
- Großhans, S.; Wang, G.; Hubbuch, J. Water on hydrophobic surfaces: Mechanistic modeling of polyethylene glycol-induced protein precipitation. Bioprocess Biosyst. Eng. 2019, 42, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Thakor, N.S.; Amrutkar, S.V. Implementing Quality by Design (QbD) in Chromatography: Review Article. Austin J. Anal. Pharm. Chem. 2017, 4, 1–5. [Google Scholar]
- EMA-FDA. EMA-FDA Pilot Program for Parallel Assessment of Quality-by-Design Applications: Lessons Learnt and Q&A Resulting from the First Parallel Assessment; FDA: Silver Spring, MD, USA, 2013.
- FDA. Guidance for Industry: Q9 Quality Risk Management; FDA: Silver Spring, MD, USA, 2006.
- Ich Harmonised Tripartite Guideline. 2009. Available online: https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf (accessed on 2 January 2020).
- Sixt, M.; Uhlenbrock, L.; Strube, J. Toward a Distinct and Quantitative Validation Method for Predictive Process Modelling—On the Example of Solid-Liquid Extraction Processes of Complex Plant Extracts. Processes 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Uhlenbrock, L.; Sixt, M.; Strube, J. Quality-by-Design (QbD) process evaluation for phytopharmaceuticals on the example of 10-deacetylbaccatin III from yew. Resour. Effic. Technol. 2017, 3, 137–143. [Google Scholar] [CrossRef]
- Kornecki, M.; Mestmäcker, F.; Zobel-Roos, S.; Heikaus de Figueiredo, L.; Schlüter, H.; Strube, J. Host Cell Proteins in Biologics Manufacturing: The Good, the Bad, and the Ugly. Antibodies 2017, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Gronemeyer, P. Entwicklung Einer Methode zur Integration Von Upstream und Downstream Processing am Beispiel der Herstellung Monoklonaler Antikörper. Available online: https://www.shaker.de/de/content/catalogue/index.asp?lang=de&ID=8&ISBN=978-3-8440-5438-5&search=yes (accessed on 2 January 2020).
- Schmidt, A.; Richter, M.; Rudolph, F.; Strube, J. Integration of Aqueous Two-Phase Extraction as Cell Harvest and Capture Operation in the Manufacturing Process of Monoclonal Antibodies. Antibodies 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Strube, J. Application and Fundamentals of Liquid-Liquid Extraction Processes: Purification of Biologicals, Botanicals, and Strategic Metals. In Encyclopedia of Chemical Technology; Kirk, R.E., Othmer, D.F., Eds.; Wiley: New York, NY, USA, 2003; pp. 1–52. ISBN 9780471238966. [Google Scholar]
- Schmidt, A.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modeling with Examples of Liquid-Liquid Extraction Processes of Complex Feed Mixtures. Processes 2019, 7, 298. [Google Scholar] [CrossRef] [Green Version]
- Zobel-Roos, S.; Stein, D.; Strube, J. Evaluation of Continuous Membrane Chromatography Concepts with an Enhanced Process Simulation Approach. Antibodies 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornecki, M.; Strube, J. Accelerating Biologics Manufacturing by Upstream Process Modelling. Processes 2019, 7, 166. [Google Scholar] [CrossRef] [Green Version]
- Cohn, E.J. The Physical Chemistry of the Proteins. Physiol. Rev. 1925, 5, 349–437. [Google Scholar] [CrossRef]
- Tscheliessnig, A.; Satzer, P.; Hammerschmidt, N.; Schulz, H.; Helk, B.; Jungbauer, A. Ethanol precipitation for purification of recombinant antibodies. J. Biotechnol. 2014, 188, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Zauner, R.; Jones, A.G. Scale-up of Continuous and Semibatch Precipitation Processes. Ind. Eng. Chem. Res. 2000, 39, 2392–2403. [Google Scholar] [CrossRef]
- Su, Q.; Nagy, Z.K.; Rielly, C.D. Pharmaceutical crystallisation processes from batch to continuous operation using MSMPR stages: Modelling, design, and control. Chem. Eng. Process. Process Intensif. 2015, 89, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Roland, M. Numerische Simulation Von Fällungsprozessen Mittels Populationsbilanzen. Ph.D. Thesis, Universität des Saarlandes, Saarbrücken, Germany, 2010. [Google Scholar]
- McPherson, A.; Gavira, J.A. Introduction to protein crystallization. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Huter, M.; Schmidt, A.; Mestmäcker, F.; Sixt, M.; Strube, J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part IV: Crystallization. Processes 2018, 6, 181. [Google Scholar] [CrossRef] [Green Version]
- Lucke, M.; Koudous, I.; Sixt, M.; Huter, M.J.; Strube, J. Integrating crystallization with experimental model parameter determination and modeling into conceptual process design for the purification of complex feed mixtures. Chem. Eng. Res. Des. 2018, 133, 264–280. [Google Scholar] [CrossRef]
- Huter, M.J.; Strube, J. Model-Based Design and Process Optimization of Continuous Single Pass Tangential Flow Filtration Focusing on Continuous Bioprocessing. Processes 2019, 7, 317. [Google Scholar] [CrossRef] [Green Version]
- Kornecki, M.; Strube, J. Process Analytical Technology for Advanced Process Control in Biologics Manufacturing with the Aid of Macroscopic Kinetic Modeling. Bioengineering 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornecki, M.; Schmidt, A.; Lohmann, L.; Huter, M.; Mestmäcker, F.; Klepzig, L.; Mouellef, M.; Zobel-Roos, S.; Strube, J. Accelerating Biomanufacturing by Modeling of Continuous Bioprocessing—Piloting Case Study of Monoclonal Antibody Manufacturing. Processes 2019, 7, 495. [Google Scholar] [CrossRef] [Green Version]
- Zobel-Roos, S.; Schmidt, A.; Mestmäcker, F.; Mouellef, M.; Huter, M.; Uhlenbrock, L.; Kornecki, M.; Lohmann, L.; Ditz, R.; Strube, J. Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable Without a Digital-Twin? Processes 2019, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Sixt, M.; Schmidt, A.; Mestmäcker, F.; Huter, M.; Uhlenbrock, L.; Strube, J. Systematic and Model-Assisted Process Design for the Extraction and Purification of Artemisinin from Artemisia annua L.—Part I: Conceptual Process Design and Cost Estimation. Processes 2018, 6, 161. [Google Scholar] [CrossRef] [Green Version]
- Sixt, M.; Strube, J. Systematic and Model-Assisted Evaluation of Solvent Based- or Pressurized Hot Water Extraction for the Extraction of Artemisinin from Artemisia annua L. Processes 2017, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Thiess, H.; Leuthold, M.; Grummert, U.; Strube, J. Module design for ultrafiltration in biotechnology: Hydraulic analysis and statistical modeling. J. Membr. Sci. 2017, 540, 440–453. [Google Scholar] [CrossRef]
- Ultrafiltration and Microfiltration Handbook, 2nd ed.; Cheryan, M.; Strauss, S. (Eds.) Chapman and Hall/CRC: Boca Raton, FL, USA, 1998; ISBN 9781566765985. [Google Scholar]
- Wijmans, J.G. Process performance = membrane properties + operating conditions. J. Membr. Sci. 2003, 220, 1–3. [Google Scholar] [CrossRef]
- Rautenbach, R. Modulauslegung und -optimierung. In Membranverfahren: Grundlagen der Modul- und Anlagenauslegung; Rautenbach, R., Ed.; Springer: Berlin/Heidelberg, Germany, 1997; pp. 97–113. ISBN 978-3-662-08656-8. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lohmann, L.J.; Strube, J. Accelerating Biologics Manufacturing by Modeling: Process Integration of Precipitation in mAb Downstream Processing. Processes 2020, 8, 58. https://doi.org/10.3390/pr8010058
Lohmann LJ, Strube J. Accelerating Biologics Manufacturing by Modeling: Process Integration of Precipitation in mAb Downstream Processing. Processes. 2020; 8(1):58. https://doi.org/10.3390/pr8010058
Chicago/Turabian StyleLohmann, Lara Julia, and Jochen Strube. 2020. "Accelerating Biologics Manufacturing by Modeling: Process Integration of Precipitation in mAb Downstream Processing" Processes 8, no. 1: 58. https://doi.org/10.3390/pr8010058
APA StyleLohmann, L. J., & Strube, J. (2020). Accelerating Biologics Manufacturing by Modeling: Process Integration of Precipitation in mAb Downstream Processing. Processes, 8(1), 58. https://doi.org/10.3390/pr8010058