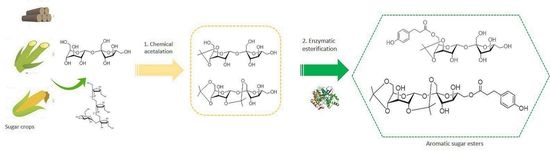
Chemoenzymatic Synthesis of New Aromatic Esters of Mono- and Oligosaccharides


 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Enzymatic Esterification/Transesterification of 1-octanol with Aromatic Substrates
2.3. Enzymatic Transesterification of Sugars Using HPPME
2.4. Chemical Synthesis of Sugar Acetals
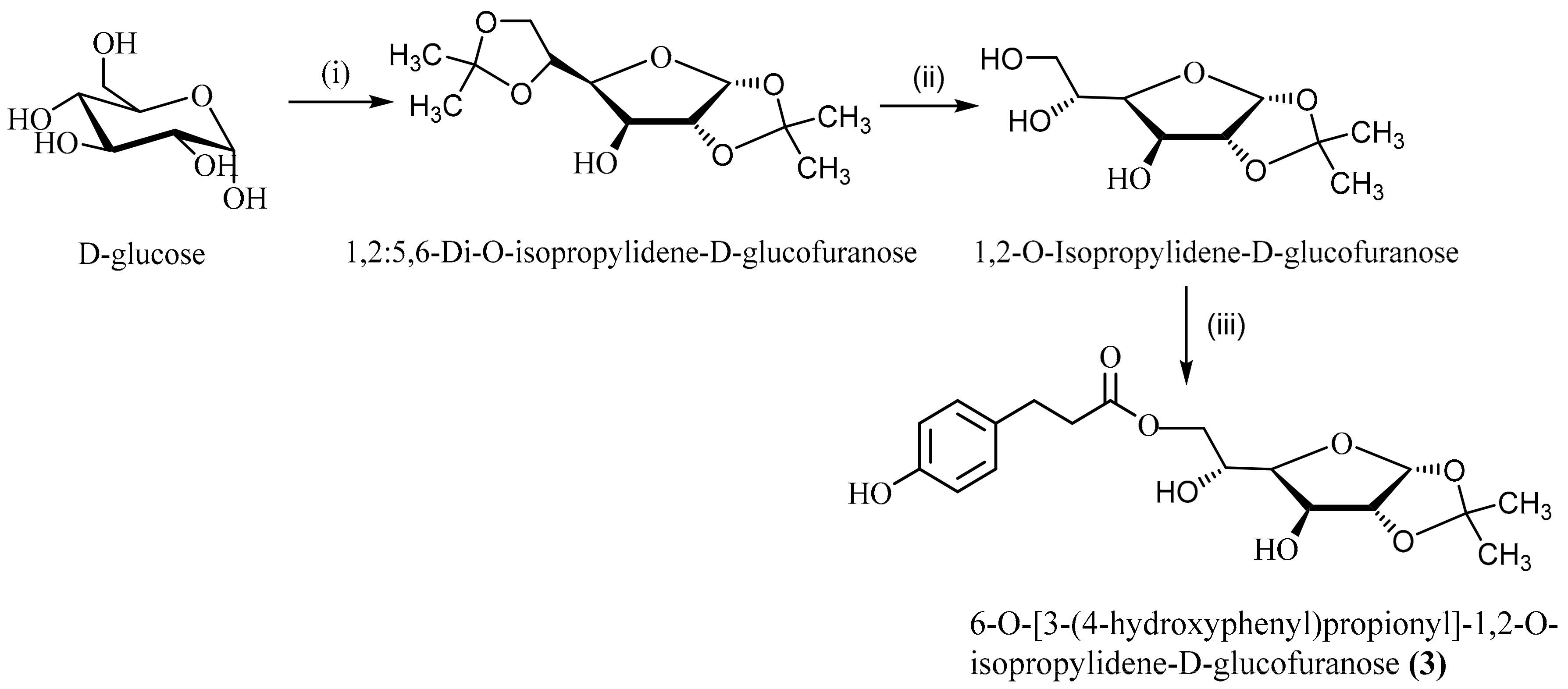
2.4.1. Synthesis of 1,2-O-isopropylidene-d-glucofuranose
2.4.2. Acetalation of Sucrose, Lactose and Inulin
2.5. Enzymatic Reactions
2.5.1. Enzymatic Esterification of Alkyl Glycosides and Sugar Acetals by HPPA
2.5.2. Separation and Purification of Aromatic Sugar Esters
2.5.3. HPLC Analysis
2.5.4. UHPLC Analysis
2.5.5. Gas Chromatography (GC) Analysis
2.5.6. Thin-Layer Chromatography
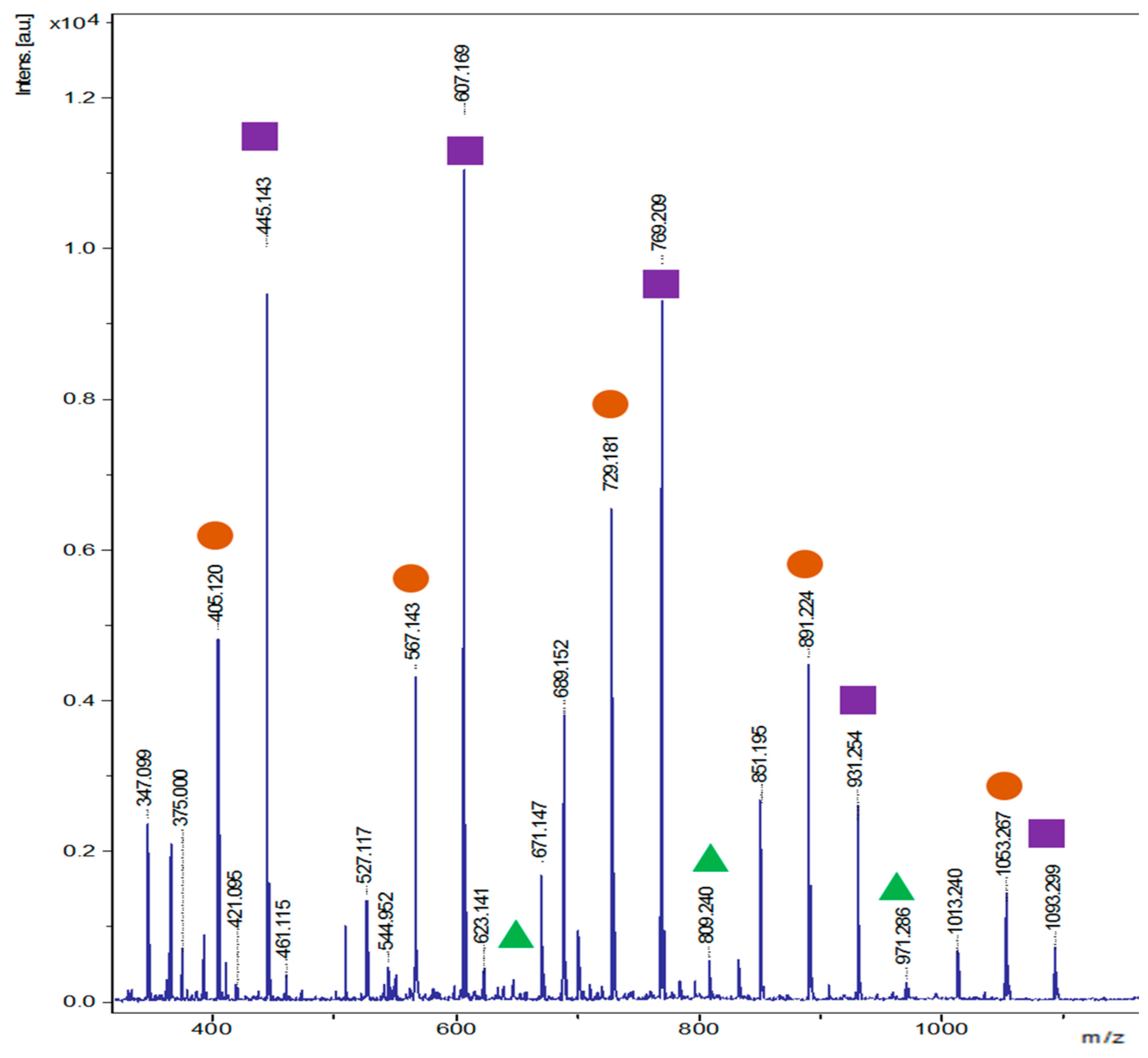
2.6. Structural Analysis
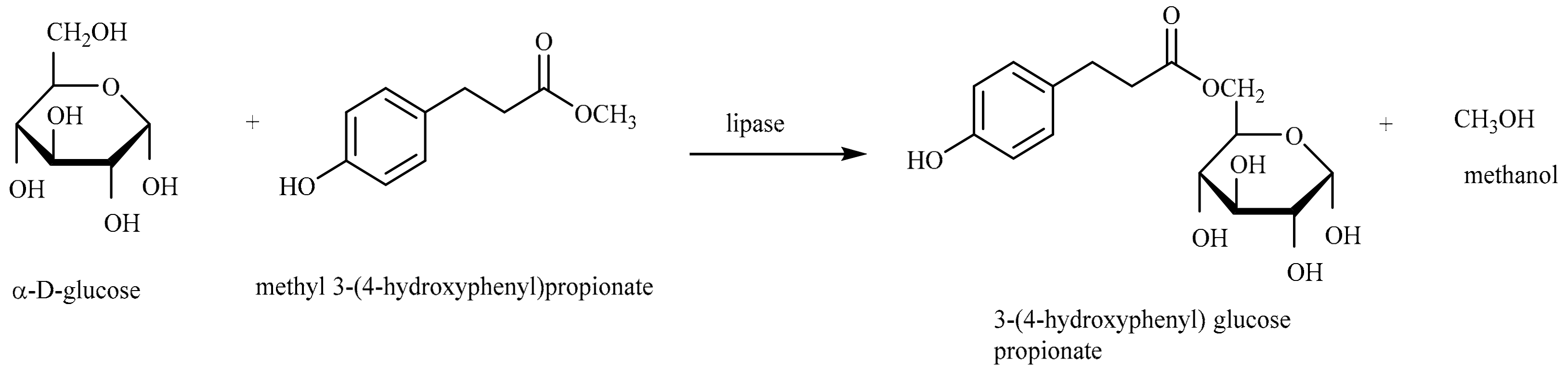
2.6.1. 6-O-[3-(4-hydroxyphenyl)propionyl] methyl-α-d-glucoside (1)
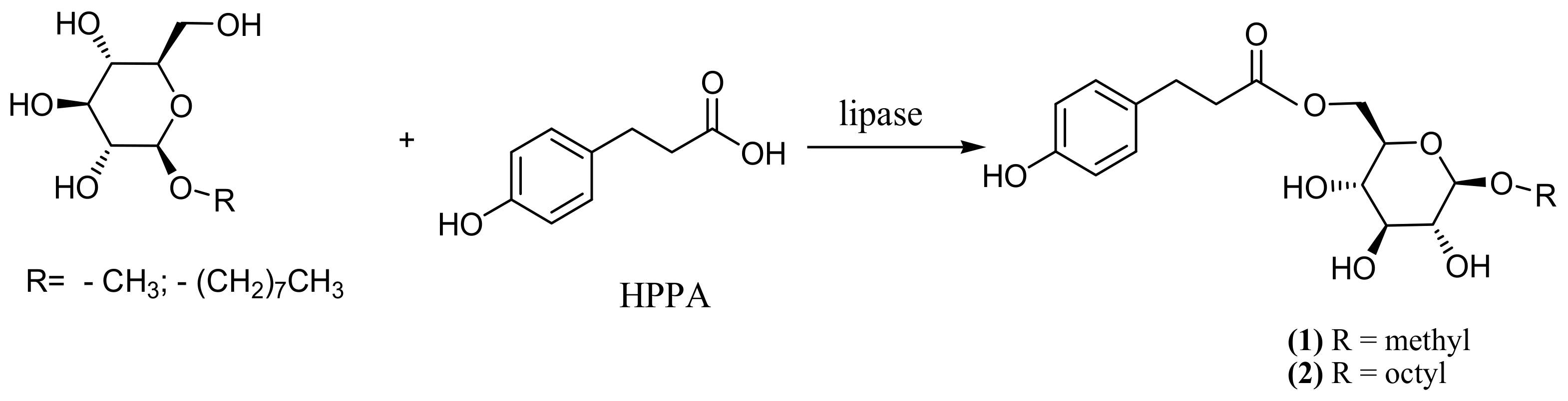
2.6.2. 6-O-[3-(4-hydroxyphenyl)propionyl] octyl-β-d-glucoside (2)
2.6.3. 6-O-[3-(4-hydroxyphenyl)propionyl]-1,2-O-isopropylidene-d-glucofuranose (3)
2.6.4. 2,1:4,6-di-O-isopropylidene sucrose (4)
2.6.5. 2,3: 5,6: 3′, 4′-tri-O-isopropylidene dimethyl lactose (5)
3. Results and Discussion
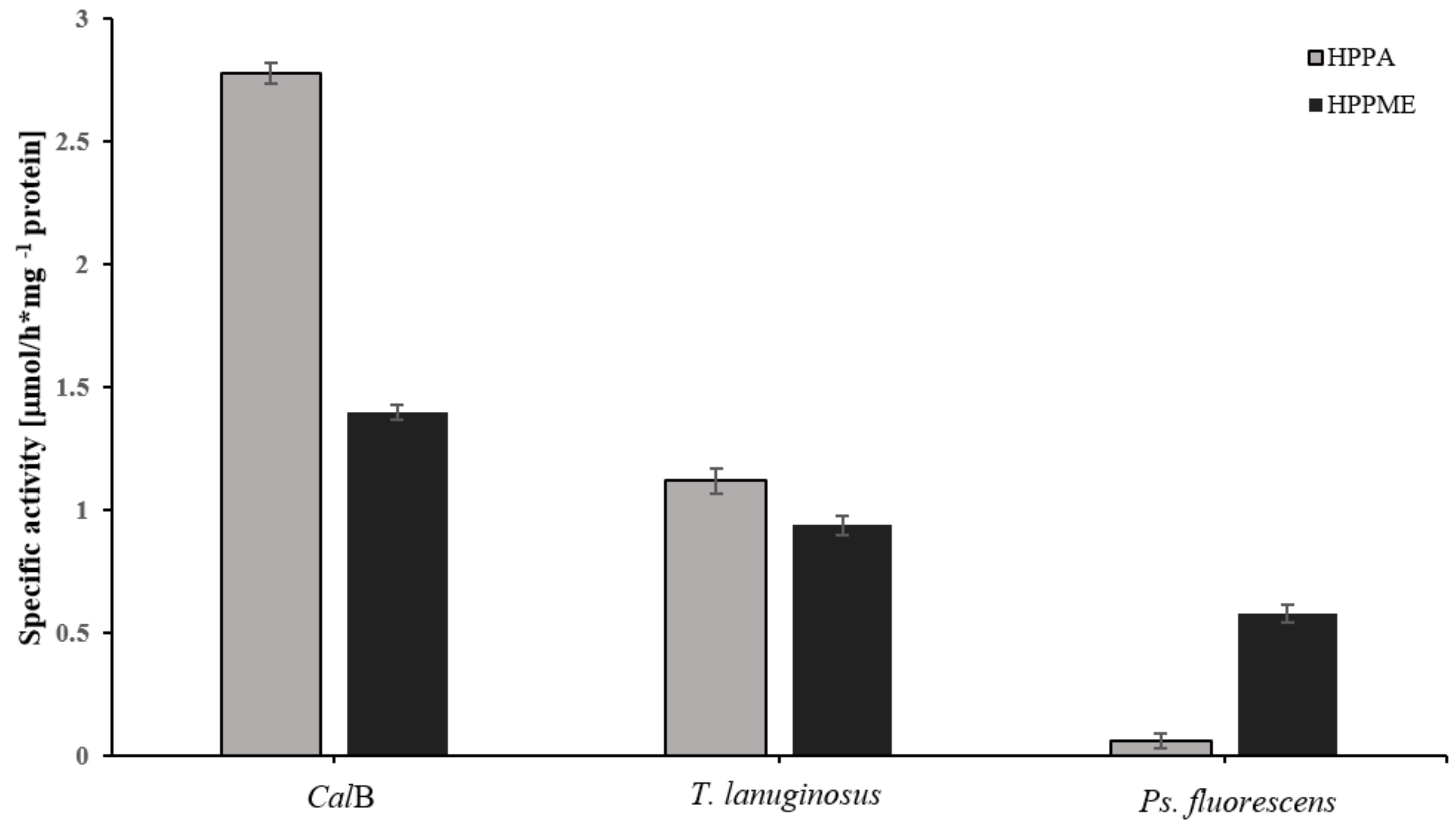
3.1. Specificity of Native Lipases for Aromatic Acids and Esters
3.2. Specificity of Native Lipases for Aromatic Esters of Carbohydrates
3.3. Enzymatic Synthesis and Characterization of Aromatic Esters of Alkyl Glycosides
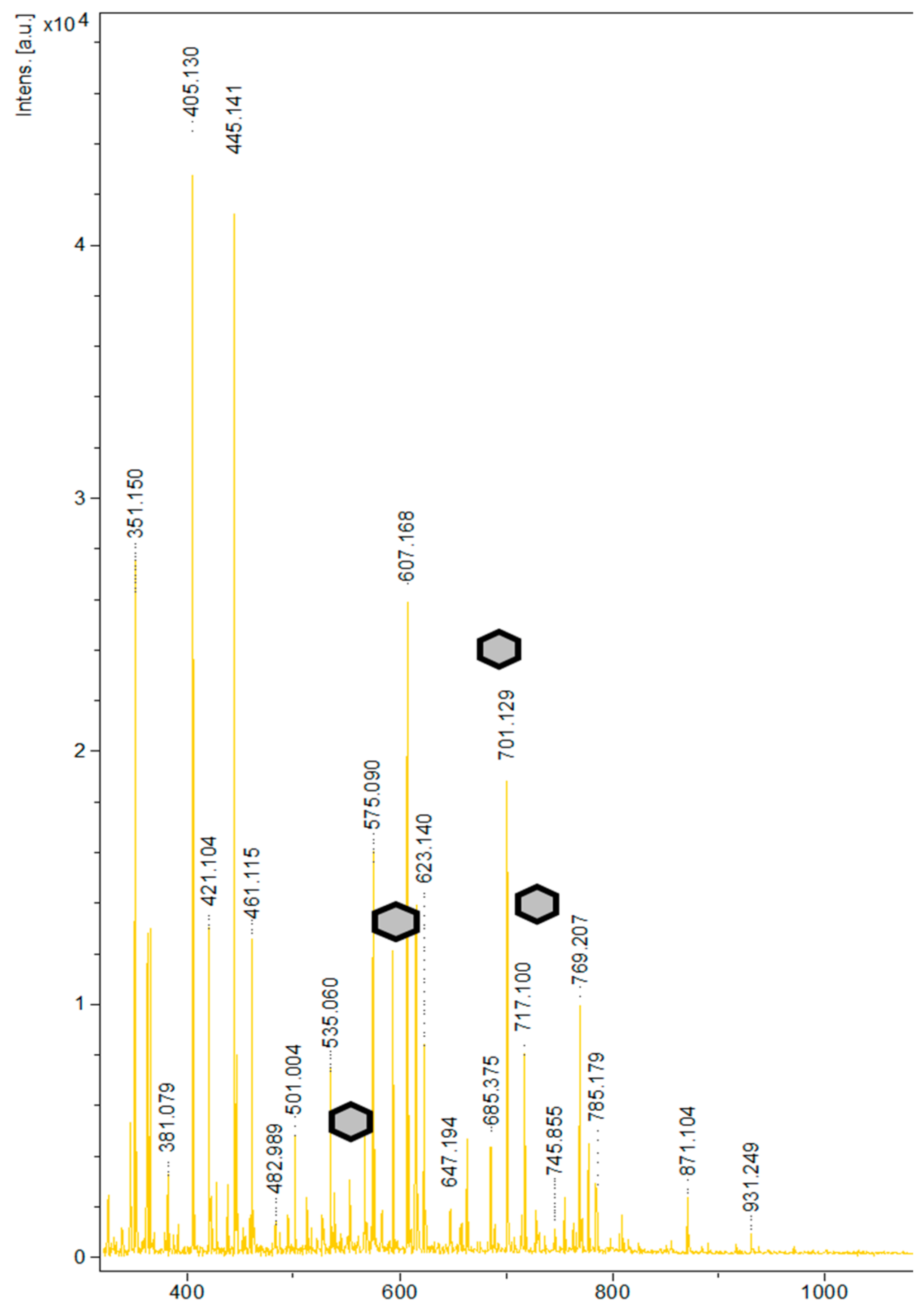
3.4. Chemoenzymatic Synthesis and Characterization of 6-O-[3-(4-hydroxyphenyl)propionyl]-1,2-O-isopropylidene-d-glucofuranose
3.5. Chemoenzymatic Synthesis and Characterization of Aromatic Esters of Disaccharides
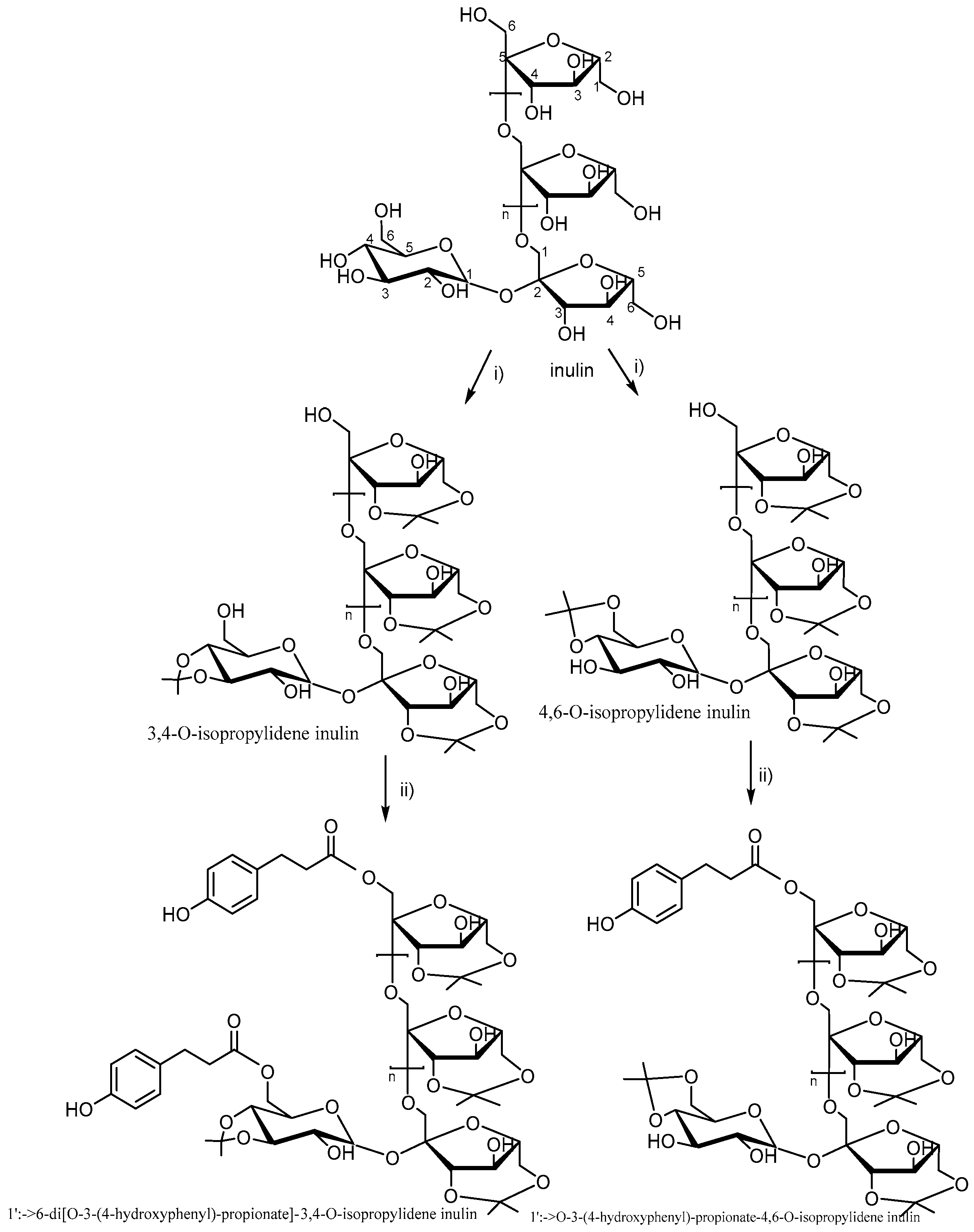
3.6. Synthesis and Characterization of Aromatic Esters of Inulin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ghosh, B.; Jones, A.D. Profiling, characterization, and analysis of natural and synthetic acylsugars (sugar esters). Anal. Methods 2017, 9, 892–905. [Google Scholar] [CrossRef]
- Rajendran, A.; Palanisamy, A.; Thangavelu, V. Lipase catalyzed ester synthesis for food processing industries. Braz. Arch. Biol. Technol. 2009, 52, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Gumel, A.M.; Annuar, M.S.M.; Heidelberg, T.; Chisti, Y. Lipase mediated synthesis of sugar fatty acid esters. Process Biochem. 2011, 46, 2079–2090. [Google Scholar] [CrossRef]
- Kennedy, J.F.; Kumar, H.; Panesar, P.S.; Marwaha, S.S.; Goyal, R.; Parmar, A.; Kaur, S. Enzyme-catalyzed regioselective synthesis of sugar esters and related compounds. J. Chem. Technol. Biotechnol. 2006, 81, 866–876. [Google Scholar] [CrossRef]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; dos Santos, J.C.S.; Rodrigues, R.C.; Berenguer-Murcia, A.; Briand, L.E.; Fernandez-Lafuente, R. Novozym 435: The “perfect” lipase immobilized biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–2420. [Google Scholar] [CrossRef] [Green Version]
- Stergiou, P.Y.; Foukis, A.; Filippou, M.; Koukouritaki, M.; Parapoul, M.; Theodorou, L.G.; Hatziloukas, E.; Afendra, A.; Pandey, A.; Papamichael, E.M. Advances in lipase-catalyzed esterification reactions. Biotechnol. Adv. 2013, 31, 1846–1859. [Google Scholar] [CrossRef]
- Stamatis, H.; Sereti, V.; Kolisis, F.N. Enzymatic synthesis of hydrophilic and hydrophobic derivatives of natural phenolic acids in organic media. J. Mol. Cat. B Enzym. 2001, 11, 323–328. [Google Scholar] [CrossRef]
- Croitoru, R.; van den Broek, L.A.M.; Frissen, A.E.; Davidescu, C.M.; Peter, F.; Boeriu, C.G. Lipase catalyzed synthesis of aromatic esters of sugar alcohols. World Acad. Sci. Eng. Technol. 2011, 76, 484–489. [Google Scholar]
- Vasilescu, C.; Todea, A.; Nan, A.; Circu, M.; Turcu, R.; Benea, I.-C.; Peter, F. Enzymatic synthesis of short-chain flavor esters from natural sources using tailored magnetic biocatalysts. Food Chem. 2019, 296, 1–8. [Google Scholar] [CrossRef]
- Facin, B.R.; Melchiors, M.; Valerio, A.; Oliveira, J.V.; de Oliveira, D. Driving immobilized lipases as biocatalysts:10 years state of the art and future prospects. Ind. Eng. Chem. Res. 2019, 58, 5358–5378. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, Q.; Shao, L.; Jia, Y.; Zhang, X. Microfluidic immobilized enzyme reactors for continuous biocatalysis. React. Chem. Eng. 2020, 5, 9–32. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, M.; Ma, Z.; Xin, B.; Guo, R.; Xu, X. Sugar fatty acid esters. In Polar Lipids. Biology, Chemistry, and Technology; Ahmad, M.U., Xu, X., Eds.; AOCS Press: Urbana, IL, USA, 2015; pp. 215–243. [Google Scholar] [CrossRef]
- Croitoru, R.; Fitigau, F.; van den Broek, L.A.M.; Frissen, A.E.; Davidescu, C.M.; Boeriu, C.G.; Peter, F. Biocatalytic acylation of sugar alcohols by 3-(4-hydroxyphenyl)propionic acid. Process Biochem. 2012, 47, 1894–1902. [Google Scholar] [CrossRef]
- Krawczyk, J. Aggregation properties of sucrose fatty acid esters and some other sugar-based surfactants at different temperatures. J. Mol. Liq. 2018, 271, 610–620. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, W.; Lu, Y.; Wang, Y.; Chen, X.; Bai, S.; Zhao, Y.; He, T.; Lao, F.; Shang, Y.; et al. Naturally occurring cinnamic acid sugar ester derivatives. Molecules 2016, 21, 1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.; Zzaman, A.; Rahman, M.; Rahman, M.A.; Kawsar, S.M.A. Novel methyl 4,6-O-benzylidene-α-d-glucopyranoside derivatives: Synthesis, structural characterization and evaluation of antibacterial activities. Hacettepe. J. Biol. Chem. 2019, 47, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Y.; Wang, R.F.; Liu, B. An update on oligosaccharides and their esters from traditional Chinese medicines: Chemical structures and biological activities. Evid. Based Complement. Altern. Med. 2015, 512675, 1–23. [Google Scholar] [CrossRef]
- do Amaral Sampaio Neta, N.; dos Santos, J.C.S.; de Oliveira Sancho, S.; Rodrigues, S.; Gonçalves, L.R.B.; Rodrigues, L.R.; Teixeira, J.A. Enzymatic synthesis of sugar esters and their potential as surface-active stabilizers of coconut milk emulsions. Food Hydrocoll. 2012, 27, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Campana, R.; Merli, A.; Verboni, M.; Biondo, F.; Favi, G.; Duranti, A.; Lucarini, S. Synthesis and evaluation of saccharide-based aliphatic and aromatic esters as antimicrobial and antibiofilm agents. Pharmaceuticals 2019, 12, 186. [Google Scholar] [CrossRef] [Green Version]
- Duarte-Almeida, M.J.; Novoa, A.V.; Linares, A.F.; Lajolo, F.M.; Genovese, M.I. Antioxidant activity of phenolics compounds from sugar cane (Saccharum officinarum L.) juice. Plant Foods Hum. Nutr. 2006, 61, 187–192. [Google Scholar] [CrossRef]
- Akong, F.O.; Bouquillon, S. Efficient syntheses of bolaform surfactants from L-rhamnose and/or 3-(4-hydroxyphenyl) propionic acid. Green Chem. 2015, 17, 3290–3300. [Google Scholar] [CrossRef]
- Bassanini, I.; Kapešov&á, J.; Petrásková, L.; Pelantová, H.; Markošová, K.; Rebroš, M.; Valentová, K.; Kotik, M.; Káňová, K.; Bojarová, P.; et al. Glycosidase-catalyzed synthesis of glycosyl esters and phenolic glycosides of aromatic acids. Adv. Synth. Catal. 2019, 361, 2627–2637. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.G.; de Meneses, A.C.; de Araújo, P.H.H.; de Oliveira, D. A review on enzymatic synthesis of aromatic esters used as flavor ingredients for food, cosmetics and pharmaceuticals industries. Trends Food Sci. Technol. 2017, 69, 95–105. [Google Scholar]
- van Kempen, S.E.H.J.; Boeriu, C.G.; Schols, H.A.; de Waard, P.; van der Linden, E.; Sagis, L.M.C. Novel surface-active oligofructose fatty acid mono-esters by enzymatic esterification. Food Chem. 2013, 138, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Sarney, D.B.; Kapeller, H.; Fregapane, G.; Vulfson, E.N. Chemo-enzymatic synthesis of disaccharide fatty acid esters. J. Am. Oil. Chem. Soc. 1994, 71, 711–714. [Google Scholar] [CrossRef]
- Kumar, A.; Dhar, K.; Kanwar, S.S.; Arora, P.K. Lipase catalysis in organic solvents: Advantages and applications. Biol. Proced. Online 2016, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yaacob, N.; Kamarudin, N.H.A.; Leow, A.T.C.; Salleh, A.B.; Rahman, R.N.Z.R.A.; Ali, M.S.M. Effects of lid 1 mutagenesis on lid displacement, catalytic performances and thermostability of cold-active Pseudomonas AMS8 lipase in toluene. Comput. Struct. Biotechnol. J. 2019, 17, 215–228. [Google Scholar] [CrossRef]
- Gihaz, S.; Kanteev, M.; Pazy, Y.; Fishman, A. Filling the void: Introducing aromatic interactions into solvent tunnels to enhance lipase stability in methanol. Appl. Environ. Microbiol. 2018, 84, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Garcia, R.; Hough, L.; Richardson, A.C. Acetalation of sucrose by acetal exchange with concomitant fission of the glycosidic bond. Some new acetals of D-glucose and methyl α-d-fructofuranoside. J. Chem. Soc. Perkin Trans. 1981, 1, 3176–3181. [Google Scholar] [CrossRef]
- Hanaya, T.; Sato, N.; Yamamoto, H. An efficient synthesis of methyl 1,3-O-isopropylidene-α-d-fructofuranoside and 2,3:5,6-di-O-isopropylidene-d-glucose dimethyl acetal derivatives from sucrose. Carbohydr. Res. 2005, 340, 2494–2501. [Google Scholar] [CrossRef]
- Rong, Y.W.; Zhang, Q.H.; Wang, W.; Li, B.L. Simple and clean method for O-isopropylidenation of carbohydrates. Bull. Korean Chem. Soc. 2014, 35, 2165–2168. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.S.; Chambers, E.S.; Preston, T.; Tedford, C.; Brignardello, J.; Garcia-Perez, I.; Holmes, E.; Wallis, G.A.; Morrison, D.J.; Frost, G.S. Effects of inulin propionate ester incorporated into palatable food products on appetite and resting energy expenditure: A randomised crossover study. Nutrients 2019, 11, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afinjuomo, F.; Barclay, T.G.; Parikh, A.; Song, Y.; Chung, R.; Wang, L.; Liu, L.; Hayball, J.D.; Petrovsky, N.; Garg, S. Design and characterization of inulin conjugate for improved intracellular and targeted delivery of pyrazinoic acid to monocytes. Pharmaceutics 2019, 11, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oostveen, E.A.; Weijnen, J.; Van Haveren, J.; Gillard, M. Polysaccharide Esters and their Use as Binders in Coating. Patent No. WO03064477, EP 1470163 B1, 7 August 2003. [Google Scholar]
- Torlopov, M.A.; Udoratina, E.V.; Kuchin, A.V. Synthesis of inulin esters of phenylcarboxylic acids. Russ. J. Org. Chem. 2013, 49, 702–706. [Google Scholar] [CrossRef]
- Schmidt, O.T. Reactions of carbohydrates. In Methods in Carbohydrate Chemistry; Whistler, R.L., Wolfrom, M.L., Eds.; Academic Press: New York, NY, USA, 1963; Volume 2, pp. 318–325. [Google Scholar]
- Naika, S.; Basu, A.; Saikia, R.; Madan, B.; Pritish, P.; Chaterjee, R.; Brask, J.; Svendsen, A. Lipases for use in industrial biocatalysis: Specificity of selected structural groups of lipases. J. Mol. Catal B Enzym. 2010, 65, 18–23. [Google Scholar] [CrossRef]
- Compton, D.L.; Laszlo, J.A.; Berhow, M.A. Lipase-catalyzed synthesis of ferulate esters. J. Am. Oil Chem. Soc. 2000, 77, 513–519. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Rodriguez-Ordonez, G.; Hidalgo, A.; Gollin, A.; Lyon, J.; Hitchman, T.S.; Weiner, D.P. Selectivity of lipases and esterases towards phenol esters. J. Mol. Catal B Enzym. 2005, 36, 8–13. [Google Scholar] [CrossRef]
- Ter Haar, R.; Schols, H.A.; van den Broek, L.A.M.; Saglam, D.; Frissen, A.E.; Boeriu, C.G.; Gruppen, H. Molecular sieves provoke multiple substitutions in the enzymatic synthesis of fructose oligosaccharide–lauryl esters. J. Mol. Catal. B 2010, 62, 183–189. [Google Scholar] [CrossRef]
- Redmann, I.; Pina, M.; Guyot, B.; Blaise, P.; Farines, M.; Graille, J. Chemoenzymatic synthesis of glucose fatty esters. Carbohydr. Res. 1997, 300, 103–108. [Google Scholar] [CrossRef]
- Fregapane, G.; Sarney, D.B.; Greenberg, S.G.; Knight, D.J.; Vulfson, E.N. Chemo-enzymatic synthesis of monosaccharide fatty acid esters and their preliminary characterization. Prog. Biotechnol. 1992, 8, 563–568. [Google Scholar]
- Rufino, A.R.; Biaggio, F.; Santos, J.C.; de Castro, H.F. Chemoenzymatic synthesis: A strategy to obtain xylitol monoesters. J. Chem. Technol. Biotechnol. 2009, 84, 957–960. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buzatu, A.R.; Frissen, A.E.; van den Broek, L.A.M.; Todea, A.; Motoc, M.; Boeriu, C.G. Chemoenzymatic Synthesis of New Aromatic Esters of Mono- and Oligosaccharides. Processes 2020, 8, 1638. https://doi.org/10.3390/pr8121638
Buzatu AR, Frissen AE, van den Broek LAM, Todea A, Motoc M, Boeriu CG. Chemoenzymatic Synthesis of New Aromatic Esters of Mono- and Oligosaccharides. Processes. 2020; 8(12):1638. https://doi.org/10.3390/pr8121638
Chicago/Turabian StyleBuzatu, Alina Ramona, August E. Frissen, Lambertus A. M. van den Broek, Anamaria Todea, Marilena Motoc, and Carmen Gabriela Boeriu. 2020. "Chemoenzymatic Synthesis of New Aromatic Esters of Mono- and Oligosaccharides" Processes 8, no. 12: 1638. https://doi.org/10.3390/pr8121638
APA StyleBuzatu, A. R., Frissen, A. E., van den Broek, L. A. M., Todea, A., Motoc, M., & Boeriu, C. G. (2020). Chemoenzymatic Synthesis of New Aromatic Esters of Mono- and Oligosaccharides. Processes, 8(12), 1638. https://doi.org/10.3390/pr8121638