Microbial Communities and Sulfate-Reducing Microorganisms Abundance and Diversity in Municipal Anaerobic Sewage Sludge Digesters from a Wastewater Treatment Plant (Marrakech, Morocco)

 ,
,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sampling Strategy and Operating Conditions
2.2. Extraction of Genomic DNA and High Throughput Sequencing
2.3. Processing of Sequencing Data and Statistical Analyses
2.4. Growth, Isolation of Sulfate-Reducing Microorganisms and Phylogenetic Identification
2.5. Quantitative PCR
3. Results
3.1. Global Analysis of Microbial Communities
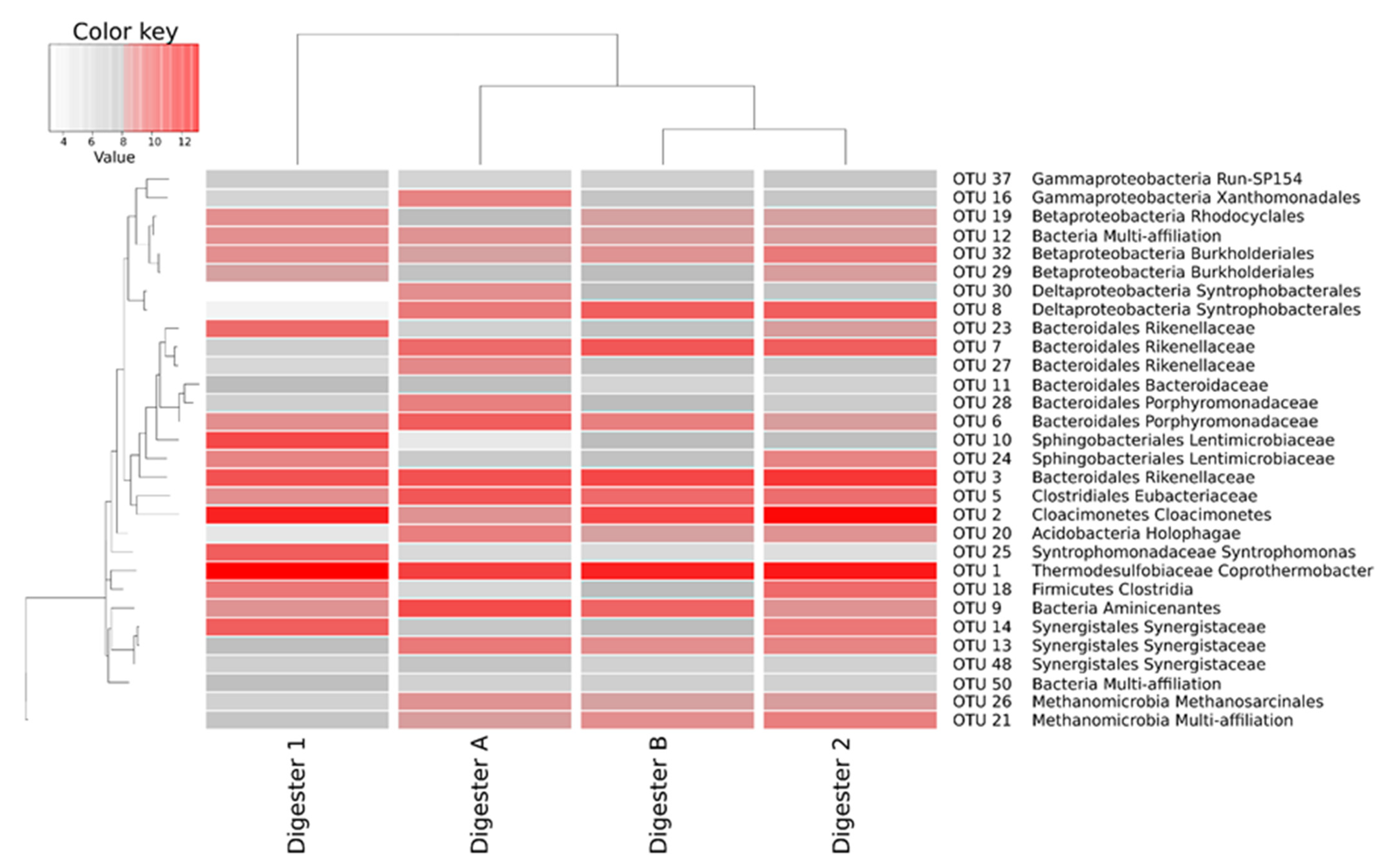
3.2. Microbial Communities in Anaerobic Digesters
3.3. Methanogens Communities Structure in Anaerobic Sewage Sludge
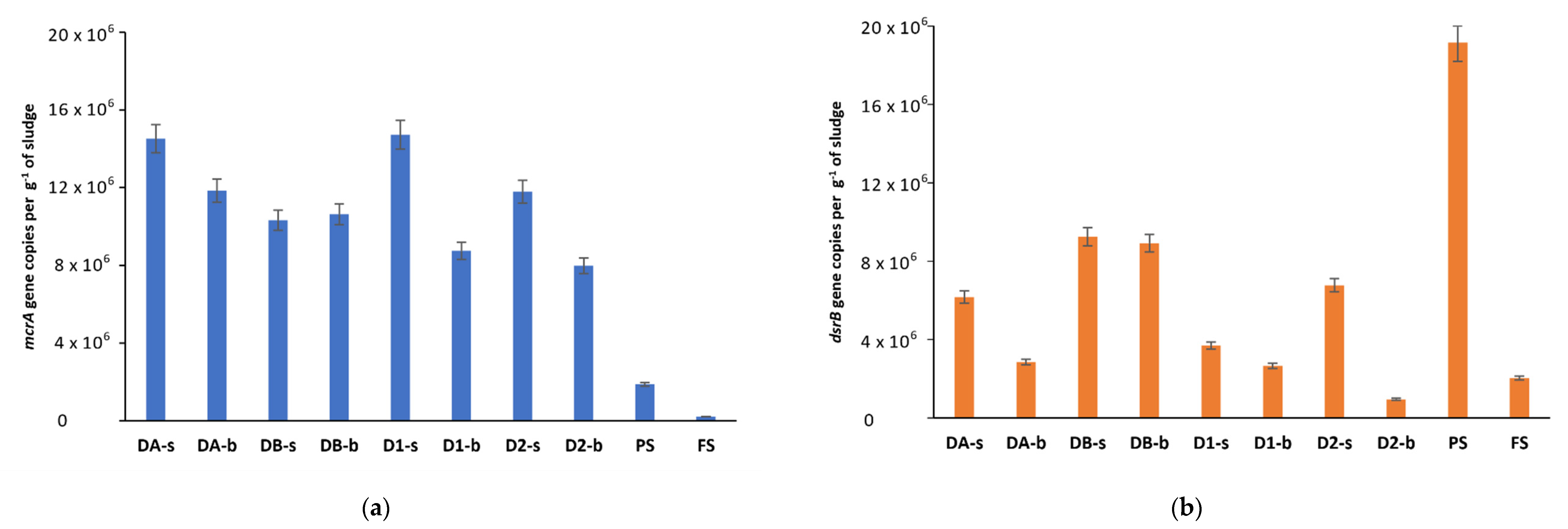
3.4. Diversity of Sulfate-Reducing Bacteria in Anaerobic Digesters
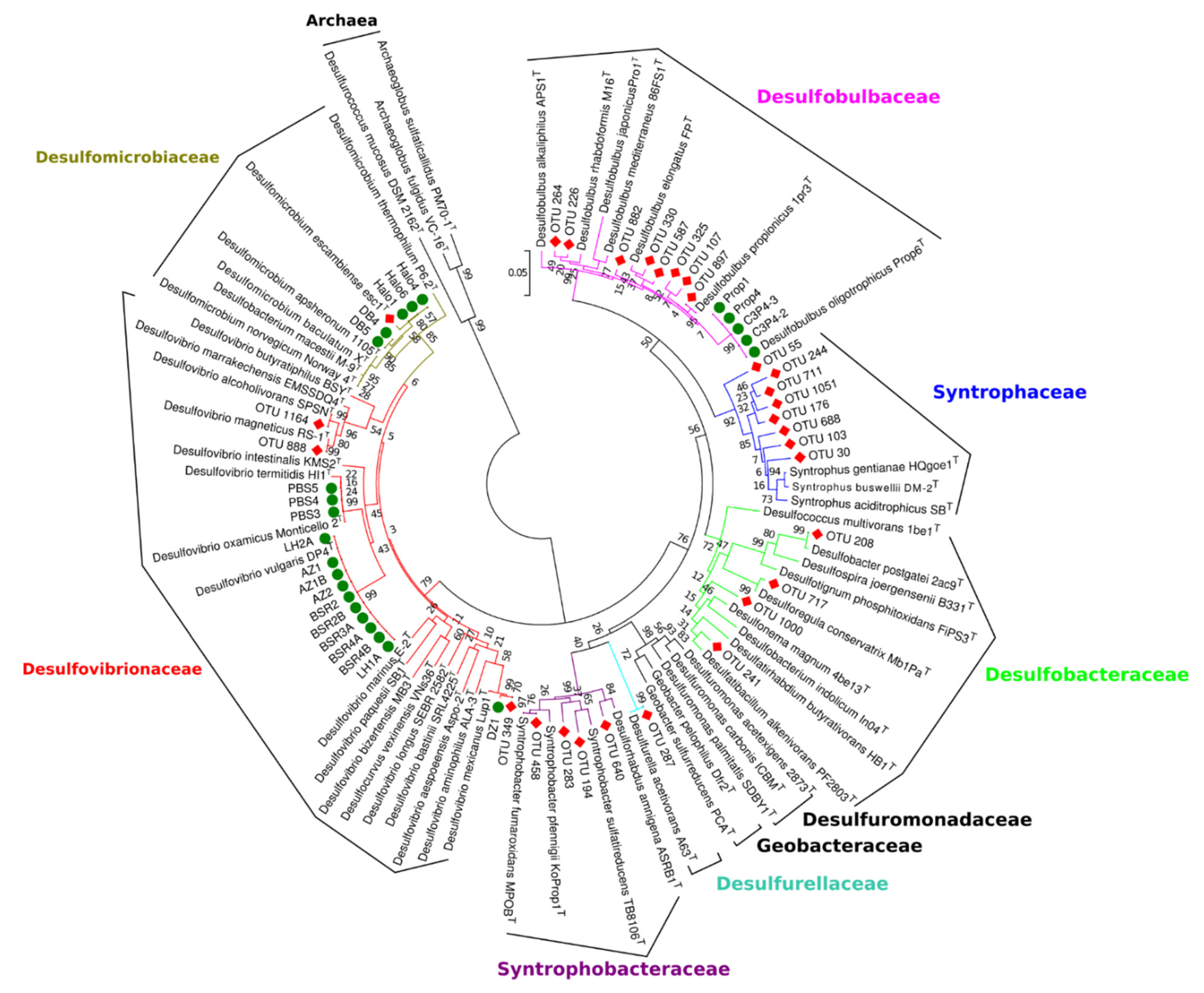
3.5. Relationship of Sulfate-Reducing Communities in Anaerobic Digesters with Isolated Sulfate-Reducing Strains from Anaerobic Digesters Sludge
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zeeman, G.; Lettinga, G. The role of anaerobic digestion of domestic sewage in closing the water and nutrient cycle at community level. Water Sci. Technol. 1999, 39, 187–194. [Google Scholar] [CrossRef]
- Ward, A.J.; Hobbs, P.J.; Holliman, P.J.; Jones, D.L. Optimisation of the anaerobic digestion of agricultural resources. Bioresour. Technol. 2008, 99, 7928–7940. [Google Scholar] [CrossRef] [PubMed]
- Narihiro, T.; Sekiguchi, Y. Microbial communities in anaerobic digestion processes for waste and wastewater treatment: A microbiological update. Curr. Opin. Biotechnol. 2007, 18, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Weiland, P. Biogas production: Current state and perspectives. Appl. Microbiol. Biotechnol. 2010, 85, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.; Huang, S.; Seston, S.; Xing, J.; Hickey, R.; Criddle, C.; Tiedje, J. How stable is stable? Function versus community composition. Appl. Environ. Microbiol. 1999, 65, 3697–3704. [Google Scholar] [CrossRef] [Green Version]
- Zumstein, E.; Moletta, R.; Godon, J. Examination of two years of community dynamics in an anaerobic bioreactor using fluorescence polymerase chain reaction (PCR) single-strand conformation polymorphism analysis. Environ. Microbiol. 2000, 2, 69–78. [Google Scholar] [CrossRef]
- Lee, S.H.; Kang, H.J.; Lee, Y.H.; Lee, T.J.; Han, K.; Choi, Y.; Park, H.D. Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters. J. Environ. Monit. 2012, 14, 1893–1905. [Google Scholar] [CrossRef]
- Werner, J.J.; Knights, D.; Garcia, M.L.; Scalfone, N.B.; Smith, S.; Yarasheski, K.; Cummings, A.T.; Beers, A.R.; Knight, B.; Angenent, L.T. Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc. Natl. Acad. Sci. USA 2011, 108, 4158–4163. [Google Scholar] [CrossRef] [Green Version]
- Colin, Y.; Goñi-Urriza, M.; Caumette, P.; Guyoneaud, R. Combination of high throughput cultivation and dsr A sequencing for assessment of sulfate-reducing bacteria diversity in sediments. FEMS Microbiol. Ecol. 2012, 83, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Gerardi, M.H. The Microbiology of Anaerobic Digesters; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Kushkevych, I.; Leščanová, O.; Dordević, D.; Jančíková, S.; Hošek, J.; Vítězová, M.; Buňková, L.; Drago, L. The Sulfate-Reducing Microbial Communities and Meta-Analysis of Their Occurrence during Diseases of Small–Large Intestine Axis. J. Clin. Med. 2019, 8, 1656. [Google Scholar] [CrossRef] [Green Version]
- Kováč, J.; Vítězová, M.; Kushkevych, I. Metabolic activity of sulfate-reducing bacteria from rodents with colitis. Open Med. 2018, 13, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Rabus, R.; Venceslau, S.S.; Wöhlbrand, L.; Voordouw, G.; Wall, J.D.; Pereira, I.A. A Post-Genomic View of the Ecophysiology, Catabolism and Biotechnological Relevance of Sulphate-Reducing Prokaryotes. In Advances in Microbial Physiology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 66, pp. 55–321. [Google Scholar]
- Dordević, D.; Jančíková, S.; Vítězová, M.; Kushkevych, I. Hydrogen sulfide toxicity in the gut environment: Meta-analysis of sulfate-reducing and lactic acid bacteria in inflammatory processes. J. Adv. Res. Mar. 2020. [Google Scholar] [CrossRef]
- Pokorna, D.; Zabranska, J. Sulfur-oxidizing bacteria in environmental technology. Biotechnol. Adv. 2015, 33, 1246–1259. [Google Scholar] [CrossRef]
- Okabe, S.; Odagiri, M.; Ito, T.; Satoh, H. Succession of sulfur-oxidizing bacteria in the microbial community on corroding concrete in sewer systems. Appl. Environ. Microbiol. 2007, 73, 971–980. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; de Schryver, P.; de Gusseme, B.; de Muynck, W.; Boon, N.; Verstraete, W. Chemical and biological technologies for hydrogen sulfide emission control in sewer systems: A review. Water Res. 2008, 42, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gayh, U.; Stooß, A.; Behrendt, J.; Otterpohl, R. Desulphurisation of biogas analysis, evaluation and optimization. In Proceedings of the Third International Symposium on Energy from Biomass and Waste, Venice, Italy, 8–11 November 2010; pp. 8–10. [Google Scholar]
- Barton, L.L.; Fauque, G.D. Biochemistry, physiology and biotechnology of sulfate-reducing bacteria. Adv. Appl. Microbiol. 2009, 68, 41–98. [Google Scholar] [CrossRef] [PubMed]
- Kushkevych, I.; Dordević, D.; Kollar, P.; Vítězová, M.; Drago, L. Hydrogen Sulfide as a Toxic Product in the Small–Large Intestine Axis and its Role in IBD Development. J. Clin. Med. 2019, 8, 1054. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.J.; Lens, P.N.; Stams, A.J.; Plugge, C.M.; Sorokin, D.Y.; Muyzer, G.; Dijkman, H.; van Zessen, E.; Luimes, P.; Buisman, C.J. Application of bacteria involved in the biological sulfur cycle for paper mill effluent purification. Sci. Total Environ. 2009, 407, 1333–1343. [Google Scholar] [CrossRef]
- Quijano, G.; Figueroa-González, I.; Buitrón, G. Fully aerobic two-step desulfurization process for purification of highly H2S-laden biogas. J. Chem. Technol. Biotechnol. 2018, 93, 3553–3561. [Google Scholar] [CrossRef]
- St-Pierre, B.; Wright, A.-D.G. Implications from distinct sulfate-reducing bacteria populations between cattle manure and digestate in the elucidation of H 2 S production during anaerobic digestion of animal slurry. Appl. Microbiol. Biotechnol. 2017, 101, 5543–5556. [Google Scholar] [CrossRef]
- Struk, M.; Vítězová, M.; Vítěz, T.; Bartoš, M.; Kushkevych, I. Modřice Plant Anaerobic Digester: Microbial Distribution and Biogas Production. Water Air Soil Pollut. 2019, 230, 240. [Google Scholar] [CrossRef]
- El Houari, A.; Ranchou-Peyruse, M.; Ranchou-Peyruse, A.; Dakdaki, A.; Guignard, M.; Idouhammou, L.; Bennisse, R.; Bouterfass, R.; Guyoneaud, R.; Qatibi, A.-I. Desulfobulbus oligotrophicus sp. nov. a sulfate-reducing and propionate-oxidizing bacterium isolated from a municipal anaerobic sewage sludge digester. Int. J. Syst. Evol. Microbiol. 2017, 67, 275–281. [Google Scholar] [CrossRef]
- Anderson, I.; Ulrich, L.E.; Lupa, B.; Susanti, D.; Porat, I.; Hooper, S.D.; Lapidus, A.; Sieprawska-Lupa, M.; Dharmarajan, L.; Goltsman, E.; et al. Genomic characterization of methanomicrobiales reveals three classes of methanogens. PLoS ONE 2009, 4, e5797. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Whitman, W.B. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N.Y. Acad. Sci. 2008, 1125, 171–189. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, P.-Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [Green Version]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, rapidly, OTUs with galaxy solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Chao, A.; Gotelli, N.J.; Hsieh, T.C.; Sander, E.L.; Ma, K.H.; Colwell, R.K.; Ellison, A.M. Rarefaction and extrapolation with Hill numbers: A framework for sampling and estimation in species diversity studies. Ecol. Monogr. 2014, 84, 45–67. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Hungate, R. Chapter IV A Roll Tube Method for Cultivation of Strict Anaerobes. In Methods in Microbiology; Elsevier: Amsterdam, The Netherlands, 1969; Volume 3, pp. 117–132. [Google Scholar]
- Dias, M.; Salvado, J.C.; Monperrus, M.; Caumette, P.; Amouroux, D.; Duran, R.; Guyoneaud, R. Characterization of Desulfomicrobium salsuginis sp. nov. and Desulfomicrobium aestuarii sp. nov. two new sulfate-reducing bacteria isolated from the Adour estuary (French Atlantic coast) with specific mercury methylation potentials. Syst. Appl. Microbiol. 2008, 31, 30–37. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Geets, J.; Borremans, B.; Diels, L.; Springael, D.; Vangronsveld, J.; van der Lelie, D.; Vanbroekhoven, K. DsrB gene-based DGGE for community and diversity surveys of sulfate-reducing bacteria. J. Microbiol. Methods 2006, 66, 194–205. [Google Scholar] [CrossRef]
- Steinberg, L.M.; Regan, J.M. Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl. Environ. Microbiol. 2008, 74, 6663–6671. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cao, A.; Zhao, G.; Zhou, C.; Xu, R. Microbial community structure and diversity in a municipal solid waste landfill. Waste Manag. 2017, 66, 79–87. [Google Scholar] [CrossRef]
- Wu, L.; Yang, Y.; Chen, S.; Zhao, M.; Zhu, Z.; Yang, S.; Qu, Y.; Ma, Q.; He, Z.; Zhou, J.; et al. Long-term successional dynamics of microbial association networks in anaerobic digestion processes. Water Res. 2016, 104, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Treu, L.; Campanaro, S.; Kougias, P.G.; Sartori, C.; Bassani, I.; Angelidaki, I. Hydrogen-fueled microbial pathways in biogas upgrading systems revealed by genome-centric metagenomics. Front. Microbiol. 2018, 9, 1079. [Google Scholar] [CrossRef] [Green Version]
- Ziels, R.M.; Svensson, B.H.; Sundberg, C.; Larsson, M.; Karlsson, A.; Yekta, S.S. Microbial rRNA gene expression and co-occurrence profiles associate with biokinetics and elemental composition in full-scale anaerobic digesters. Microb. Biotechnol. 2018, 11, 694–709. [Google Scholar] [CrossRef]
- Tian, Z.; Cabrol, L.; Ruiz-Filippi, G.; Pullammanappallil, P. Microbial ecology in anaerobic digestion at agitated and non-agitated conditions. PLoS ONE 2014, 9, e109769. [Google Scholar] [CrossRef]
- Jegede, A.O.; Zeeman, G.; Bruning, H. A review of mixing, design and loading conditions in household anaerobic digesters. Crit. Rev. Environ. Sci. Technol. 2019, 49, 2117–2153. [Google Scholar] [CrossRef]
- Bhandari, V.; Gupta, R.S. Molecular signatures for the phylum Synergistetes and some of its subclades. Antonie Van Leeuwenhoek 2012, 102, 517–540. [Google Scholar] [CrossRef] [PubMed]
- Cirne, D.; Lehtomäki, A.; Björnsson, L.; Blackall, L. Hydrolysis and microbial community analyses in two-stage anaerobic digestion of energy crops. J. Appl. Microbiol. 2007, 103, 516–527. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, C.A.; Burrell, P.C.; Clarke, W.P.; Blackall, L.L. Structure of a cellulose degrading bacterial community during anaerobic digestion. Biotechnol. Bioeng. 2005, 92, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Sträuber, H.; Schröder, M.; Kleinsteuber, S. Metabolic and microbial community dynamics during the hydrolytic and acidogenic fermentation in a leach-bed process. Energy Sustain. Soc. 2012, 2, 13. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Dai, X.; Zhou, J.; Xu, X. The stability of aerobic granular sludge under 4-chloroaniline shock in a sequential air-lift bioreactor (SABR). Bioresour. Technol. 2013, 140, 126–130. [Google Scholar] [CrossRef]
- Mei, R.; Narihiro, T.; Nobu, M.K.; Kuroda, K.; Liu, W.-T. Evaluating digestion efficiency in full-scale anaerobic digesters by identifying active microbial populations through the lens of microbial activity. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, L.; Xiang, F.; Zhao, L.; Qiao, Z. Activated Sludge Microbial Community and Treatment Performance of Wastewater Treatment Plants in Industrial and Municipal Zones. Int. J. Environ. Res. Public. Health 2020, 17, 436. [Google Scholar] [CrossRef] [Green Version]
- Calusinska, M.; Goux, X.; Fossépré, M.; Muller, E.E.; Wilmes, P.; Delfosse, P. A year of monitoring 20 mesophilic full-scale bioreactors reveals the existence of stable but different core microbiomes in bio-waste and wastewater anaerobic digestion systems. Biotechnol. Biofuels 2018, 11, 196. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. New insights from the biogas microbiome by comprehensive genome-resolved metagenomics of nearly 1600 species originating from multiple anaerobic digesters. Biotechnol. Biofuels 2020, 13, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Kim, H.; Kakegawa, T.; Hanada, S. A novel lineage of sulfate-reducing microorganisms: Thermodesulfobiaceae fam. nov. Thermodesulfobium narugense, gen. nov. sp. nov. a new thermophilic isolate from a hot spring. Extremophiles 2003, 7, 283–290. [Google Scholar] [CrossRef]
- Moset, V.; Poulsen, M.; Wahid, R.; Højberg, O.; Møller, H.B. Mesophilic versus thermophilic anaerobic digestion of cattle manure: Methane productivity and microbial ecology. Microb. Biotechnol. 2015, 8, 787–800. [Google Scholar] [CrossRef]
- Sasaki, D.; Hori, T.; Haruta, S.; Ueno, Y.; Ishii, M.; Igarashi, Y. Methanogenic pathway and community structure in a thermophilic anaerobic digestion process of organic solid waste. J. Biosci. Bioeng. 2011, 111, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Peng, Y.; Ni, B.-J.; Han, X.; Fan, L.; Yuan, Z. Dissecting microbial community structure and methane-producing pathways of a full-scale anaerobic reactor digesting activated sludge from wastewater treatment by metagenomic sequencing. Microb. Cell Factories 2015, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavergne, C.; Bovio-Winkler, P.; Etchebehere, C.; García-Gen, S. Towards centralized biogas plants: Co-digestion of sewage sludge and pig manure maintains process performance and active microbiome diversity. Bioresour. Technol. 2020, 297, 122442. [Google Scholar] [CrossRef]
- García-Lozano, M.; Lira, H.-D.; Omar, I.; Huber, D.H.; Balagurusamy, N. Spatial Variations of Bacterial Communities of an Anaerobic Lagoon-Type Biodigester Fed with Dairy Manure. Processes 2019, 7, 408. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Pope, P.B.; Eijsink, V.G.H.; Schnürer, A. Characterization of microbial community structure during continuous anaerobic digestion of straw and cow manure: Community structure during anaerobic digestion. Microb. Biotechnol. 2015, 8, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Chu, Y.N.; Wang, X.; Ren, L.; Yu, J.; Liu, X.; Yan, J.; Zhang, L.; Wu, S.; Li, S. A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol. Biofuels 2013, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Westerholm, M.; Liu, T.; Schnürer, A. Comparative study of industrial-scale high-solid biogas production from food waste: Process operation and microbiology. Bioresour. Technol. 2020, 304, 122981. [Google Scholar] [CrossRef]
- Kushkevych, I.; Dordević, D.; Vítězová, M. Possible synergy effect of hydrogen sulfide and acetate produced by sulfate-reducing bacteria on inflammatory bowel disease development. J. Adv. Res. Mar. 2020. [Google Scholar] [CrossRef]
- Gao, J.; Liu, G.; Li, H.; Xu, L.; Du, L.; Yang, B. Predictive functional profiling using marker gene sequences and community diversity analyses of microbes in full-scale anaerobic sludge digesters. Bioprocess Biosyst. Eng. 2016, 39, 1115–1127. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, K.; Xia, Y.; Lau, F.T.; Tang, D.T.; Fung, W.C.; Fang, H.H.P.; Zhang, T. Metagenomic analysis of sludge from full-scale anaerobic digesters operated in municipal wastewater treatment plants. Appl. Microbiol. Biotechnol. 2014, 98, 5709–5718. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, T.; Tsushima, I.; Tsumori, J. Comparative analyses of microbial structures and gene copy numbers in the anaerobic digestion of various types of sewage sludge. Bioresour. Technol. 2018, 253, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Traversi, D.; Villa, S.; Lorenzi, E.; Degan, R.; Gilli, G. Application of a real-time qPCR method to measure the methanogen concentration during anaerobic digestion as an indicator of biogas production capacity. J. Environ. Manag. 2012, 111, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Taylor, M.W.; Turner, S.J. dsrAB-based analysis of sulphate-reducing bacteria in moving bed biofilm reactor (MBBR) wastewater treatment plants. Appl. Microbiol. Biotechnol. 2014, 98, 7211–7222. [Google Scholar] [CrossRef]
- Klein, M.; Friedrich, M.; Roger, A.J.; Hugenholtz, P.; Fishbain, S.; Abicht, H.; Blackall, L.L.; Stahl, D.A.; Wagner, M. Multiple lateral transfers of dissimilatory sulfite reductase genes between major lineages of sulfate-reducing prokaryotes. J. Bacteriol. 2001, 183, 6028–6035. [Google Scholar] [CrossRef] [Green Version]
- Stahl, D.A.; Loy, A.; Wagner, M.; Barton, L.; Hamilton, W. Molecular Strategies for Studies of Natural Populations of Sulphate-Reducing Microorganisms; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Moura AD, L.; Centurion, V.B.; Okada, D.Y.; Motteran, F.; Delforno, T.P.; Oliveira, V.M.; Varesche, M.B.A. Laundry wastewater and domestic sewage pilot-scale anaerobic treatment: Microbial community resilience regarding sulfide production. J. Environ. Manag. 2019, 251, 109495. [Google Scholar] [CrossRef]
- Dar, S.A.; Yao, L.; van Dongen, U.; Kuenen, J.G.; Muyzer, G. Analysis of diversity and activity of sulfate-reducing bacterial communities in sulfidogenic bioreactors using 16S rRNA and dsrB genes as molecular markers. Appl. Environ. Microbiol. 2007, 73, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Dar, S.A.; Kleerebezem, R.; Stams, A.J.; Kuenen, J.G.; Muyzer, G. Competition and coexistence of sulfate-reducing bacteria, acetogens and methanogens in a lab-scale anaerobic bioreactor as affected by changing substrate to sulfate ratio. Appl. Microbiol. Biotechnol. 2008, 78, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Kushkevych, I.; Kováč, J.; Vítězová, M.; Vítěz, T.; Bartoš, M. The diversity of sulfate-reducing bacteria in the seven bioreactors. Arch. Microbiol. 2018, 200, 945–950. [Google Scholar] [CrossRef]
- Santegoeds, C.M.; Ferdelman, T.G.; Muyzer, G.; de Beer, D. Structural and functional dynamics of sulfate-reducing populations in bacterial biofilms. Appl. Environ. Microbiol. 1998, 64, 3731–3739. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Santegoeds, C.M.; Nielsen, H.K.; Ploug, H.; Wagner, M.; Pribyl, M.; Wanner, J.; Amann, R.; de Beer, D. On the occurrence of anoxic microniches, denitrification, and sulfate reduction in aerated activated sludge. Appl. Environ. Microbiol. 1999, 65, 4189–4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sass, A.; Rütters, H.; Cypionka, H.; Sass, H. Desulfobulbus mediterraneus sp. nov. a sulfate-reducing bacterium growing on mono-and disaccharides. Arch. Microbiol. 2002, 177, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Widdel, F.; Pfennig, N. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids II. Incomplete oxidation of propionate by Desulfobulbus propionicus gen. nov. sp. nov. Arch. Microbiol. 1982, 131, 360–365. [Google Scholar] [CrossRef]
- Dannenberg, S.; Kroder, M.; Dilling, W.; Cypionka, H. Oxidation of H2, organic compounds and inorganic sulfur compounds coupled to reduction of O2 or nitrate by sulfate-reducing bacteria. Arch. Microbiol. 1992, 158, 93–99. [Google Scholar] [CrossRef]
- Jung, H.; Kim, J.; Lee, C. Temperature effects on methanogenesis and sulfidogenesis during anaerobic digestion of sulfur-rich macroalgal biomass in sequencing batch reactors. Microorganisms 2019, 7, 682. [Google Scholar] [CrossRef] [Green Version]
- Ariesyady, H.D.; Ito, T.; Yoshiguchi, K.; Okabe, S. Phylogenetic and functional diversity of propionate-oxidizing bacteria in an anaerobic digester sludge. Appl. Microbiol. Biotechnol. 2007, 75, 673–683. [Google Scholar] [CrossRef]
- Manz, W.; Eisenbrecher, M.; Neu, T.R.; Szewzyk, U. Abundance and spatial organization of Gram-negative sulfate-reducing bacteria in activated sludge investigated by in situ probing with specific 16S rRNA targeted oligonucleotides. FEMS Microbiol. Ecol. 1998, 25, 43–61. [Google Scholar] [CrossRef]
- Cypionka, H.; Widdel, F.; Pfennig, N. Survival of sulfate-reducing bacteria after oxygen stress, and growth in sulfate-free oxygen-sulfide gradients. FEMS Microbiol. Lett. 1985, 31, 39–45. [Google Scholar] [CrossRef]
- Marschall, C.; Frenzel, P.; Cypionka, H. Influence of oxygen on sulfate reduction and growth of sulfate-reducing bacteria. Arch. Microbiol. 1993, 159, 168–173. [Google Scholar] [CrossRef]
- Burgess, J.E.; Parsons, S.A.; Stuetz, R.M. Developments in odour control and waste gas treatment biotechnology: A review. Biotechnol. Adv. 2001, 19, 35–63. [Google Scholar] [CrossRef]
- Biswas, K.; Turner, S.J. Microbial community composition and dynamics of moving bed biofilm reactor systems treating municipal sewage. Appl. Environ. Microbiol. 2012, 78, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabari, L.; Gannoun, H.; Khelifi, E.; Cayol, J.L.; Godon, J.J.; Hamdi, M.; Fardeau, M.L. Bacterial ecology of abattoir wastewater treated by an anaerobic digestor. Braz. J. Microbiol. 2016, 47, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Biswal, B.K.; Chen, G.-H.; Wu, D. Sulfidogenic anaerobic digestion of sulfate-laden waste activated sludge: Evaluation on reactor performance and dynamics of microbial community. Bioresour. Technol. 2020, 297, 122396. [Google Scholar] [CrossRef] [PubMed]
- Jantharadej, K.; Mhuantong, W.; Limpiyakorn, T.; Mongkolsuk, S.; Sirikanchana, K.; Suwannasilp, B.B. Identification of sulfate-reducing and methanogenic microbial taxa in anaerobic bioreactors from industrial wastewater treatment plants using next-generation sequencing and gene clone library analyses. J. Environ. Sci. Health Part A 2020, 1–11. [Google Scholar] [CrossRef]
- Isaksen, M.F.; Teske, A. Desulforhopalus vacuolatus gen. nov. sp. nov. a new moderately psychrophilic sulfate-reducing bacterium with gas vacuoles isolated from a temperate estuary. Arch. Microbiol. 1996, 166, 160–168. [Google Scholar] [CrossRef]
- Kremer, D.; Hansen, T. Pathway of propionate degradation in Desulfobulbus propionicus. FEMS Microbiol. Lett. 1988, 49, 273–277. [Google Scholar] [CrossRef]
- Promnuan, K.; Higuchi, T.; Imai, T.; Kongjan, P.; Reungsang, A.; Sompong, O. Simultaneous biohythane production and sulfate removal from rubber sheet wastewater by two-stage anaerobic digestion. Int. J. Hydrog. Energy 2020, 45, 263–274. [Google Scholar] [CrossRef]
- Gittel, A.; Mußmann, M.; Sass, H.; Cypionka, H.; Könneke, M. Identity and abundance of active sulfate-reducing bacteria in deep tidal flat sediments determined by directed cultivation and CARD-FISH analysis. Environ. Microbiol. 2008, 10, 2645–2658. [Google Scholar] [CrossRef]
- Leloup, J.; Fossing, H.; Kohls, K.; Holmkvist, L.; Borowski, C.; Jørgensen, B.B. Sulfate-reducing bacteria in marine sediment (Aarhus Bay, Denmark): Abundance and diversity related to geochemical zonation. Environ. Microbiol. 2009, 11, 1278–1291. [Google Scholar] [CrossRef]
- Braga, J.K.; Motteran, F.; Silva, E.L.; Varesche, M.B.A. Evaluation of bacterial community from anaerobic fluidized bed reactor for the removal of linear alkylbenzene sulfonate from laundry wastewater by 454-pyrosequence. Ecol. Eng. 2015, 82, 231–240. [Google Scholar] [CrossRef]
- Shi, X.; Zuo, J.; Li, B.; Yu, H. Two-stage anaerobic digestion of food waste coupled with in situ ammonia recovery using gas membrane absorption: Performance and microbial community. Bioresour. Technol. 2020, 297, 122458. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Okabe, S.; Satoh, H.; Watanabe, Y. Successional development of sulfate-reducing bacterial populations and their activities in a wastewater biofilm growing under microaerophilic conditions. Appl. Environ. Microbiol. 2002, 68, 1392–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, X.; Zhen, Y.; Mi, T.; He, H.; Yu, Z. Microbial Diversity and Community Structure of Sulfate-Reducing and Sulfur-Oxidizing Bacteria in Sediment Cores from the East China Sea. Front. Microbiol. 2017, 8, 2133. [Google Scholar] [CrossRef] [PubMed]
- Jr, W.P.K.; Scholten, J.C.; Culley, D.; Hickey, R.; Zhang, W.; Brockman, F.J. Microbial dynamics in upflow anaerobic sludge blanket (UASB) bioreactor granules in response to short-term changes in substrate feed. Microbiology 2010, 156, 2418–2427. [Google Scholar]
- Boone, D.R.; Bryant, M.P. Propionate-degrading bacterium, Syntrophobacter wolinii sp. nov. gen. nov. from methanogenic ecosystems. Appl. Environ. Microbiol. 1980, 40, 626–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmsen, H.J.; van Kuijk, B.L.; Plugge, C.M.; Akkermans, A.D.; de Vos, W.M.; Stams, A.J. Syntrophobacter fumaroxidans sp. nov. a syntrophic propionate-degrading sulfate-reducing bacterium. Int. J. Syst. Evol. Microbiol. 1998, 48, 1383–1387. [Google Scholar] [CrossRef] [Green Version]
- Wallrabenstein, C.; Hauschild, E.; Schink, B. Syntrophobacter pfennigii sp. nov. new syntrophically propionate-oxidizing anaerobe growing in pure culture with propionate and sulfate. Arch. Microbiol. 1995, 164, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.A.; Albers, S.-V.; Atomi, H.; Allers, T. Model organisms for genetics in the domain Archaea: Methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 2011, 35, 577–608. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organic Loading Rate (Kg VDM/day) | Temperature (°C) | pH | VFAs (mg.L−1) | H2S (ppm) | Alkalinity (g.L−1) | |
|---|---|---|---|---|---|---|
| Digester A | 6801 ± 931 | 29.7 ± 0.5 | 7.0 ± 0.1 | 183 ± 24 | n.a | 1.8 ± 0.3 |
| Digester B | 6193 ± 1035 | 32.1 ± 1.3 | 7.1 ± 0.1 | 227 ± 73 | n.a | 2.2 ± 0.3 |
| Digester 1 | 6802 ± 951 | 41.1 ± 2.3 | 7.3 ± 0.1 | 620 ± 361 | 219 | 2.5 ± 0.4 |
| Digester 2 | 5717 ± 960 | 35.5 ± 0.4 | 7.2 ± 0.1 | 276 ± 70 | 2.1 | 2.4 ± 0.4 |
| Samples | Sequences Number | OTUs | Chao1 | Exponential (H-Shannon) | 1/Simpson |
|---|---|---|---|---|---|
| FS | 30,103 ± 13,365 | 566 ± 57 | 628.1 | 118.99 | 35.52 |
| PS | 24,177 ± 9086 | 593 ± 43 | 669.61 | 110.76 | 36.87 |
| D1-b | 41,212 ± 22,030 | 722 ± 72 | 785.64 | 56.05 | 11.81 |
| D1-s | 37,788 ± 15,262 | 744 ± 37 | 802.61 | 58.11 | 11.41 |
| D2-b | 45,925 ± 9174 | 771 ± 27 | 810.89 | 63.97 | 14.67 |
| D2-s | 40,254 ± 18,170 | 741 ± 74 | 812.35 | 58.55 | 12.89 |
| DA-b | 31,097 ± 11,116 | 748 ± 25 | 794.42 | 135.44 | 51.61 |
| DA-s | 40,134 ± 18,067 | 785 ± 36 | 819.27 | 133.28 | 45.44 |
| DB-b | 46,781 ± 7138 | 816 ± 13 | 854.7 | 105.12 | 26.73 |
| DB-s | 18,698 ± 472 | 695 ± 13 | 767.26 | 93.55 | 21.57 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Houari, A.; Ranchou-Peyruse, M.; Ranchou-Peyruse, A.; Bennisse, R.; Bouterfas, R.; Goni Urriza, M.S.; Qatibi, A.-I.; Guyoneaud, R. Microbial Communities and Sulfate-Reducing Microorganisms Abundance and Diversity in Municipal Anaerobic Sewage Sludge Digesters from a Wastewater Treatment Plant (Marrakech, Morocco). Processes 2020, 8, 1284. https://doi.org/10.3390/pr8101284
El Houari A, Ranchou-Peyruse M, Ranchou-Peyruse A, Bennisse R, Bouterfas R, Goni Urriza MS, Qatibi A-I, Guyoneaud R. Microbial Communities and Sulfate-Reducing Microorganisms Abundance and Diversity in Municipal Anaerobic Sewage Sludge Digesters from a Wastewater Treatment Plant (Marrakech, Morocco). Processes. 2020; 8(10):1284. https://doi.org/10.3390/pr8101284
Chicago/Turabian StyleEl Houari, Abdelaziz, Magali Ranchou-Peyruse, Anthony Ranchou-Peyruse, Rhizlane Bennisse, Radia Bouterfas, Maria Soledad Goni Urriza, Abdel-Ilah Qatibi, and Rémy Guyoneaud. 2020. "Microbial Communities and Sulfate-Reducing Microorganisms Abundance and Diversity in Municipal Anaerobic Sewage Sludge Digesters from a Wastewater Treatment Plant (Marrakech, Morocco)" Processes 8, no. 10: 1284. https://doi.org/10.3390/pr8101284
APA StyleEl Houari, A., Ranchou-Peyruse, M., Ranchou-Peyruse, A., Bennisse, R., Bouterfas, R., Goni Urriza, M. S., Qatibi, A. -I., & Guyoneaud, R. (2020). Microbial Communities and Sulfate-Reducing Microorganisms Abundance and Diversity in Municipal Anaerobic Sewage Sludge Digesters from a Wastewater Treatment Plant (Marrakech, Morocco). Processes, 8(10), 1284. https://doi.org/10.3390/pr8101284