In this section, a rather simple model of the separation process is constructed. The aim is to obtain a representation of the process that allows for an analytical solution and gives insight into essential qualitative properties of the process.
2.1. Model Assumptions
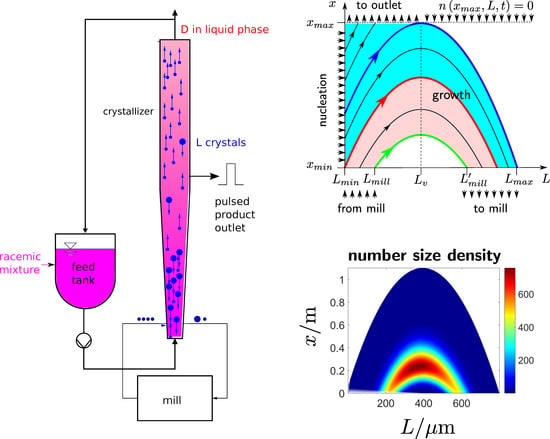
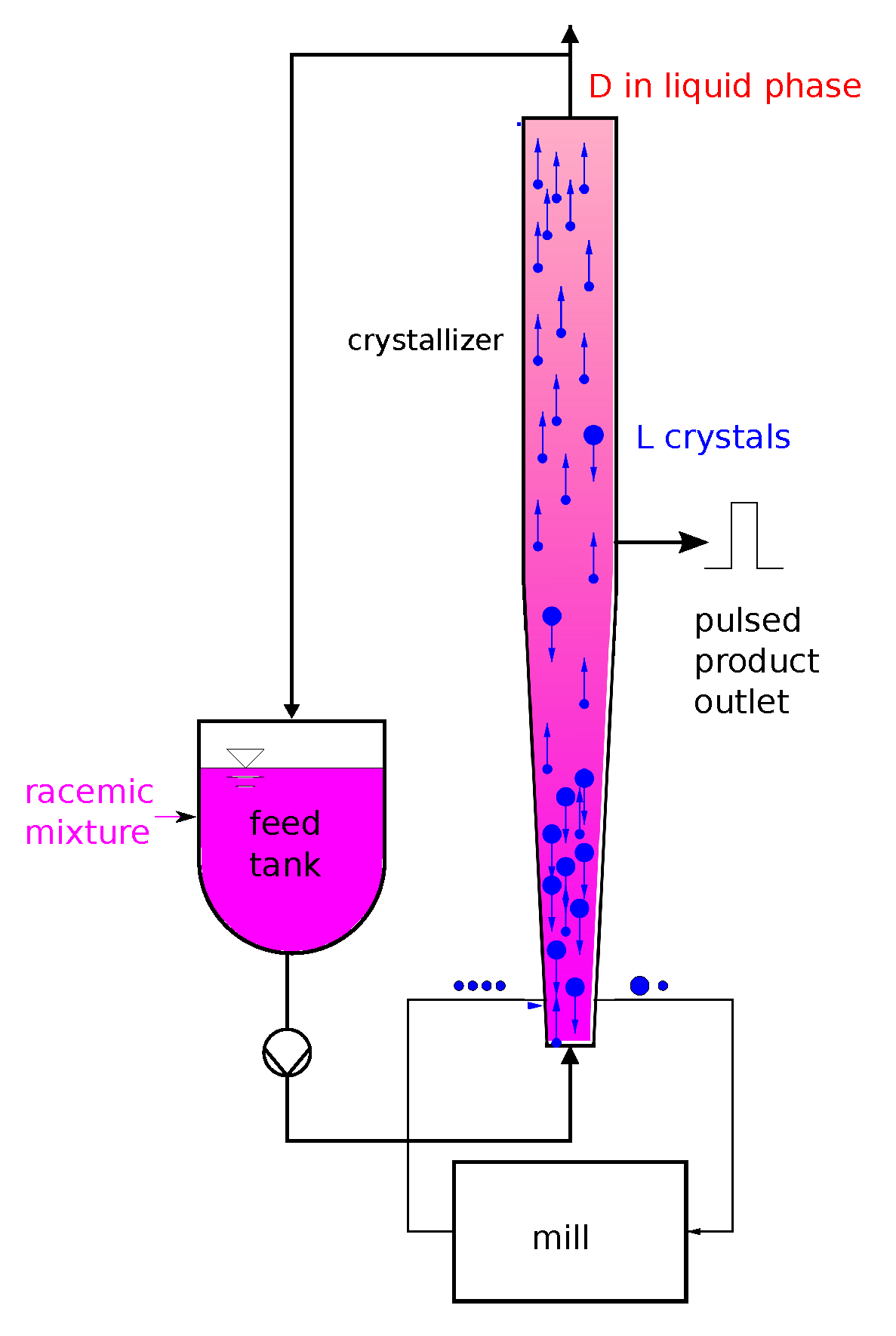
The geometry of the crystallizer is described by a single space coordinate x in the direction of the fluid flow. The fluid inlet point at the bottom of the crystallizer and the fluid outlet point at the top are denoted as and , respectively. Gradients perpendicular to the x coordinate are neglected.
The properties of the crystal population depend on a single property coordinate
L representing a characteristic crystal length. The number size density
n of the crystals (number of crystals per crystal size and volume) is a function of
L,
x, and of time
t. The computational domain of
n is shown in
Figure 2.
Gravity and drag forces cause the movement of the crystals in space. For simplicity, the interaction between crystals is neglected. Hence, the crystal velocity v is only a function of L and x. For small crystals, the drag force dominates and pushes the particles upwards (), whereas gravity causes the larger crystals to sink to the bottom (). For a certain crystal size , gravity and drag force cancel each other, and the crystal is in mechanical equilibrium and not moving, .
As a strong simplification, it is assumed that the composition of the liquid phase does not change, and that the supersaturation in the liquid phase is approximately constant. This is only valid for high liquid flow rates. More detailed models for the liquid phase have been published in [
7,
9,
10,
11,
12]. As a consequence of this assumption, the crystal growth rate
and the nucleation rate
B are constant over space. Nuclei have a characteristic length
. The same growth rate and nucleation rate are assumed for the desired L enantiomer and the undesired D enantiomer.
A population balance results in the following equation for the number size density
:
Crystals sinking to the bottom of the crystallizer are ground in a mill and returned as seeds to the crystallizer. It is assumed that the share of crystals coming out of the mill with a certain size L is always the same and is equal to . The maximum size of the crystals out of the mill is limited to , which, for example, may be the gap between the mill’s rotating blade and its stationary parts.
The number size density at the mill outlet reads
with a time dependent scaling factor
following from a mass balance of the mill. Assuming that the mill has negligible hold-up, the total mass of crystals leaving the mill must always be identical to the total mass of crystals entering the mill:
The combination of (
2) and (
3) leads to the boundary condition
The corresponding boundary condition at the crystallizer top reads
because no particles are fed to the crystallizer top.
The boundary along the
x-coordinate depends on the nucleation of crystals. It reads
An initial condition
completes the model.
2.2. Analytical Solution of the Linear Model by the Method of Characteristics
The method of characteristics is an established method for the analysis of separation processes with negligible dispersion [
13]. In this work, it provides an approach for the solution of the model system (
1) and (
4)–(
7). The size distribution is calculated along an arc with arc length parameter
s, i.e.,
. The total derivative with respect to
s reads
A comparison of (
8) with the population balance (
1) leads to the following set of equations:
From (
9), it is obvious that the arc length parameter
s is identical to the progress in time. Equation (12) shows that the number size density does not change when moving along a characteristic curve—i.e.,
where
is an arbitrary reference point on the same characteristic. Note that (
13) does not mean that at one moment in time, the number size density
n has the same value along the characteristic –
t and
are different time points. Instead, (
13) says that the
number of crystals in a small fluid element moving along a characteristic does not change, whereas the
size of the crystals in the fluid element does.
In the remainder of this subsection and in the next subsection, a very simple velocity profile is studied. It is assumed that the crystal velocity decreases linearly with the crystal size and does not depend on the space coordinate:
where
is the velocity of the nuclei identical to the fluid flow velocity, and
is a constant slope. The extension to a more general velocity profile is outlined in
Section 2.5.
In the case of this simple velocity profile, the solution of (
9)–(12) is straightforward. From (11), one gets
From (10), in combination with (
15) and (
14), one obtains
With the help of (
15), the arc length parameter
s or the time
t is eliminated from (
16). This gives an explicit equation for the characteristic curve in the
L-
x-plane:
It follows that the characteristic lines are parabolas in the
L-
x-plane (see
Figure 3a). For all of them, the vertex is at
where the particle velocity vanishes and the crystals are at mechanical equilibrium.
Figure 3b shows that four different regions may be distinguished in the
L-
x-plane. Region 1 (shaded blue in the figure) contains all characteristic curves that start on the
x-axis. The number of crystals in this region depends on the nucleation rate. To obtain the number size density at any point in the region, one has to go back in time until the characteristic curve intersects the
x-axis. The number size density is equal to the number of crystals nucleated at that time. From (
6):
. Region 1 is further divided into a subregion (a) containing crystals that escape through the top of the crystallizer, and into a subregion (b) with crystals that sink to the bottom of the crystallizer and are recycled in the mill. The distinction between subregions (a) and (b) is important for the process operation. On the one hand, as many crystals of the desired enantiomer as possible should be kept in the system. On the other hand, undesired nuclei of the counter-enantiomer should be flushed out of the crystallizer before they grow large and contaminate the product. Subregion 1 (a) and 1 (b) are separated by the characteristic curve highlighted blue in
Figure 3b. This is the characteristic of nuclei moving up to the highest point
of the crystallizer and sinking to the bottom afterwards. The end point of that characteristic on the
L-axis marks the biggest size
that the crystal can reach for the chosen operation conditions. From an elementary calculation, one obtains
Region 2 (shaded red in the figure) is formed by all characteristics starting on the
L-axis. All crystals in Region 2 were generated in the mill. In order to determine the number size density at some point
in that region, one has to go back in time to the point where the characteristic curve intersects the
L axis and where
. The number size density
is equal to the number of crystals generated in the mill at that time. The upper border of Region 2 is the characteristic curve highlighted red in
Figure 3b, which starts at
. If the vertex of this characteristic curve has an
x-coordinate smaller than
, then all crystals in Region 2 remain in the process permanently, rising from the mill, sinking to the bottom of the crystallizer and returning to the mill. The lower border of Region 2 (highlighted green in the figure) is given by the characteristic curve of the largest crystals coming out of the mill with size
.
Region 3 is the area below the green characteristic. No crystals exist in that region, except when they are introduced from outside, but even in this case, they will vanish quickly.
Region 4 in the upper right corner of the diagram is another crystal free area. The reason is that no large crystals are fed into the crystallizer from the top.
The residence time
T of the crystals in the crystallizer is easily calculated from the characteristics. It is identical to the arc length of the crystals’ characteristic curve
, where
denote the starting point of the characteristic either on the
x axis (with
) or on the
L axis (with
), and
is the end point on the
L axis. From (
16), one gets
or, after solving for
T:
The crystals with the shortest residence time
are those on the characteristic curve marked as a thick green line in
Figure 3. These are the largest crystals coming out of the mill with
. From (
21), it follows that their residence time is
The biggest residence time
belongs to the crystals on the characteristic curve marked as a thick blue line in
Figure 3. These are the crystals that rise to the highest point in the crystallizer and subsequently, after sinking to the bottom, reach the largest particle size. After some calculations, one gets
In conclusion, the solution of the process model (
1) and (
4)–(
7) is quite simple, when moving along the characteristic lines. The situation is complicated by the mill, which forms a feedback loop between large crystals sinking to the bottom and small crystals entering the crystallizer. The treatment of that feedback loop is discussed in the next section.
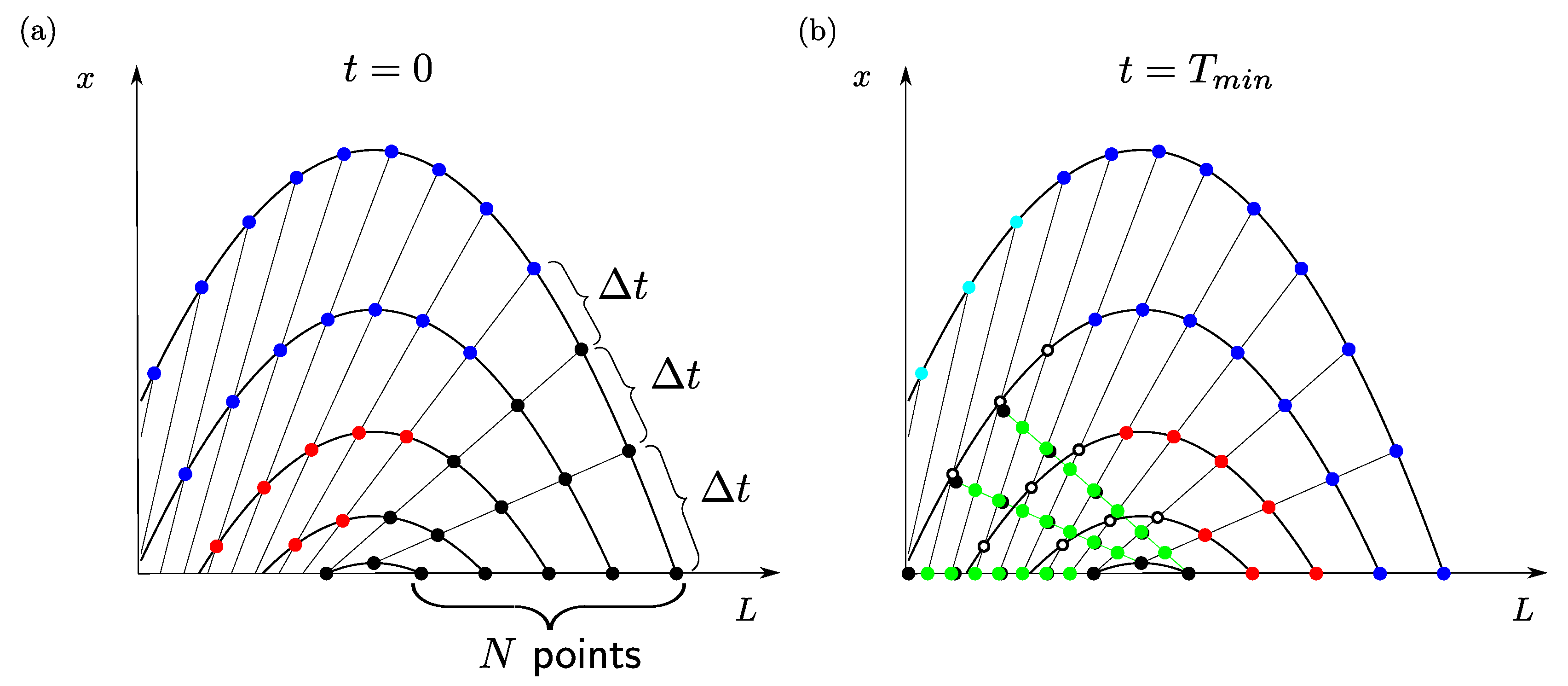
2.3. Solution of the Linear Model by Stepping Forward in Time
with a Problem Specific Step Size
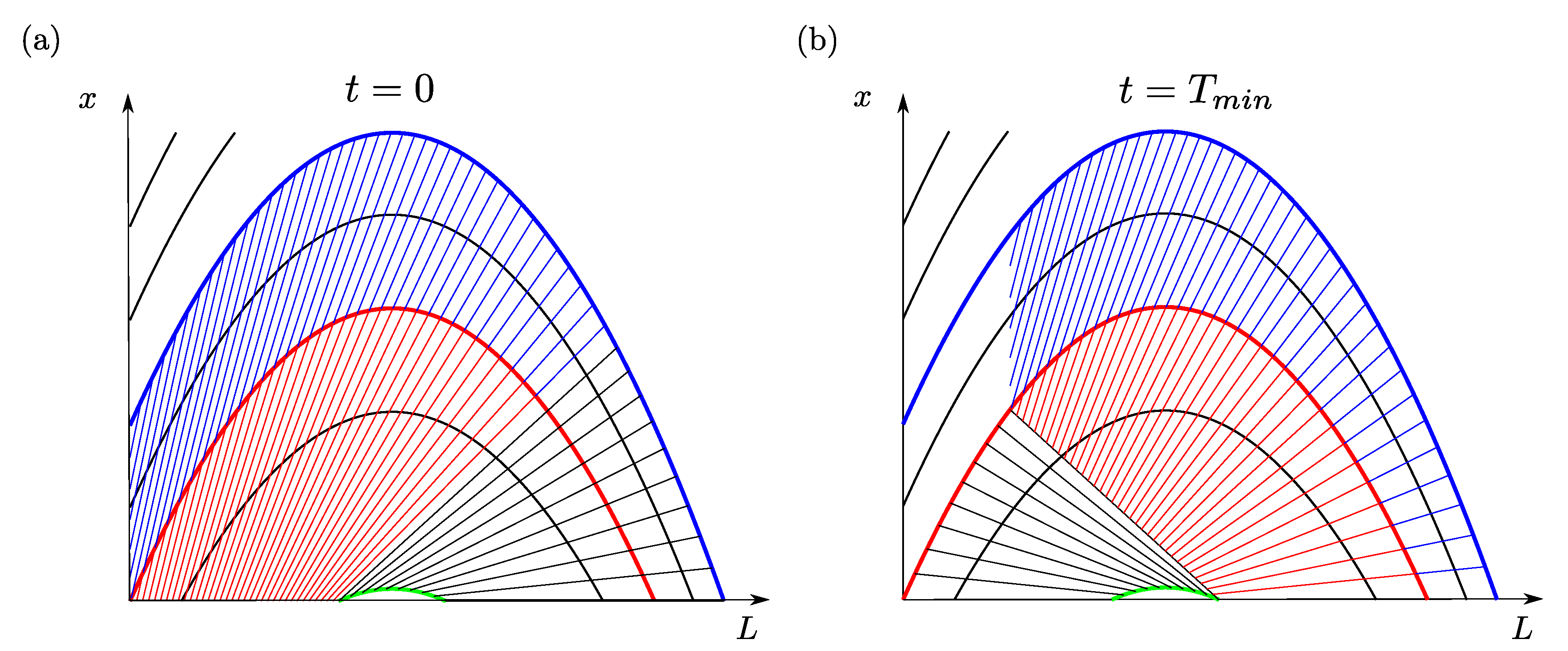
Figure 4 illustrates how the feedback loop caused by the mill can be tackled. At time
, the number size density
is completely known because of the initial condition (
7). The diagram in
Figure 4a shows straight lines representing crystals that will arrive in the mill at the same time. In a first time interval equal to the minimum residence time
of the crystals, the input to the mill only depends on the initial conditions. For a time
, the input to the mill is
In the above equation,
stands for the point on the characteristic through
that is reached when going back an arc length
t on that characteristic.
is the size of the largest crystals coming out of the mill, after they have sunk to the crystallizer bottom (see
Figure 3b). All crystals going into the mill between
and
are indicated by black lines in
Figure 4a.
The outcome of the mill for the time interval
is calculated by inserting (
24) into the integral on the right-hand side of Equation (
4). The crystals coming out of the mill before time
are represented by the black lines in the right-hand side diagram of
Figure 4.
During the same time interval, the crystals in the red region (coming out of the mill) and the blue region (generated by nucleation) in
Figure 4a move to the regions with the same color in
Figure 4b, without changing their number. The number size densities in the red and blue regions in the right-hand side diagram at time
are just copies of the number size densities at time
in the left-hand side diagram.
The number size densities in the white unshaded area of the right-hand side diagram depend on nucleation and can be determined with Equation (
6).
In summary, the number size density
can be calculated completely from the number size density
by analytical calculations, provided that the integrals in Equation (
4) have elementary antiderivatives. However, the analytical solution is not particularly useful, as it tends to become very complicated after a few steps, even for a computer algebra system. Therefore, a numerical time stepping scheme is presented in the following section. An analytical solution of a very simple case will be used at the end of this section to test the accuracy of the numerical solution.
For the numerical solution, an initial grid is chosen as shown in the diagram on the left-hand side of
Figure 5. The grid points lie on straight lines, which indicate a certain distance in time required to reach the bottom of the crystallizer and the entrance to the mill. The time interval between two neighboring straight lines is always equal to the same value
,
M, being a free parameter.
N equidistant grid points are chosen on the
L axis between
and
. The intersection between the characteristics through those points with the straight lines defines the remaining grid points at
. As previously explained, one can calculate where the crystals on the grid points at
will be at time
, just by tracing back the characteristics. Crystals at the blue and red grid points in
Figure 5 move inside the crystallizer in the considered time interval, but do not reach the mill. Their number size densities at time
are just copies of the number size densities at time
of the corresponding points in
Figure 5a. More effort is required for the crystals that pass the mill before time
. These points are marked black in
Figure 5a,b. The number size densities at the black points at time
(
Figure 5b) are computed from the number size densities at time
(
Figure 5a) with the help of Equation (
4); the trapezoidal rule is used to approximate the integrals. As a challenge for the numerical approach, the black points in
Figure 5b are no longer a part of the original grid. Instead, they are used to calculate the number size densities at the white points
Figure 5b, which are still undefined. This is done by linear interpolation in the following way:
The black points in
Figure 5b, leaving the mill at the same time, are connected by green lines. The intersection points between the green and the black grid lines are determined (green points in the figure).
The number size densities at the intersection points are computed by linear interpolation along the green lines.
The number size densities at the white points are computed by linear interpolation along the black lines.
Finally, the number size densities at some further grid points, shown as light-blue points, are given by the boundary condition (
6). This concludes a step forward in time with step length
. As the algorithm is only a linear mapping from the number size densities on the grid at
to the number size densities on the same grid at
, it can be solved very efficiently in Matlab.
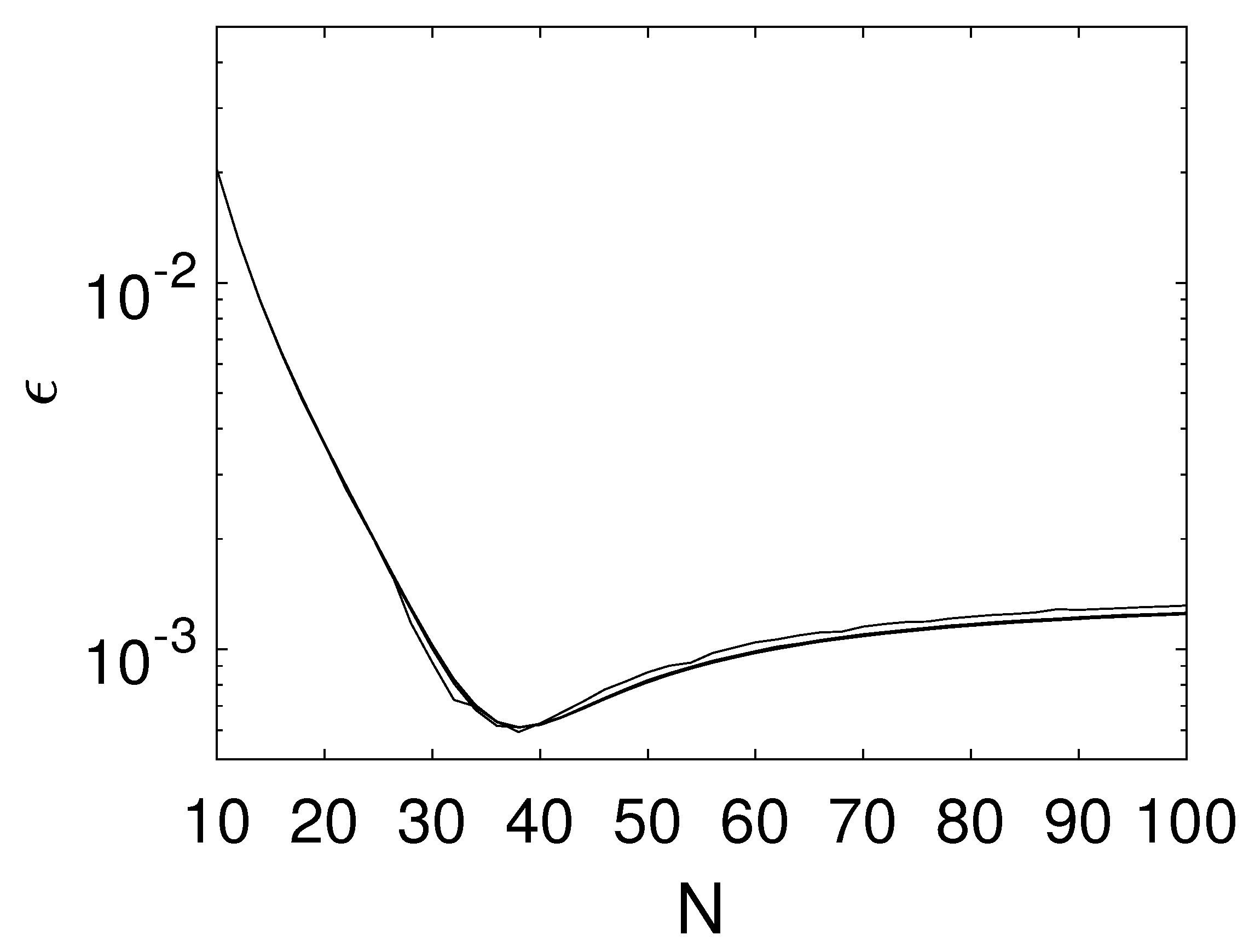
The numerical algorithm has two sources of inaccuracies: the integration by the trapezoidal rule and the linear interpolation. In order to validate the algorithm, it is compared with the analytical solution for a very simple testing case that has no physical relevance, but is only chosen because it allows an analytical solution of the integrals in (
4). The initial number size density of the testing case is chosen as
with
and
. The number size density of the crystals leaving the mill is obtained from
The numerical and the analytical solutions at
are compared for different values of the grid parameters
M and
N. The error is defined as the Frobenius norm of the difference between the numerically and the analytically computed number size density at all grid points, divided by the Frobenius norm of the analytical number size density. As can be seen from
Figure 6, the error is already quite small for a very coarse grid. A good compromise between systematic and rounding errors is achieved for
grid points per grid line. The number
M of grid lines is of a minor importance for this specific example.
2.4. Numerical Results
Numerical computations are done using the parameter values from
Table 1. The parameter values are chosen as close as possible to the simulation conditions of the more detailed model in [
7], although there are some systematic differences between the models. This will be discussed in
Section 3.
It is assumed that the crystals coming out of the mill have a bell curve-shaped distribution,
Initially, a small amount of seed crystals of the desired L enantiomer are placed in the middle of the crystallizer.
Figure 7 shows snapshots of the crystal populations at different time points that are multiples of the minimum residence time, which under the chosen conditions is equal to
. The crystal populations are represented by mass related size distributions
, defined as
At the beginning, the crystallizer is completely free from undesired D crystals. The liquid flow velocity
is chosen such that no crystals, apart from the nuclei in region 1a of
Figure 3, can leave the process, and the crystal mass grows exponentially. Additionally, nuclei of the D crystals in region 1b remain in the crystallizer. Therefore, crystals of the counter-enantiomer D gradually accumulate in the crystallizer.
After 20 time steps, the bulk of the L crystals reach the mill and the first recycled fragments of the L crystal are moved upwards. One may also recognize a small population of D crystals rising in the crystallizer (encircled in the diagram).
After 120 time steps, the first D crystals have also passed the mill. The L crystals slowly approach the final bimodal distribution.
For longer times, the L and D crystal distributions become more and more similar to each other, although the absolute numbers of L and D crystals are very different. Especially in the lower half of the crystallizer, one may observe a counter-flow of small crystals moving up and large crystals sinking down. The resulting bimodal distribution differs from the first intuition that crystals should be sorted according to their size with small crystals at the top and large crystals at the bottom. This is only true for the larger half of the crystals.
Figure 8 shows the total mass of crystals in the crystallizer over time. For the chosen conditions, hardly any crystals escape through the crystallizer top. Hence, both the mass of the L crystals and the mass of the D crystals grow exponentially. Note that this does not cause a decrease in the purity of the crystal phase. The L crystals keep their initial lead in mass over the D crystals, and the contamination of the crystal phase remains low. This is visualized in
Figure 8b containing the purity of the crystal phase, which is defined as the mass of the L crystals defined by the total crystal mass.
2.5. Possible Extensions of the Linear Model
The linear model contains a number of strongly simplifying assumptions. The topic of this section is to discuss, how some of these assumptions may be relaxed in order to bring the simulation results closer to reality.
So far, it has been assumed that
B is constant in time and space. However, in reality,
B varies due to the spatially varying supersaturation of the liquid phase. Further, experimental results in [
8] indicate that the nucleation mainly takes place at the crystallizer walls and, therefore, may be different at different points in the crystallizer and may also change over time, when crystals adhere to the walls. A time and space dependent nucleation rate
would hardly have an effect on the proposed solution approach for the linear model. All described solution steps are applicable to a varying nucleation rate, too.
In a similar way, a space and time dependent growth rate
G could be taken into account; however, this might require a numerical solution of Equations (
9)–(12).
A more detailed description of the crystal velocity is also possible, but requires a higher effort. The linear expression for the particle velocity (
14) has the advantage that it allows analytical calculations, but it neglects some aspects of experimental reality. First, the experimental crystallizer has a conical shape with a diameter increasing from the bottom to the middle of the tube. This causes a decreasing liquid flow velocity in the empty tube and also makes the particle velocity dependent on the space coordinate
x. Second, the linear dependence of the particle velocity on the characteristic size
L is a rather crude approximation. The classic model by Richardson and Zaki [
14] gives a more realistic picture of the particle velocity and has been applied successfully to the continuous enantiomer separation in [
7,
9].
To obtain the particle velocity
according to the model by Richardson and Zaki, first the Archimedes number (describing the ratio between buoyancy and friction force on a particle of size
L) is calculated from
where
is the size of an equivalent spherical particle and
is a shape dependent sphericity parameter. The fluid velocity
needed to keep a single particle of size
L in equilibrium follows from
with the Reynolds number
For higher particle densities,
has to be corrected in the following way to account for particle-particle interactions:
with the fluid volume fraction
and
Finally, the particle velocity
is computed from
where
is the cross-sectional area of the crystallizer at position
x. In this contribution, the model is simplified a bit by assuming a constant fluid volume fraction
. Due to the conical crystallizer shape, the cross-sectional area
depends on the space coordinate
x.
Figure 9a shows particle velocity profiles at different positions in the crystallizer for a constant liquid flow rate. The linear velocity profile used so far is a rough average of these profiles.
The computation of the characteristics with the particle velocity by Richardson and Zaki requires a numerical integration of (
9)–(12). From
Figure 9b, one can see that the result is not too different from the simple approach in
Figure 3. For small crystal sizes
L, the characteristics are still roughly parabola shaped, but for large crystals, there are clear deviations. Lines connecting crystals that will reach the mill at the same time (blue) or connecting crystals having left the mill at the same time (red) are no longer straight lines, but curves. Apart from that, important qualitative results obtained for the linear velocity profile are still valid for the Richardson-Zaki model. One can still distinguish one area in the
L-
x-plane, where crystals from the mill are permanently recycled, and an area in which all crystals originate from nucleation. The two areas are separated by the characteristic through
, shown as a thick green line. The highest point of that characteristic determines if nuclei can accumulate in the crystallizer or not. If the highest point lies above the crystallizer top, all nuclei escape through the outlet and high product purity is guaranteed at all times. This can be achieved by adjusting the liquid flow rate, as illustrated in
Figure 9c showing the characteristic through
for different liquid flow rates.
The numerical solution of the more detailed model by the time stepping scheme is also possible in principle. The main difference in this case is that the grid points have to be computed numerically.












{kind=link}
{kind=link}
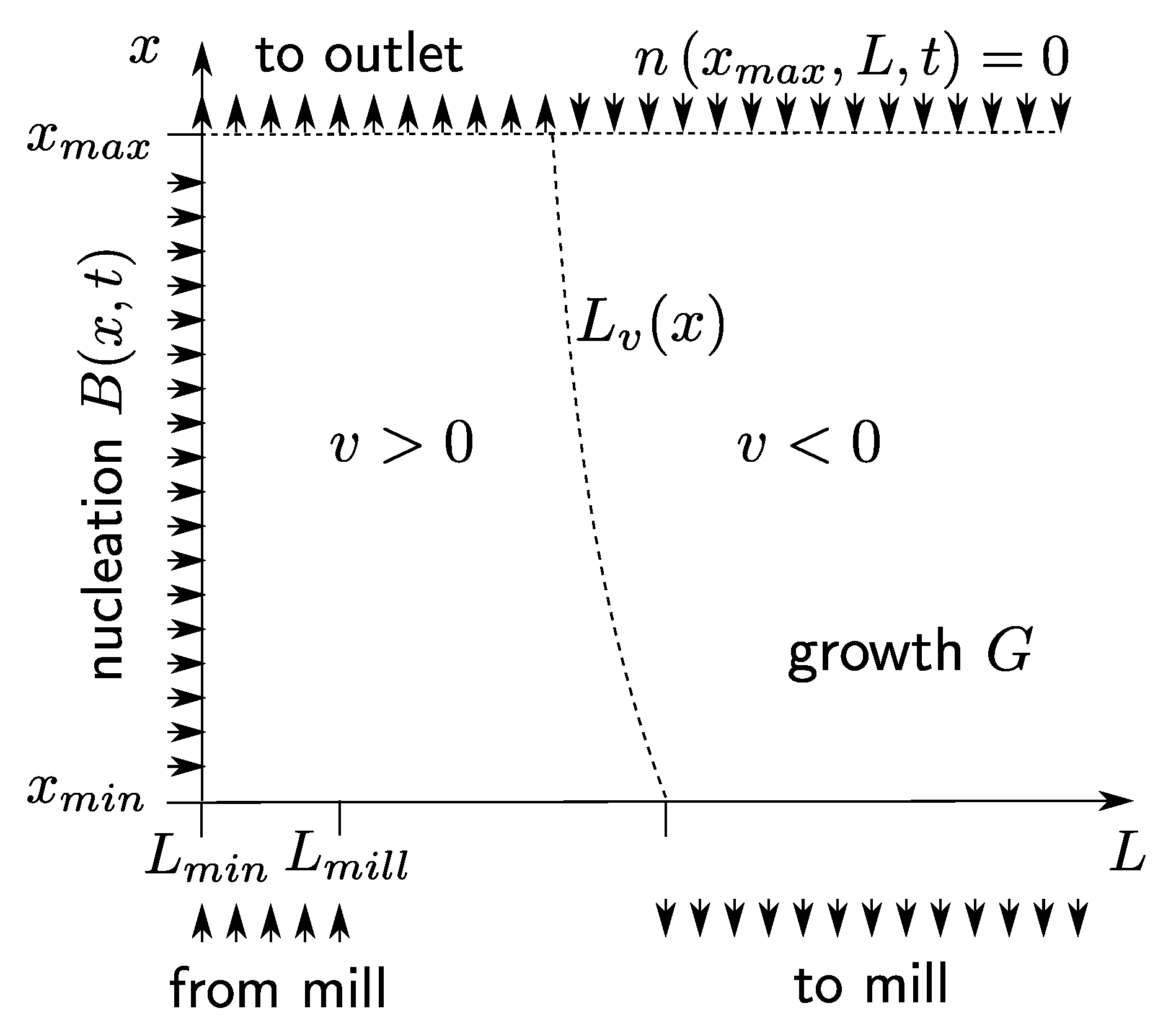{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}