TLC-Densitometric Determination of Five Coxibs in Pharmaceutical Preparations


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Methods
2.1.1. Chemicals and Apparatus
2.1.2. Standard Substances and Solutions
2.1.3. Samples Solutions
2.1.4. Chromatographic Conditions
2.2. Validation of the Method
2.2.1. Selectivity
2.2.2. Range and Linearity
2.2.3. Limit of Detection (LOD) and Limit of Quantification (LOQ)
2.2.4. Precision
2.2.5. Accuracy
2.2.6. Robustness
2.3. Stability-Indicating Study
2.4. Analysis of Pharmaceutical Formulations
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Katori, M.; Majima, M. Cyclooxygenase-2: Its rich diversity of roles and possible application if it’s selective inhibitors. Inflamm. Res. 2000, 49, 367–392. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [Green Version]
- Curtis, E.; Fuggle, N.; Shaw, S.; Spooner, L.; Ntani, G.; Parsons, C.; Corp, N.; Honvo, G.; Baird, J.; Maggi, S.; et al. Safety of cyclooxygenase-2 inhibitors in osteoarthritis: Outcomes of a systematic review and meta-analysis. Drugs Aging 2019, 36, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Martín Arias, L.H.; Martín González, A.; Sanz Fadrique, R.; Salgueiro Vázquez, E. Gastrointestinal safety of coxibs: Systematic review and meta-analysis of observational studies on selective inhibitors of cyclo-oxygenase 2. Fundam. Clin. Pharmacol. 2019, 33, 134–147. [Google Scholar] [CrossRef]
- Fu, J.Y.; Masferrer, J.L.; Seibert, K.; Raz, A.; Needleman, P. The induction and suppression of prostaglandin Hz synthase (cyclooxygenase) in human monocytes. J. Biol. Chem. 1990, 265, 16737–16740. [Google Scholar]
- Bjorkman, D.J. One hundred years of NSAID gastropathy: Are coxibs the answer? Rev. Gastroenterol. Disord. 2001, 1, 121–127. [Google Scholar]
- McMurray, R.W.; Hardy, K.J. COX-2 inhibitors: Today and tomorrow. Am. J. Med. Sci. 2002, 323, 181–189. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 inhibitors: A review of their structure-activity relationships. Iran. J. Pharm. Res. 2011, 10, 655–683. [Google Scholar]
- Dingle, J.T. The effect of NSAID on the matrix of human articular cartilages. Z. Rheumatol. 1999, 58, 125–129. [Google Scholar] [CrossRef]
- Merck Announces Voluntary Worldwide Withdrawal of VIOXX®. Available online: http://www.vioxx.com/vioxx/documents/english/vioxx_press_release.pdf (accessed on 17 November 2004).
- Braun, J.; Baraliakos, X.; Westhoff, T. Nonsteroidal anti-inflammatory drugs and cardiovascular risk—A matter of indication. Semin. Arthritis Rheum. 2020, 50, 285–288. [Google Scholar] [CrossRef]
- Meek, I.L.; Van de Laar, M.A.F.J.; Vonkeman, H.E. Non-steroidal anti-inflammatory drugs: An overview of cardiovascular risks. Pharmaceuticals 2010, 3, 2146–2162. [Google Scholar] [CrossRef] [Green Version]
- Sgambati, S.A. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial: Commentary. Dis. Colon Rectum 2005, 48, 1330–1331. [Google Scholar]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Eng. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Bergh, M.S.; Budsberg, S.C. The Coxib NSAIDs: Potential clinical and pharmacologic importance in veterinary medicine. J. Vet. Intern. Med. 2005, 9, 633–643. [Google Scholar] [CrossRef]
- Jung, M.; Lees, P.; Seewald, W.; King, J.N. Analytical determination and pharmacokinetics of robenacoxib in the dog. J. Vet. Pharmacol. Ther. 2009, 32, 41–48. [Google Scholar] [CrossRef]
- Kim, T.W.; Giorgi, M. A brief overview of the coxib drugs in the veterinary field. Am. J. Anim. Vet. Sci. 2013, 8, 89–97. [Google Scholar] [CrossRef]
- Kongara, K.; Chambers, P. Robenacoxib in the treatment of pain in cats and dogs: Safety, efficacy, and place in therapy. Vet. Med. Res. Rep. 2018, 9, 53–61. [Google Scholar] [CrossRef] [Green Version]
- FDA Guidance, Analytical Procedures and Method Validation: Chemistry, Manufacturing, and Controls Documentation, Draft Guidance, Food and Drug Administration. 2000. Available online: http://www.fda.gov/downloads/Drugs/.../Guidance/ucm122858.pdf (accessed on 20 January 2020).
- Pollmeier, M.; Toulemonde, C.; Fleishman, C.; Hanson, P.D. Clinical evaluation of firocoxib and carprofen for the treatment of dogs with osteoarthritis. Vet. Res. 2006, 159, 547–551. [Google Scholar] [CrossRef]
- Hanson, P.D.; Brooks, K.C.; Case, J.; Conzemius, M.; Gordon, W.; Schuessler, J.; Shelley, B.; Sifferman, R.; Drag, M.; Alva, R.; et al. Efficacy and safety of firacoxib in the management of canine osteoarthritis under field conditions. Vet. Ther. 2006, 7, 127–140. [Google Scholar]
- Üner, M.; Yener, G.; Ergüven, M. Design of colloidal drug carriers of celecoxib for use in treatment of breast cancer and leukemia. Mater. Sci. Eng. C 2019, 103. [Google Scholar] [CrossRef]
- Regulski, M.; Regulska, K.; Prukała, W.; Piotrowska, H.; Stanisz, B.; Murias, M. COX-2 inhibitors: A novel strategy in the management of breast cancer. Drug Discov. Today 2016, 21, 598–615. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Yarla, N.S.; Madka, V.; Rao, C.V. Clinically relevant anti-inflammatory agents for chemoprevention of colorectal cancer: New perspectives. Int. J. Mol. Sci. 2018, 19, 2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasy, N.K.A.; Ragab, D.; Sallam, M.A.; Abdelmonsif, D.A.; Aly, R.G.; Elkhodairy, K.A. A comparative study: The prospective influence of nanovectors in leveraging the chemopreventive potential of COX-2 inhibitors against skin cancer. Int. J. Nanomed. 2019, 14, 7561–7581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, S.; Villarino, N.; Sommardahl, C.; Kvaternick, V.; Zarabadipour, C.; Siger, L.; Yarbrough, J.; Amicucci, A.; Reed, K.; Breeding, D.; et al. Disposition of firocoxib in equine plasma after an oral loading dose and a multiple dose regimen. Vet. J. 2013, 198, 382–385. [Google Scholar]
- Oh, H.A.; Kim, D.; Lee, S.H.; Jung, B.H. Simultaneous quantitative determination of celecoxib and its two metabolites using liquid chromatography-tandem mass spectrometry in alternating polarity switching mode. J. Pharm. Biomed. Anal. 2015, 107, 32–39. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, N.; Ji, W.; Wen, Q. Rapid quantitative analysis of etoricoxib in human plasma by UPLC-MS/MS and application to a pharmacokinetic study in Chinese healthy volunteers. Biomed. Chromatogr. 2019, 33, e4414. [Google Scholar] [CrossRef]
- Jedziniak, P.; Szprengier-Juszkiewicz, T.; Pietruk, K.; Ledziska, E.; Żmudzki, J. Determination of non-steroidal anti-inflammatory drugs and their metabolites in milk by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 2955–2963. [Google Scholar] [CrossRef]
- Starek, M.; Komsta, Ł.; Krzek, J. Reversed-phase thin-layer chromatography technique for the comparison of the lipophilicity of selected non-steroidal anti-inflammatory drugs. J. Pharm. Biomed. Anal. 2013, 85, 132–137. [Google Scholar] [CrossRef]
- Kumar, S.; Joshi, A.; Thakur, R.S.; Pathak, A.K. Simultaneous estimation of etoricoxib and thiocolchicoside by RP-HPLC method in combined dosage forms. Acta Pol. Pharm. 2011, 68, 839–843. [Google Scholar]
- Rajmane, V.S.; Gandhi, S.V.; Patil, U.P.; Sengar, M.R. High-performance thin-layer chromatographic determination of etoricoxib and thiocolchicoside in combined tablet dosage form. J. AOAC Int. 2010, 93, 783–787. [Google Scholar]
- Singh, S.; Mishra, A.; Verma, A.; Ghosh, A.K.; Mishra, A.K. A simple ultraviolet spectrophotometric method for the determination of etoricoxib in dosage formulations. J. Adv. Pharm. Technol. Res. 2012, 3, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangoi, M.S.; Wrasse-Sangoi, M.; Oliveira, P.R.; Bernardi, L.S. Determination of lumiracoxib by a validated stability-indicating MEKC method and identification of its degradation products by LC-ESI-MS studies. J. Sep. Sci. 2011, 34, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, L.; Bao, Y.; Yang, Q.; Zhou, L.; Hao, H.; Xie, C. Confirmation of more stable polymorphic form of etoricoxib at room temperature. J. Pharm. Sci. 2018, 107, 1903–1910. [Google Scholar] [CrossRef]
- Giorgi, M.; Kim, T.-W.; Saba, A.; Rouini, M.-R.; Yun, H.; Ryschanova, R.; Owen, H. Detection and quantification of cimicoxib, a novel COX-2 inhibitor, in canine plasma by HPLC with spectrofluorimetric detection: Development and validation of a new methodology. J. Pharm. Biomed. Anal. 2013, 83, 28–33. [Google Scholar] [CrossRef]
- Knych, H.K.; Stanley, S.D.; Arthur, R.M.; Mitchell, M.M. Detection and pharmacokinetics of three formulations of firocoxib following multiple administrations to horses. Equine Vet. J. 2014, 46, 734–738. [Google Scholar] [CrossRef]
- Triñanes, S.; Casais, M.C.; Mejuto, M.C.; Cela, R. Selective determination of COXIBs in environmental water samples by mixed-mode solid phase extraction and liquid chromatography quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2015, 1420, 35–45. [Google Scholar] [CrossRef]
- ICH Guide Q2 (R1). Validation of Analytical Procedures-Text and Methodology; International Conference on Harmonization: Geneva, Switzerland, 2005; Available online: https://www.ich.org/page/quality-guidelines (accessed on 8 January 2020).
- Bakshi, M.; Singh, S. Development of validated stability-indicating assay methods—Critical review. J. Pharm. Biomed. Anal. 2002, 28, 1011–1040. [Google Scholar] [CrossRef]
- Rao, R.N.; Meena, S.; Nagaraju, D.; Rao, A.R.R. Development and validation of a reversed-phase liquid chromatographic method for separation and simultaneous determination of COX-2 inhibitors in pharmaceuticals and its application to biological fluids. Biomed. Chromatogr. 2005, 19, 362–368. [Google Scholar]
- Hamama, A.K.; Ray, J.; Day, R.O.; Brien, E. Simultaneous determination of rofecoxib and celecoxib in human plasma by high-performance liquid chromatography. J. Chromatogr. Sci. 2005, 43, 351–354. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
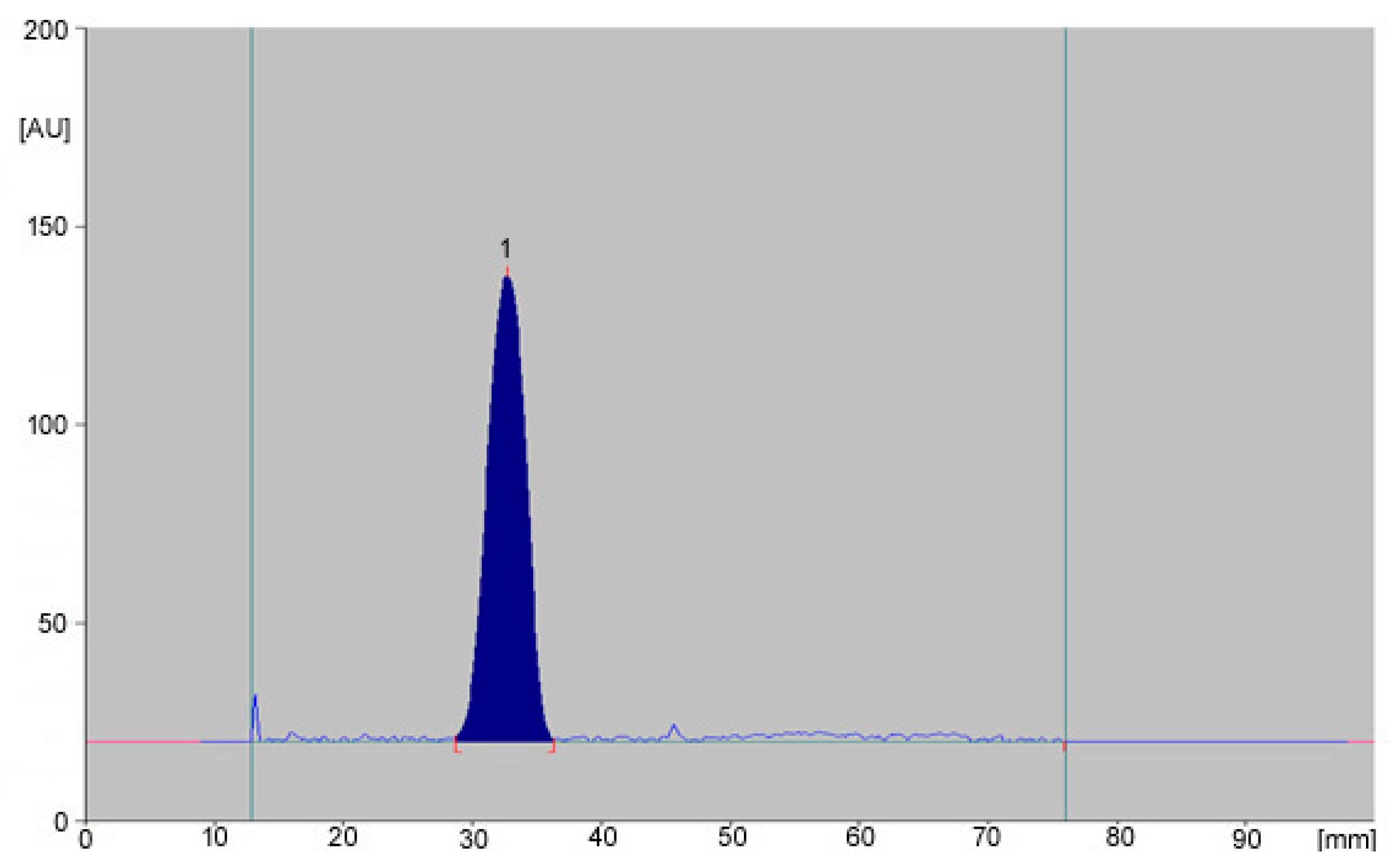{kind=link}
{kind=link}
| Substance | RF | α | R |
|---|---|---|---|
| cimicoxib | 0.25 | - | - |
| robenacoxib | 0.34 | 1.3 | 1.6 |
| etoricoxib | 0.45 | 1.3 | 2.8 |
| celecoxib | 0.71 | 3.2 | 5.9 |
| firocoxib | 0.83 | 1.4 | 1.4 |
| Parameter | Celecoxib | Etoricoxib | Firocoxib | Cimicoxib | Robenacoxib |
|---|---|---|---|---|---|
| λ = 254 (nm) | |||||
| Linearity (mg/mL) | 0.30–5.00 | 0.10–5.00 | 0.60–6.00 | 0.10–7.00 | 0.20–10.00 |
| a | 3069.28 | 3733.77 | 1598.63 | 2725.37 | 1484.92 |
| b | 1103.12 | 1169.34 | 223.43 | 1205.32 | 601.22 |
| Sa | 225.33 | 242.61 | 55.00 | 165.63 | 39.35 |
| Sb | 583.05 | 627.13 | 184.23 | 603.66 | 218.90 |
| Se | 871.41 | 960.96 | 240.71 | 1022.26 | 319.49 |
| r | 0.9894 | 0.9916 | 0.9952 | 0.9927 | 0.9986 |
| r(res) | 0.47 × 10−6 | 0.20 × 10−6 | 0.13 × 10−6 | 0.51 × 10−7 | 0.11 × 10−6 |
| CD | 0.9772 | 0.9437 | 0.4886 | 0.5102 | 0.2388 |
| LOD (ng/band) | 93.69 | 84.93 | 49.71 | 32.78 | 71.00 |
| LOQ (ng/band) | 283.91 | 257.37 | 150.63 | 99.34 | 215.16 |
| λ = 290 (nm) | |||||
| Linearity (mg/mL) | 0.10–5.00 | 0.02–3.00 | 0.40–4.00 | 0.05–3.00 | 0.10–10.00 |
| a | 1097.24 | 5483.18 | 3003.35 | 4281.88 | 1498.74 |
| b | 758.60 | 644.69 | −118.54 | −369.07 | 488.84 |
| Sa | 74.78 | 189.1 | 164.93 | 204.52 | 41.24 |
| Sb | 192.63 | 352.28 | 381.60 | 382.10 | 224.85 |
| Se | 301.07 | 492.13 | 492.39 | 528.69 | 361.14 |
| r | 0.9908 | 0.9976 | 0.9940 | 0.9954 | 0.9985 |
| r (res) | 0.20 × 10−6 | 0.40 × 10−6 | 1.10 × 10−1 | 0.40 × 10−6 | 0.38 × 10−7 |
| CD | 0.8276 | 0.4462 | 0.6400 | 0.4337 | 0.2176 |
| LOD (ng/band) | 90.55 | 29.62 | 54.10 | 40.75 | 79.52 |
| LOQ (ng/band) | 274.41 | 89.75 | 163.95 | 123.46 | 240.96 |
| Parameter | Celecoxib | Etoricoxib | Firocoxib | Cimicoxib | Robenacoxib | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ [nm] | 254 | 290 | 254 | 290 | 254 | 290 | 254 | 290 | 254 | 290 |
| Intra-day precision | ||||||||||
| xm | 3005.75 | 7420.98 | 4029,27 | 1649.43 | 2961.85 | 5134.45 | 6479.98 | 4100.50 | 5875.85 | 9630.23 |
| SD | 29.01 | 64.68 | 37.70 | 17.26 | 26.77 | 16.37 | 59.68 | 34.87 | 46.11 | 89.18 |
| Sxm | 11.85 | 26.41 | 15.39 | 7.05 | 10.93 | 6.68 | 24.37 | 14.24 | 18.83 | 36.41 |
| %RSD | 0.97 | 0.87 | 0.94 | 1.05 | 0.90 | 0.32 | 0.92 | 0.85 | 0.78 | 0.93 |
| Inter-day precision | ||||||||||
| xm | 3014.60 | 7422.07 | 4027.55 | 1656.52 | 2958.05 | 5130.57 | 6504.10 | 4117.62 | 5870.15 | 9638.03 |
| SD | 21.68 | 45.67 | 40.35 | 20.27 | 25.52 | 33.98 | 47.74 | 49.05 | 34.45 | 78.34 |
| Sxm | 8.85 | 18.64 | 16.47 | 8.28 | 10.42 | 13.87 | 19.49 | 20.02 | 14.06 | 31.98 |
| %RSD | 0.59 | 0.81 | 0.73 | 1.19 | 0.86 | 0.66 | 1.00 | 1.22 | 0.72 | 0.62 |
| Substance | λ (nm) | Level of Concentration | |||||
|---|---|---|---|---|---|---|---|
| 80% | 100% | 120% | |||||
| % Recovery | %RSD | % Recovery | %RSD | % Recovery | %RSD | ||
| celecoxib | 254 | 99.16 | 1.52 | 99.23 | 0.99 | 95.08 | 1.58 |
| 290 | 98.08 | 1.86 | 98.91 | 0.88 | 93.65 | 1.23 | |
| etoricoxib | 254 | 97.91 | 2.06 | 100.06 | 1.32 | 95.17 | 1.75 |
| 290 | 98.08 | 1.70 | 99.29 | 1.00 | 98.89 | 1.76 | |
| firecoxib | 254 | 96.07 | 1.37 | 98.16 | 0.99 | 105.95 | 1.03 |
| 290 | 99.93 | 0.95 | 99.95 | 0.90 | 101.25 | 0.48 | |
| cimicoxib | 254 | 98.68 | 0.85 | 99.98 | 0.49 | 100.33 | 0.75 |
| 290 | 98.20 | 0.90 | 98.30 | 1.01 | 101.30 | 1.10 | |
| robenacoxib | 254 | 100.85 | 1.31 | 100.67 | 1.21 | 101.64 | 1.20 |
| 290 | 99.19 | 0.73 | 99.71 | 0.78 | 98.23 | 0.92 | |
| Preparation | Declared Content | Determined Content | Statistical Analysis |
|---|---|---|---|
| (mg/Tablet or Capsule) | (mg/Tablet or Capsule) | ||
| Celebrex | 200 mg of celecoxib | 201.22 201.07 | xm = 200.12 |
| 199.97 200.08 | SD = 0.88 | ||
| 198.93 200.42 | Sxm = 0.33 | ||
| 199.14 | RSD = 0.44 | ||
| Arcoxia | 120 mg of etoricoxib | 120.33 121.11 | xm = 120.45 |
| 119.85 120.56 | SD = 0.48 | ||
| 121.02 110.98 | Sxm = 0.18 | ||
| 120.27 | RSD = 0.40 | ||
| Previcox | 227 mg of firocoxib | 226.72 226.44 | xm = 227.06 |
| 226.90 227.15 | SD = 0.60 | ||
| 228.08 227.61 | Sxm = 0.23 | ||
| 226.53 | RSD = 0.26 | ||
| Cimalgex | 80 mg of cimicoxib | 79.23 80.14 | xm = 79.49 |
| 79.37 78.98 | SD = 0.53 | ||
| 79.20 80.36 | Sxm = 0.20 | ||
| 79.17 | RSD = 0.67 | ||
| Onsior | 40 mg of robenacoxib | 40.24 39.77 | xm = 39.77 |
| 39.50 39.41 | SD = 0.35 | ||
| 39.96 39.39 | Sxm = 0.13 | ||
| 40.13 | RSD = 0.88 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gumułka, P.; Dąbrowska, M.; Starek, M. TLC-Densitometric Determination of Five Coxibs in Pharmaceutical Preparations. Processes 2020, 8, 620. https://doi.org/10.3390/pr8050620
Gumułka P, Dąbrowska M, Starek M. TLC-Densitometric Determination of Five Coxibs in Pharmaceutical Preparations. Processes. 2020; 8(5):620. https://doi.org/10.3390/pr8050620
Chicago/Turabian StyleGumułka, Paweł, Monika Dąbrowska, and Małgorzata Starek. 2020. "TLC-Densitometric Determination of Five Coxibs in Pharmaceutical Preparations" Processes 8, no. 5: 620. https://doi.org/10.3390/pr8050620
APA StyleGumułka, P., Dąbrowska, M., & Starek, M. (2020). TLC-Densitometric Determination of Five Coxibs in Pharmaceutical Preparations. Processes, 8(5), 620. https://doi.org/10.3390/pr8050620