
Oxidative Coupling of Methane for Ethylene Production: Reviewing Kinetic Modelling Approaches, Thermodynamics and Catalysts
 ,
,  ,
, 

Abstract
:
1. Introduction
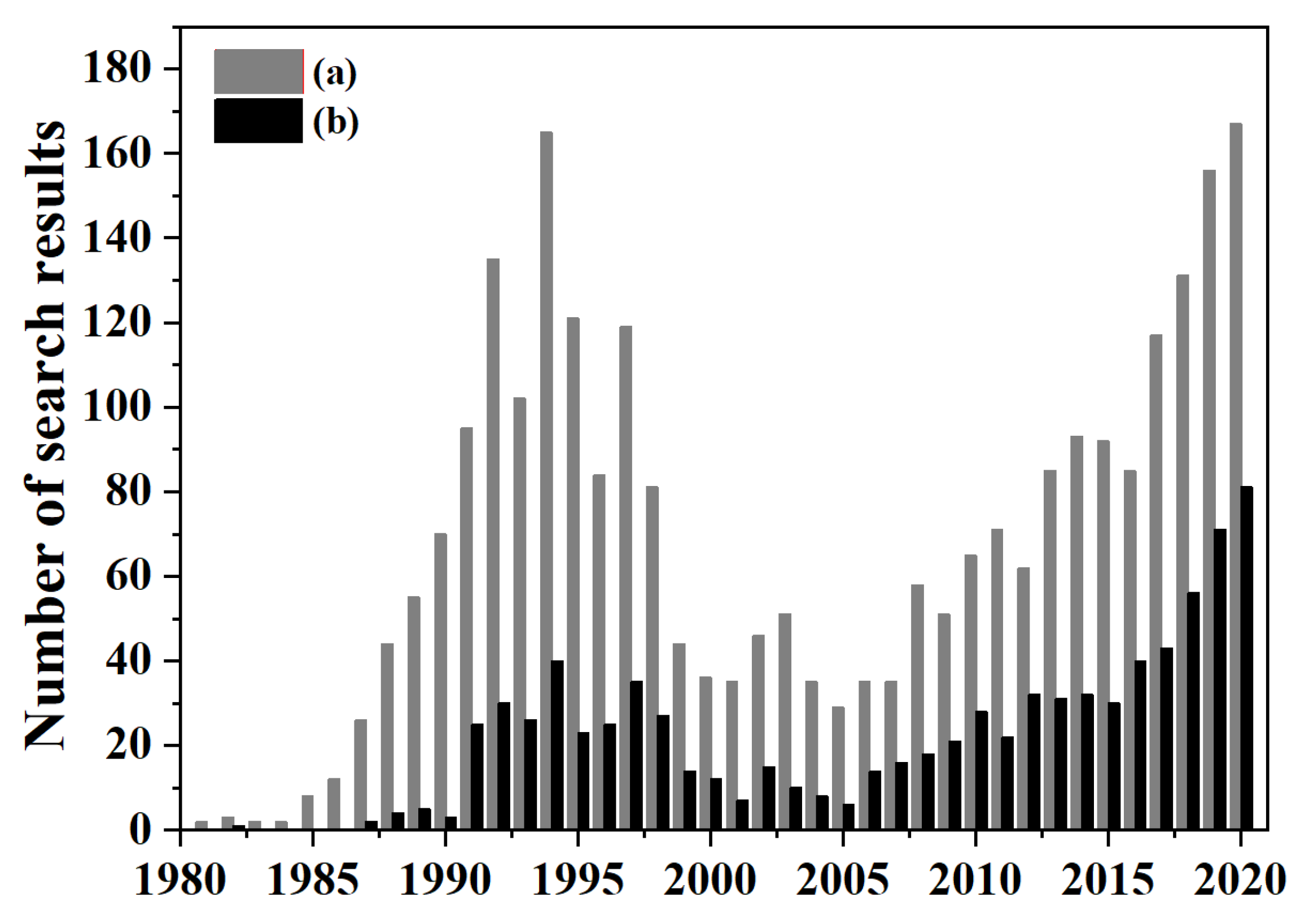
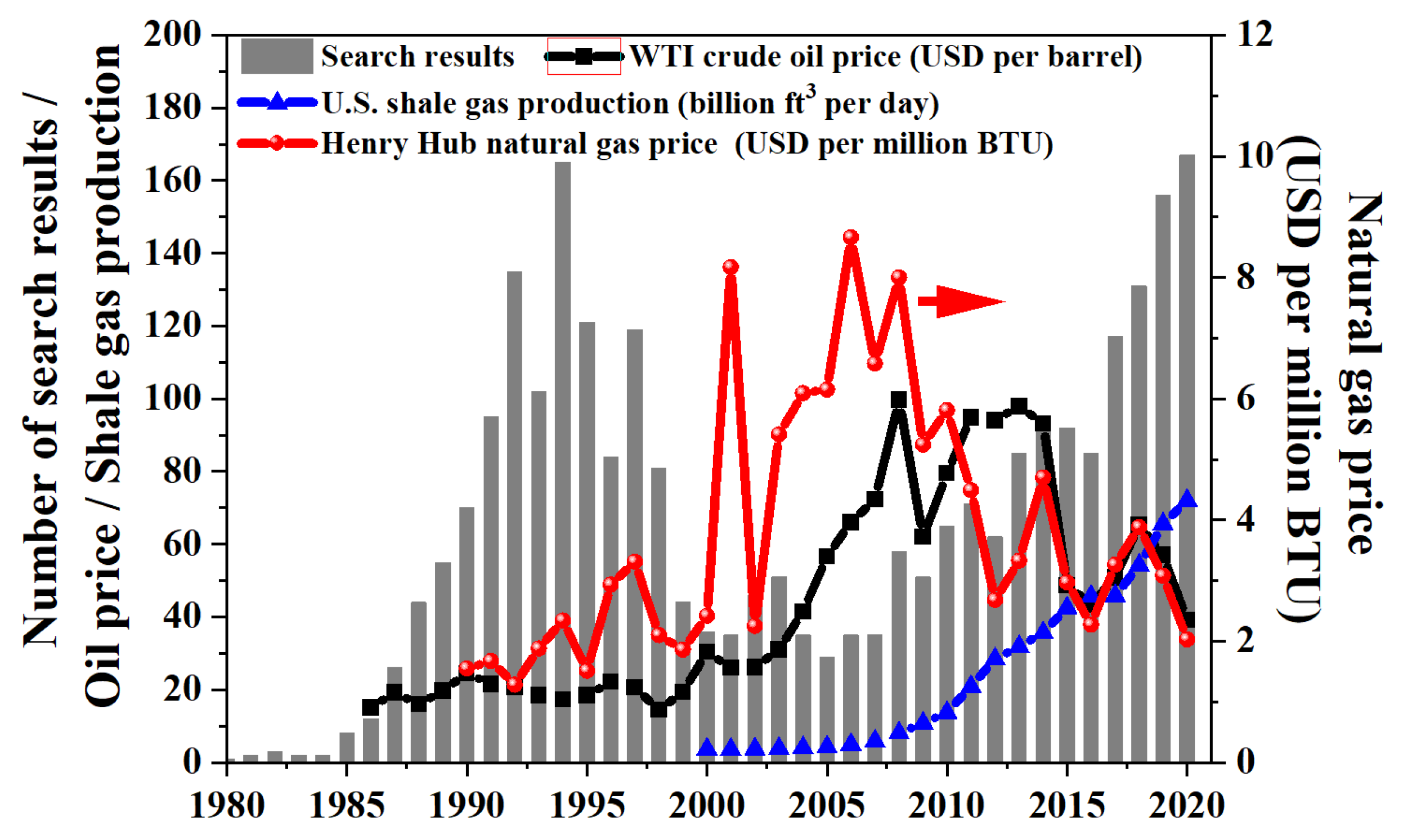
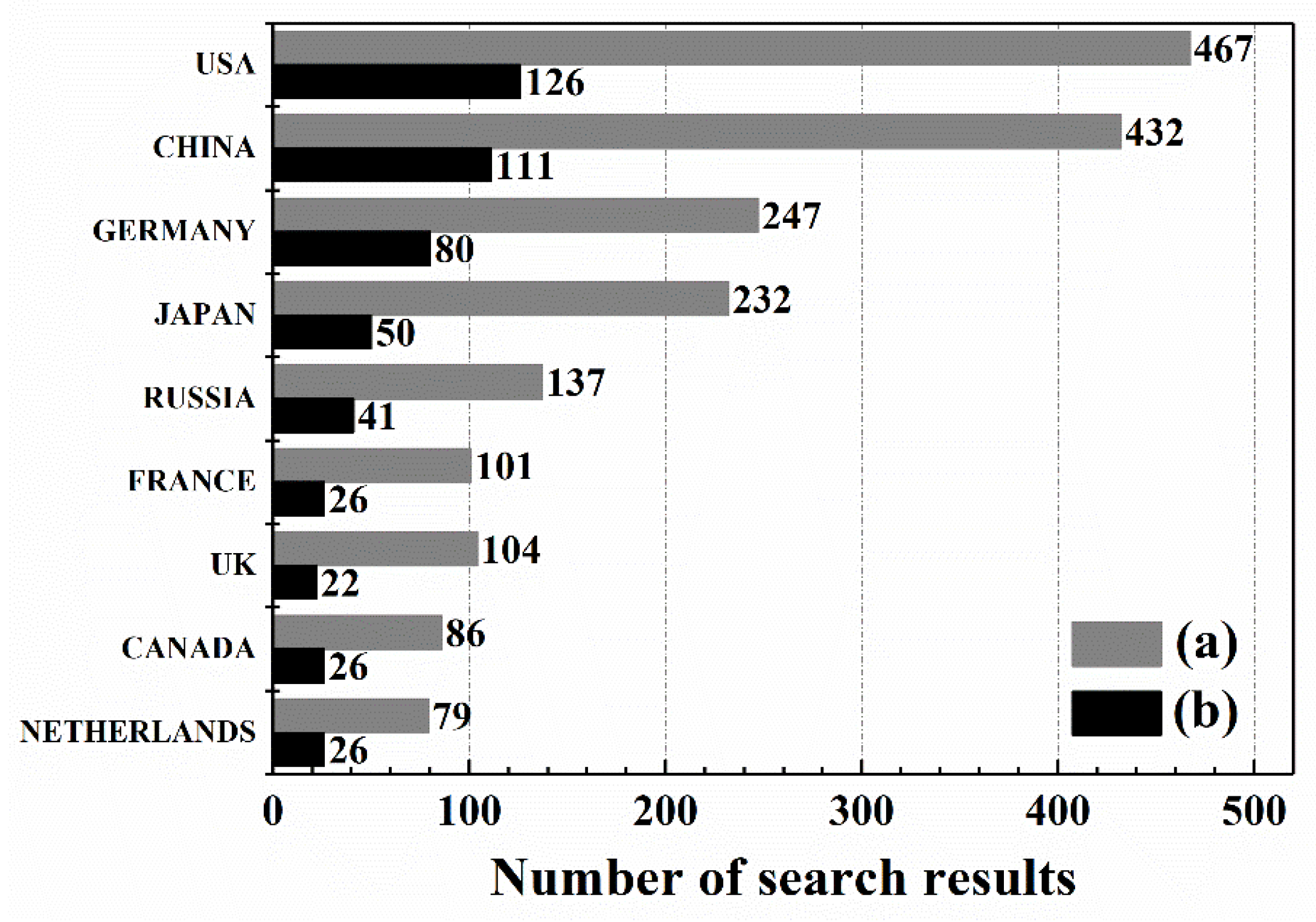
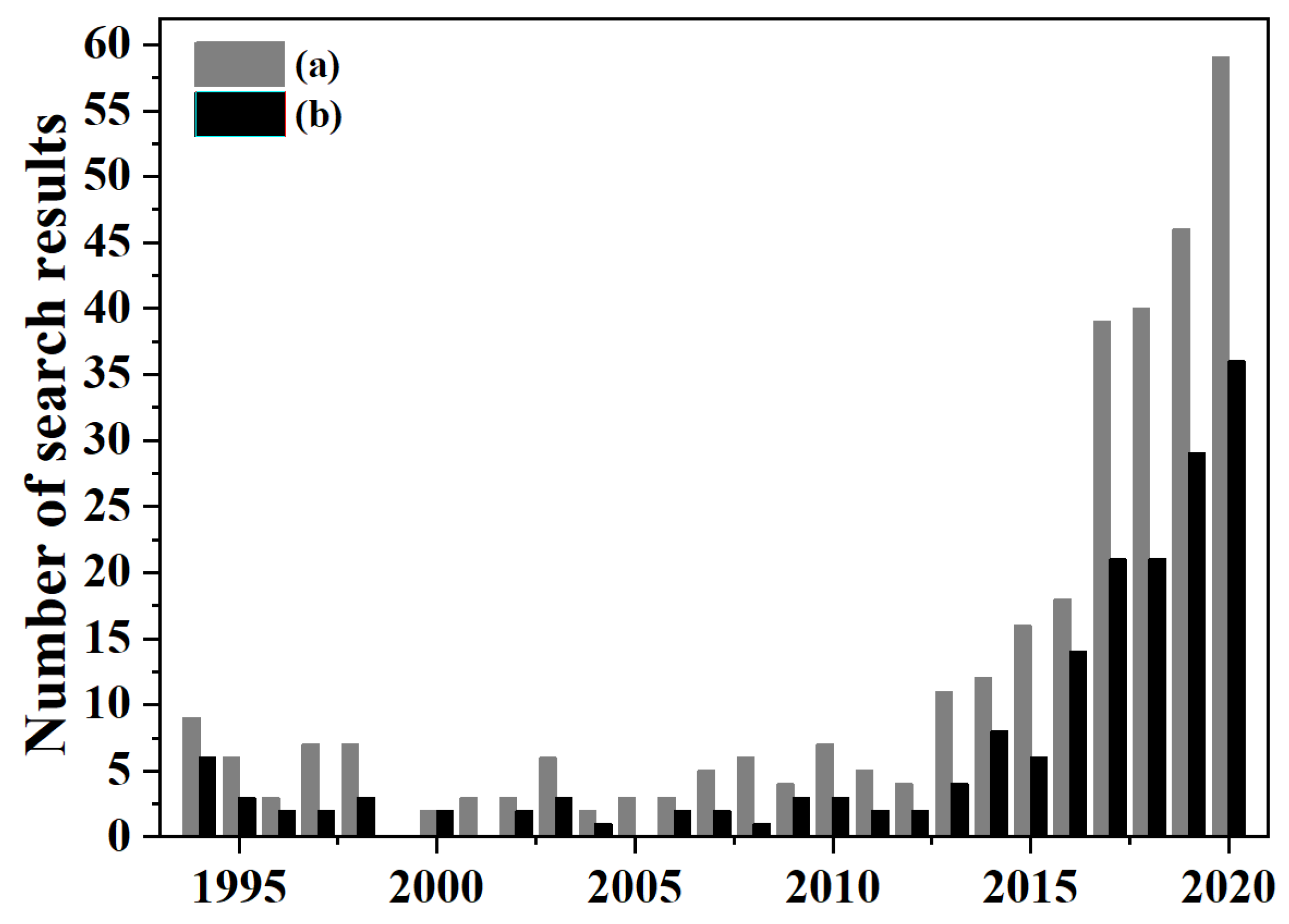
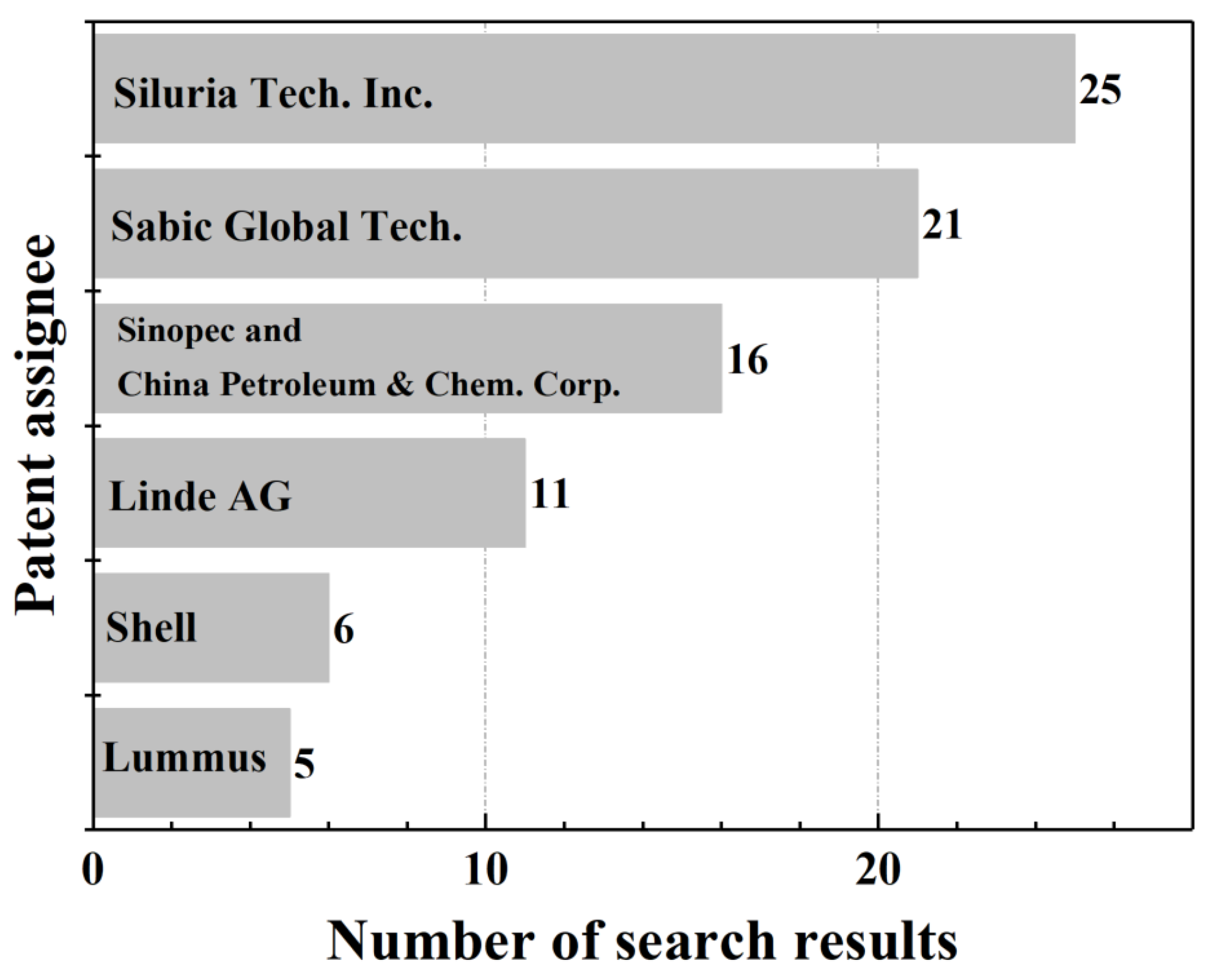
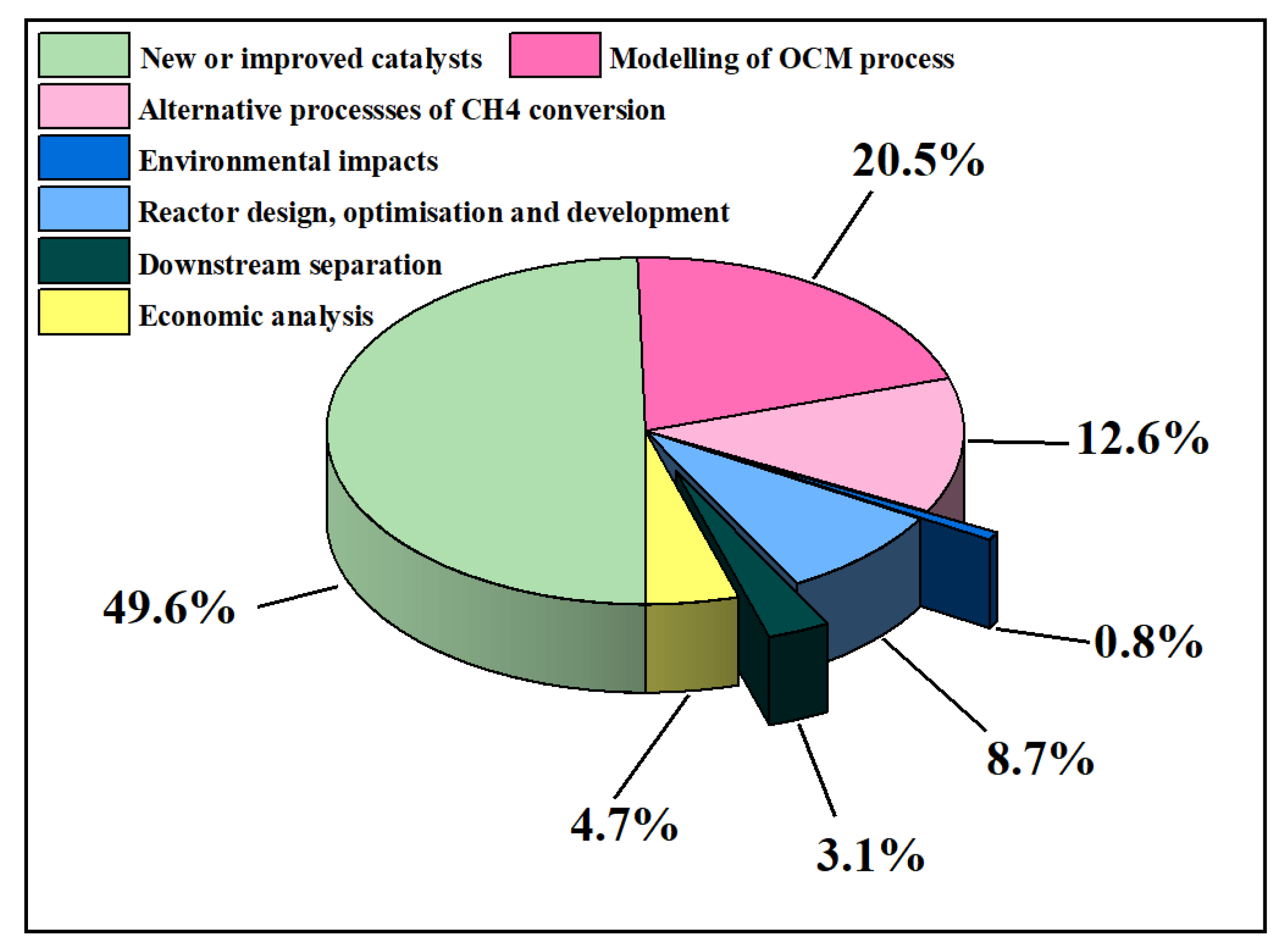
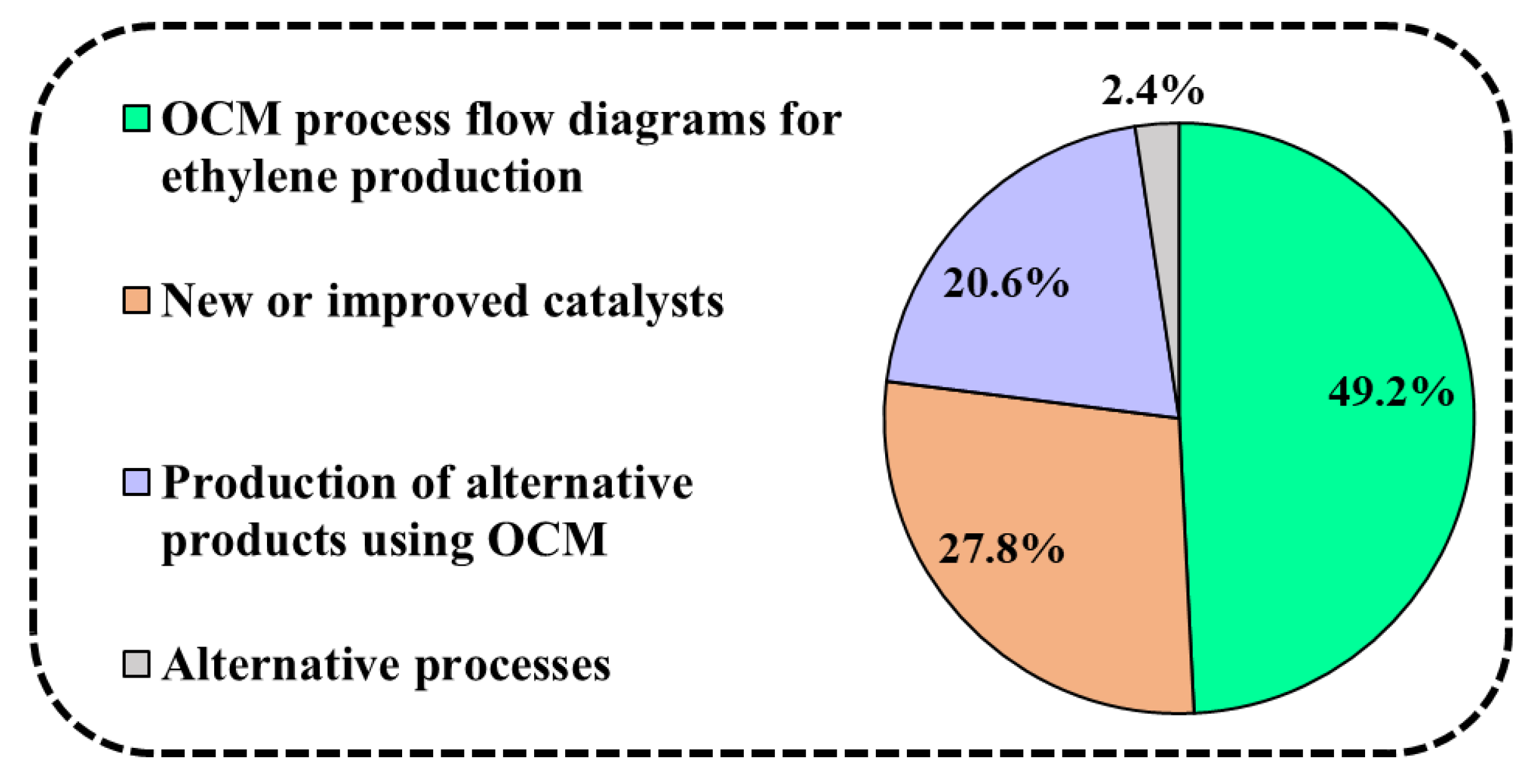
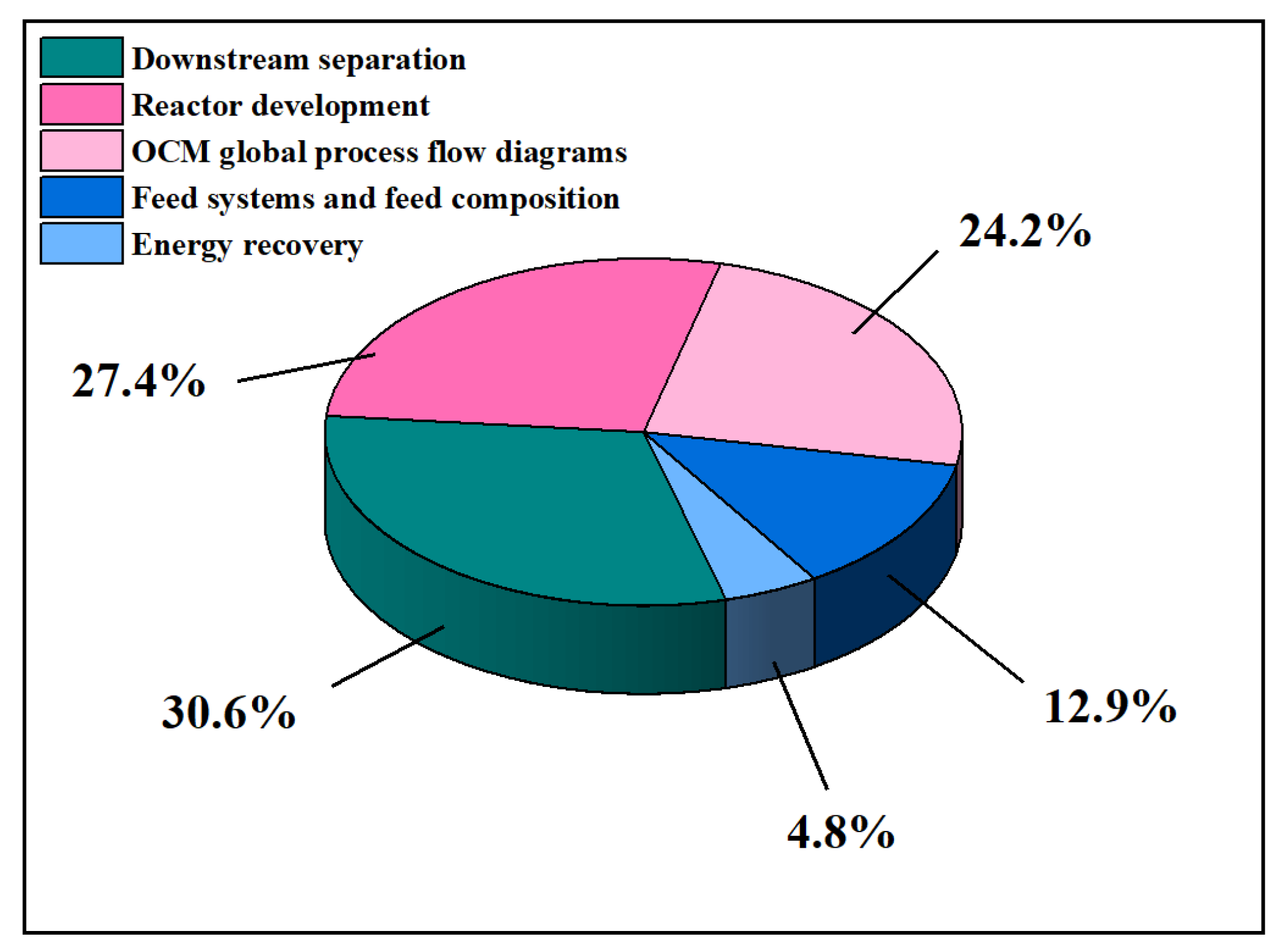
2. An Overview of OCM Scientific Publications and Patents
What Are OCM Scientific Publications and Patents about?
3. OCM Reactions and Kinetic Models
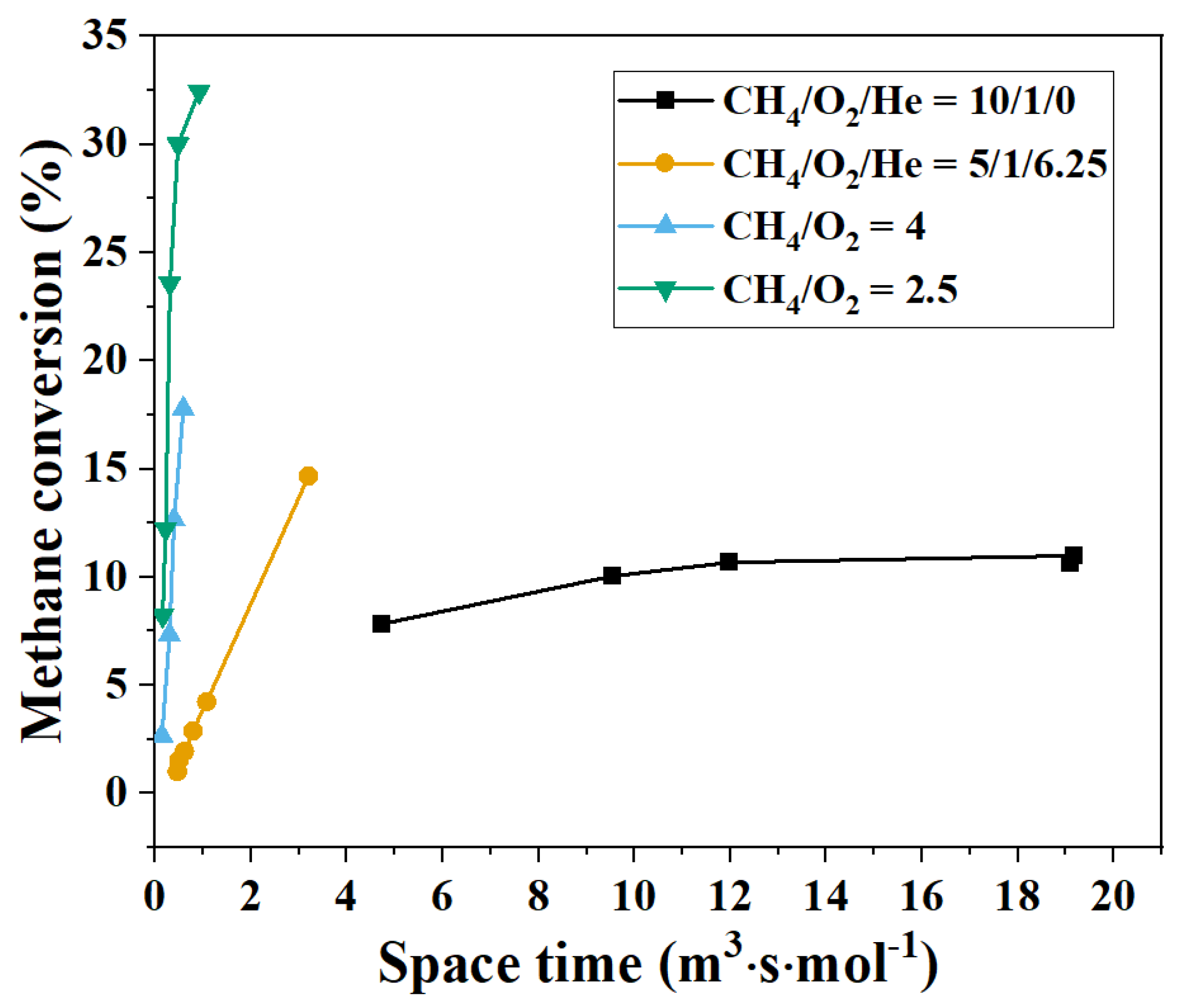
3.1. Kinetics in the Absence of Catalyst
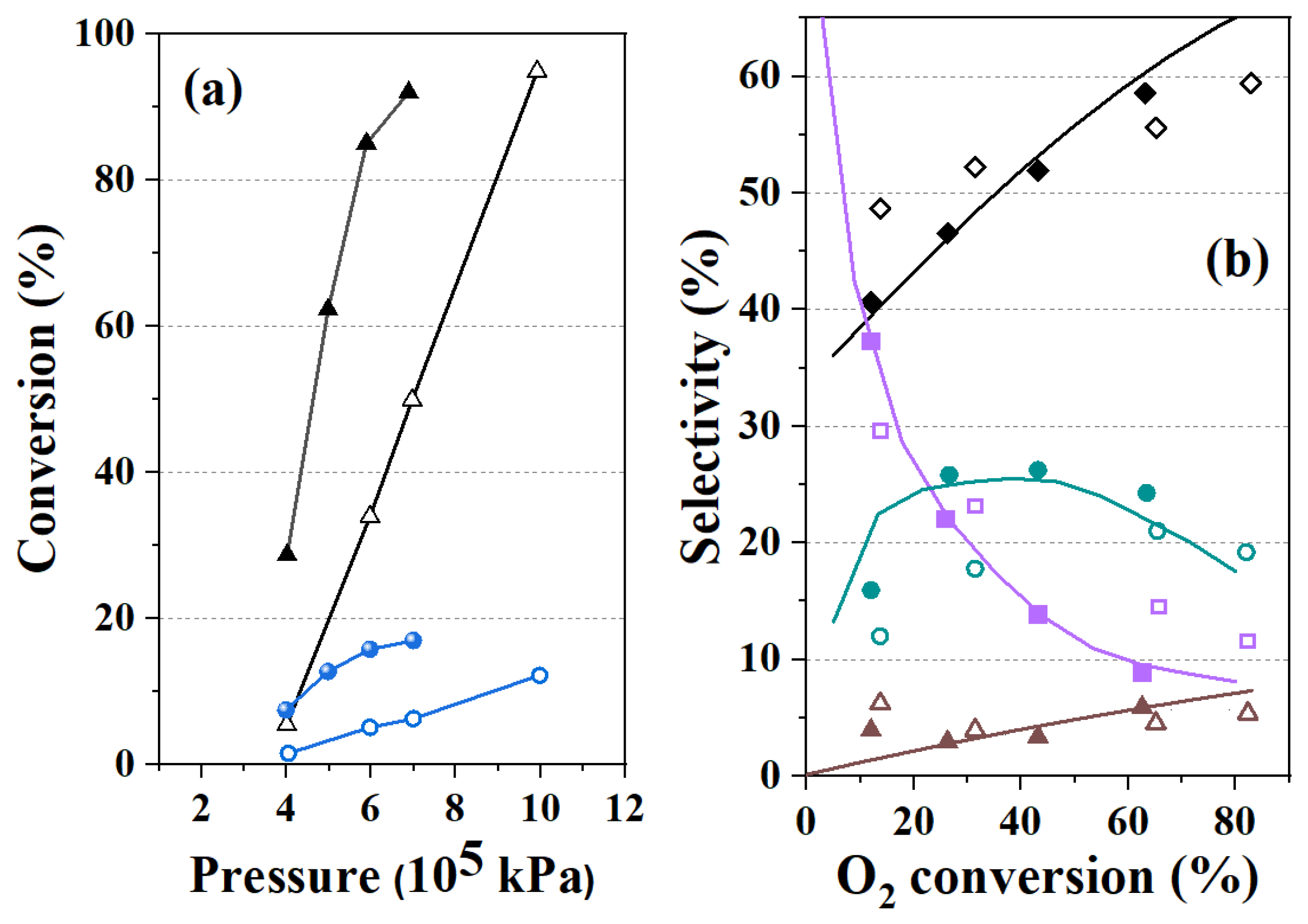
3.2. Kinetics in the Presence of Catalyst
3.3. Modelling Approaches for OCM Reactors
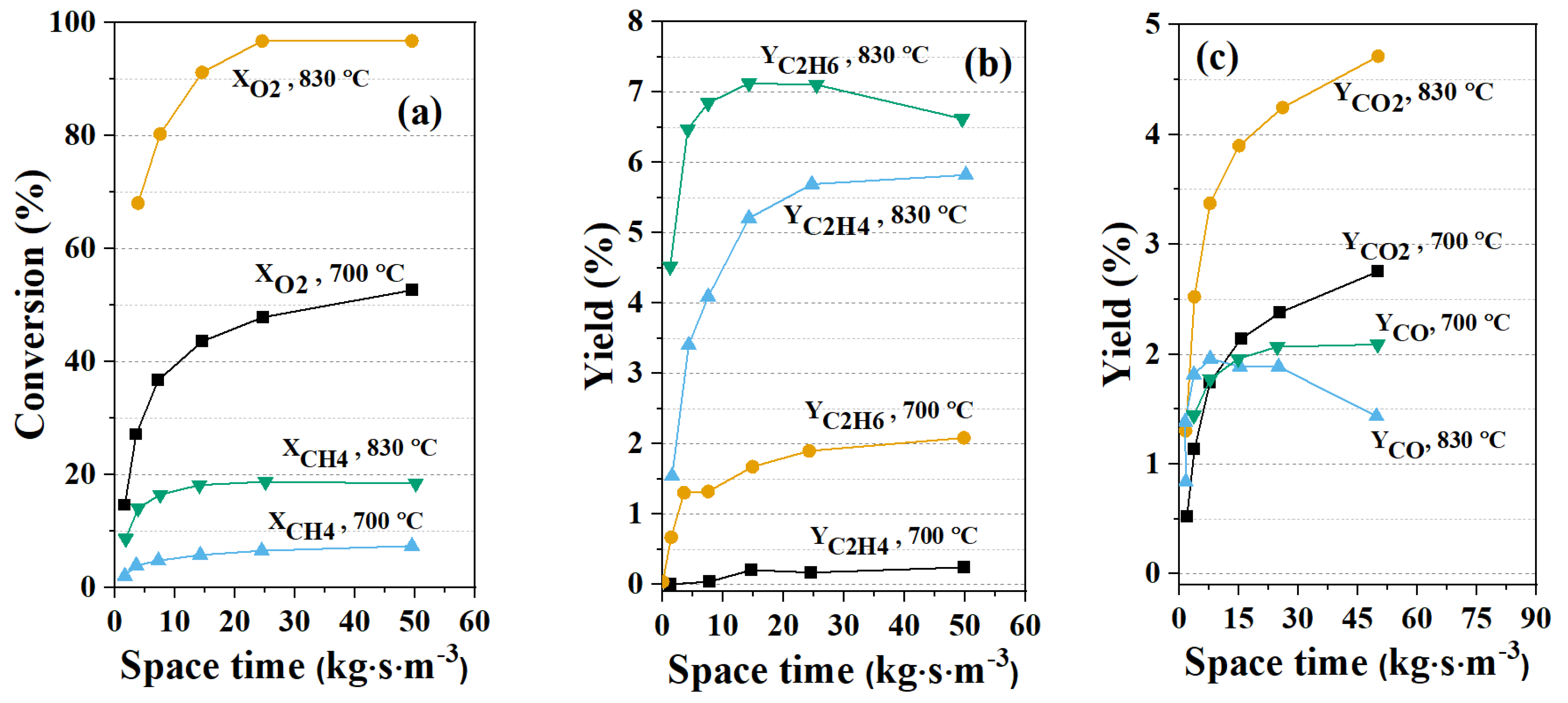
3.3.1. Modelling Approaches for Packed-Bed Reactors
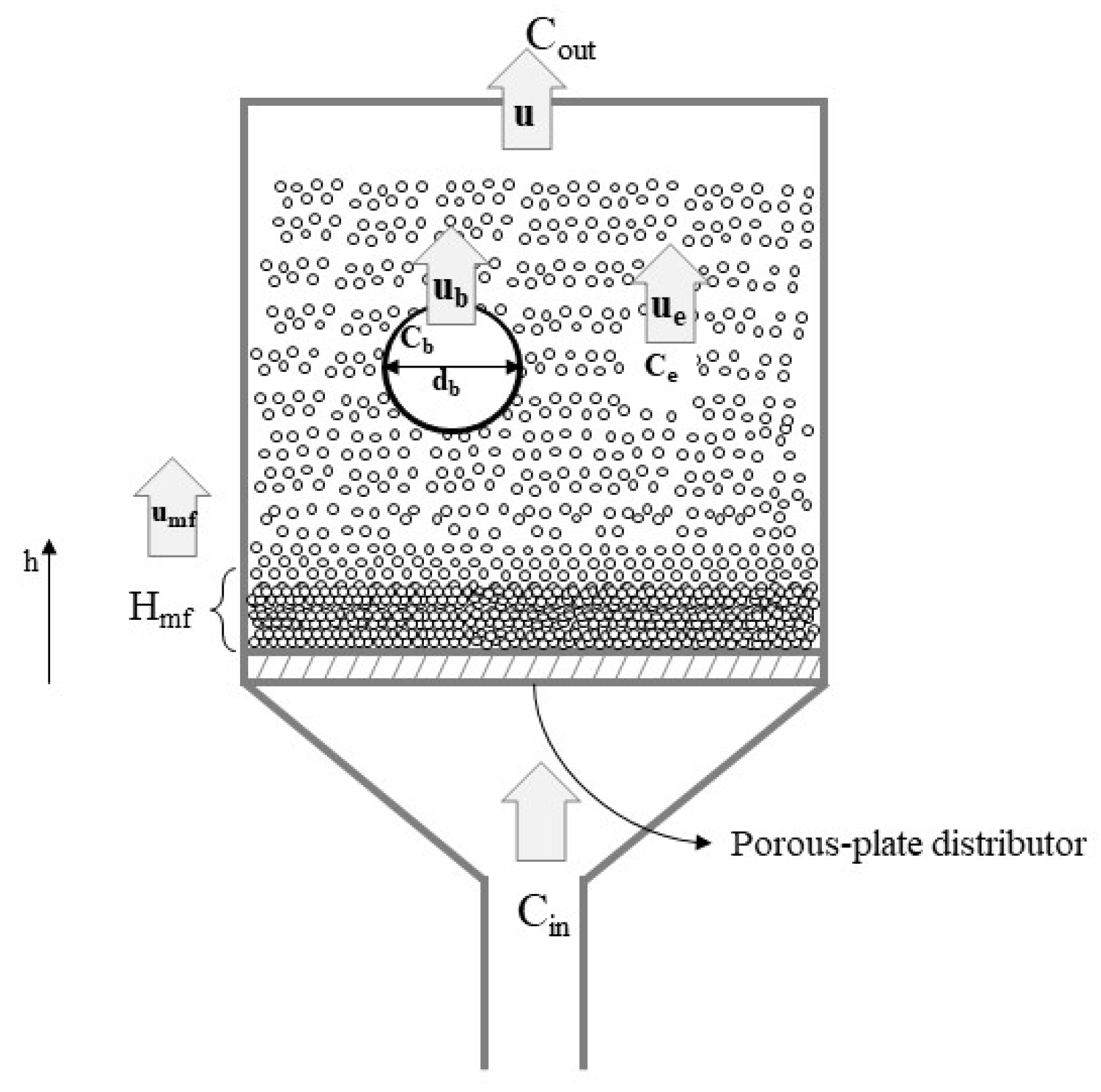
3.3.2. Modelling Approaches for Fluidized Bed Reactors
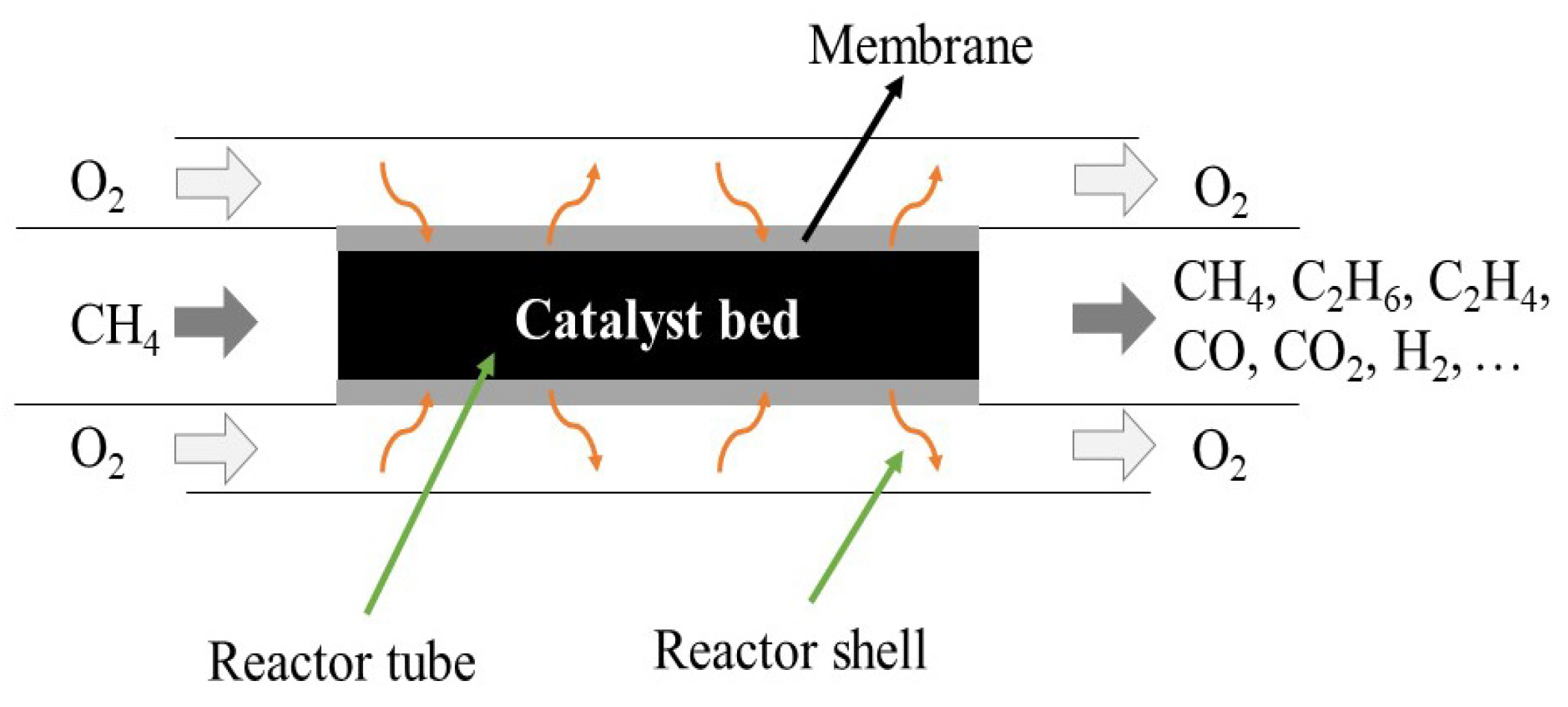
3.3.3. Modelling Approaches for Membrane Reactors
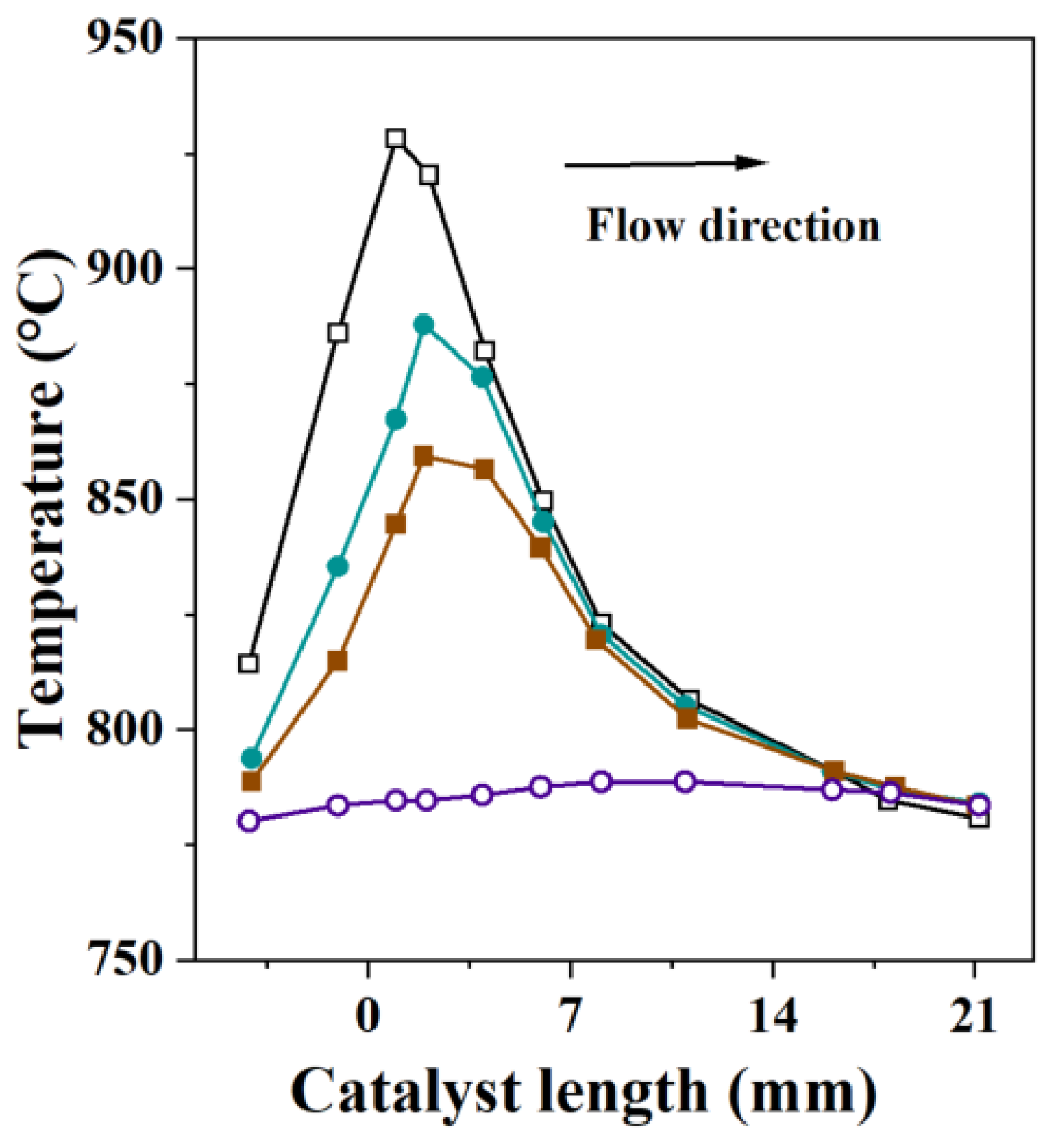
3.4. Thermal Effects
4. Thermodynamic Considerations
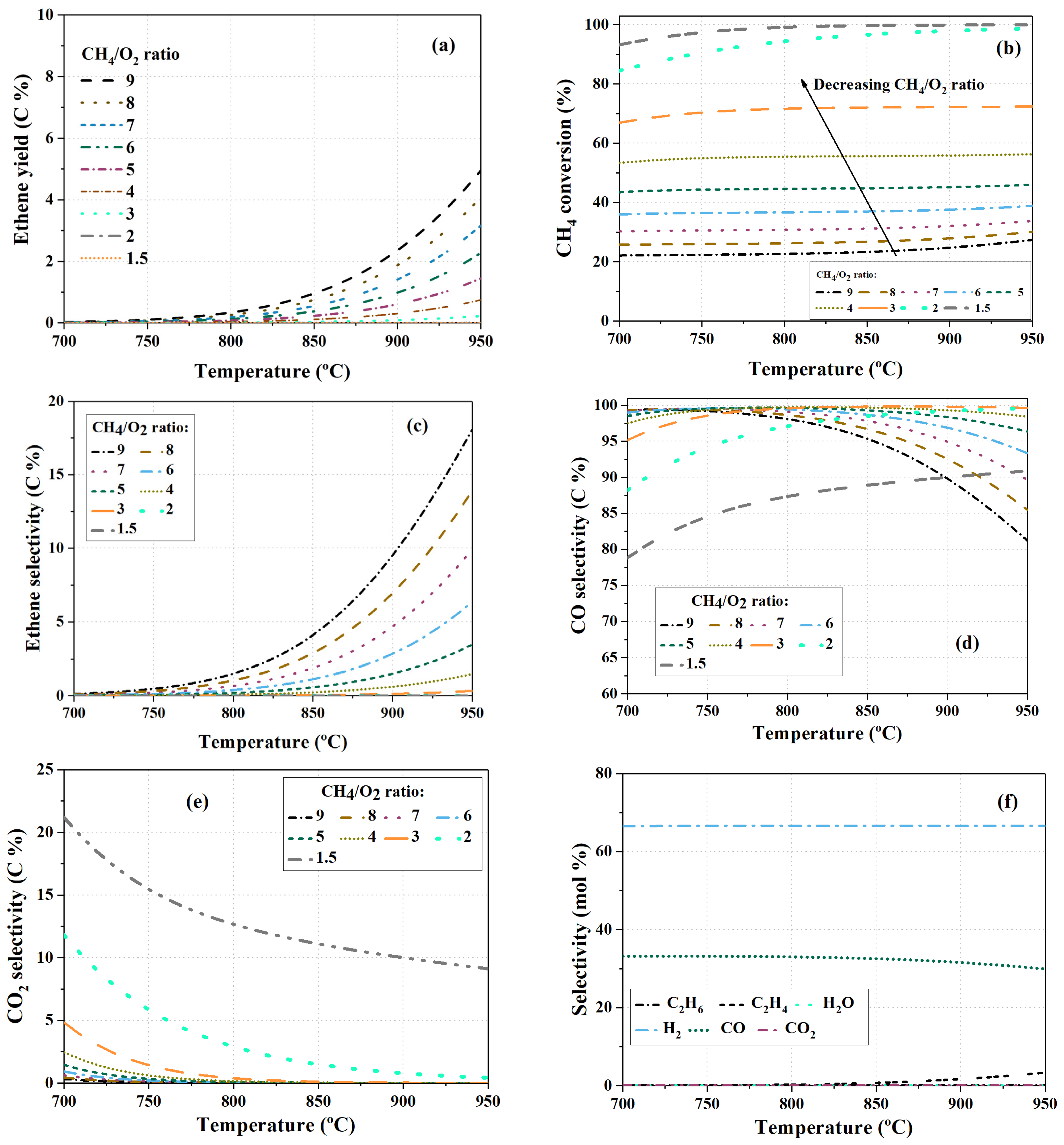
4.1. The Effect of Temperature and CH4/O2 Feed Ratio on Equilibrium Compositions
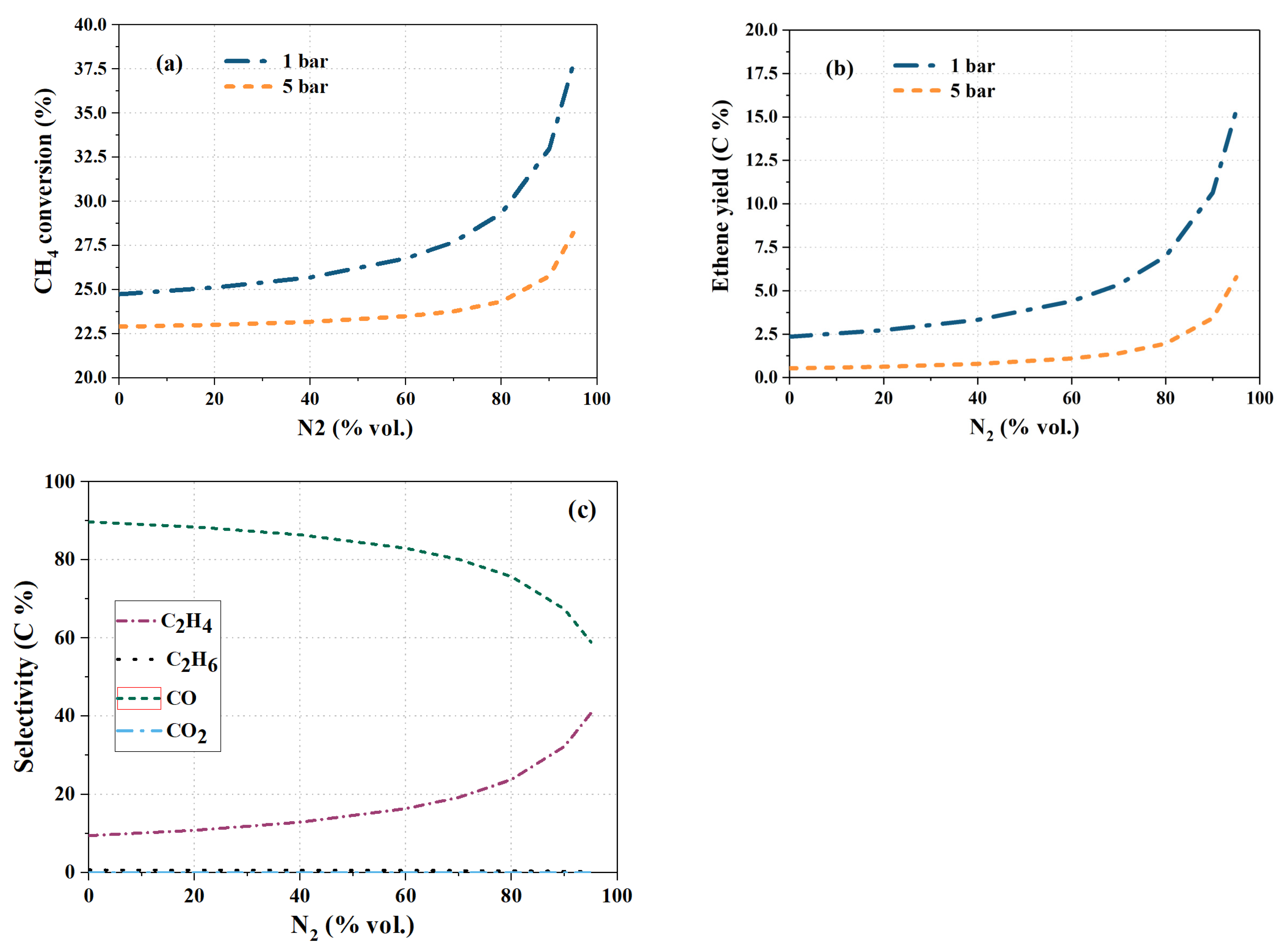
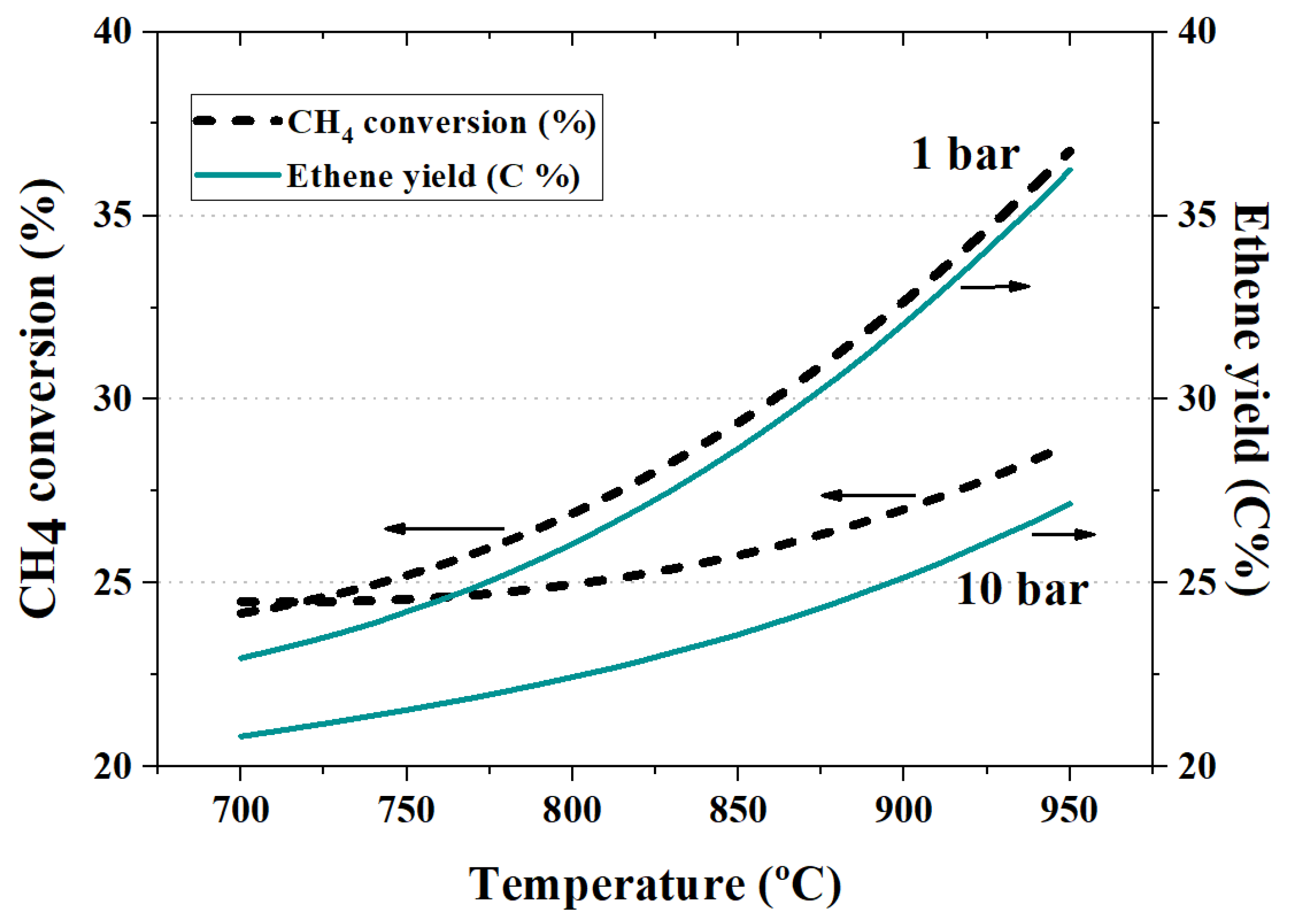
4.2. The Effect of Inert Dilution and Pressure
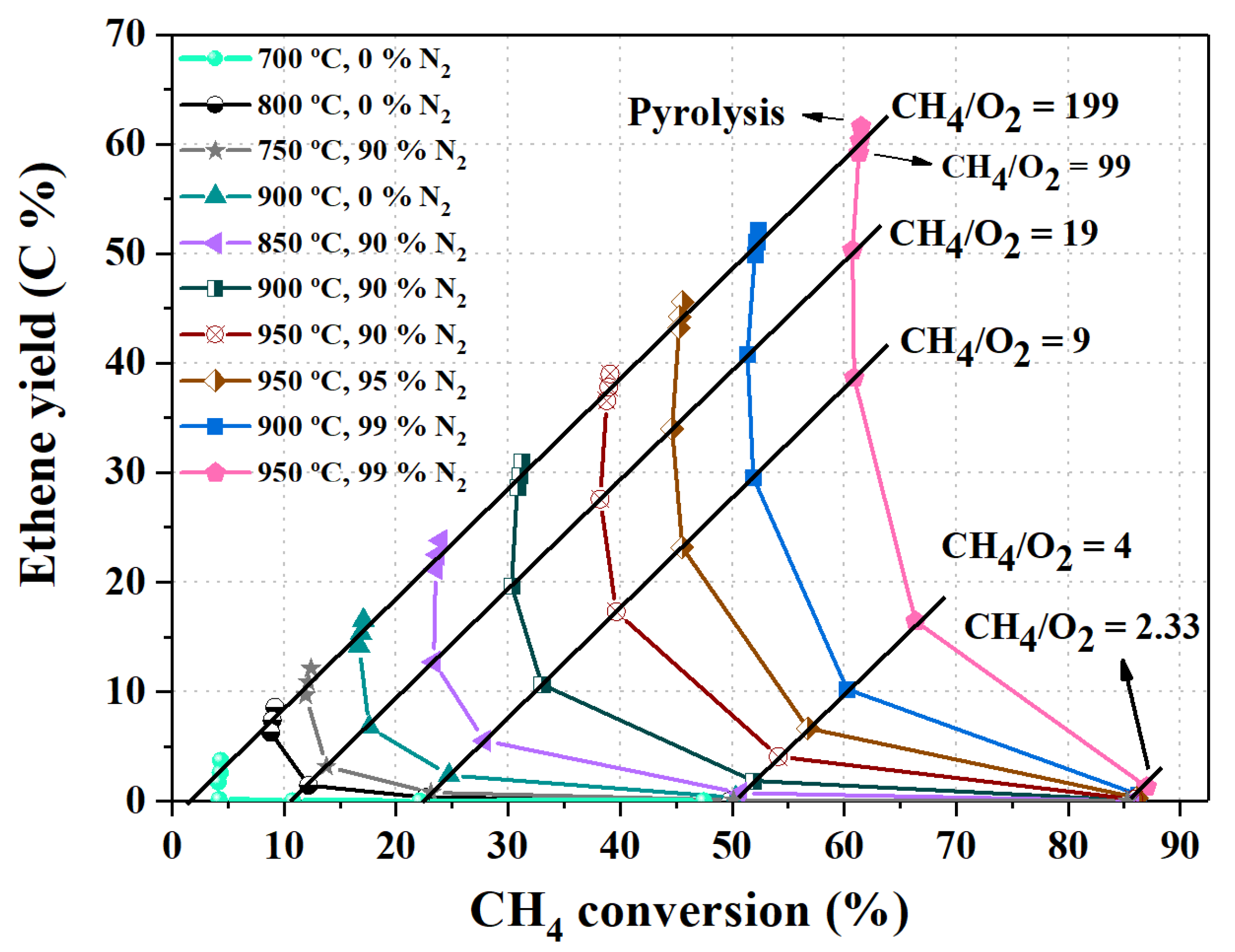
4.3. The Combined Effect of Temperature, CH4/O2 Feed Ratio and Inert Composition
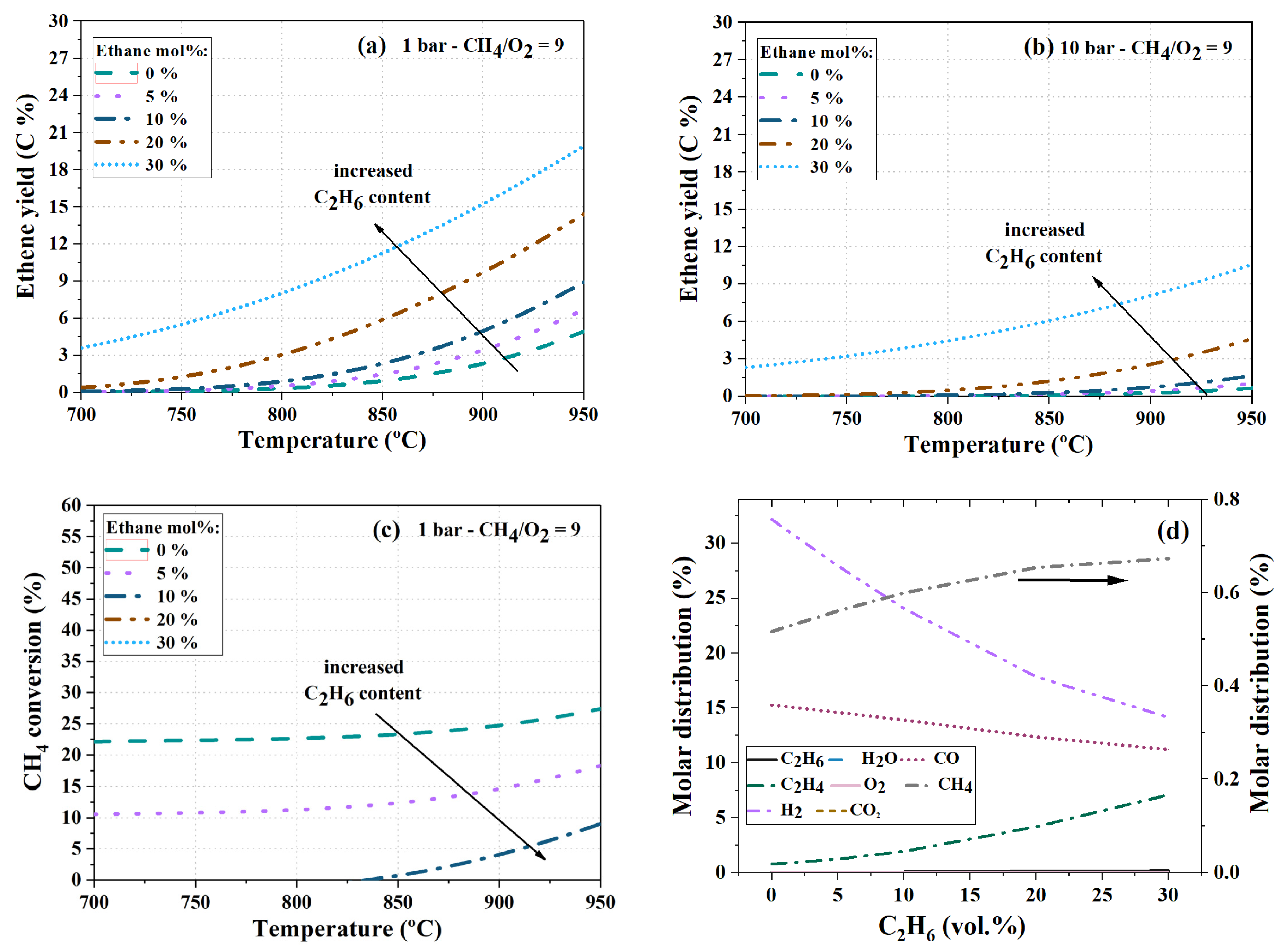
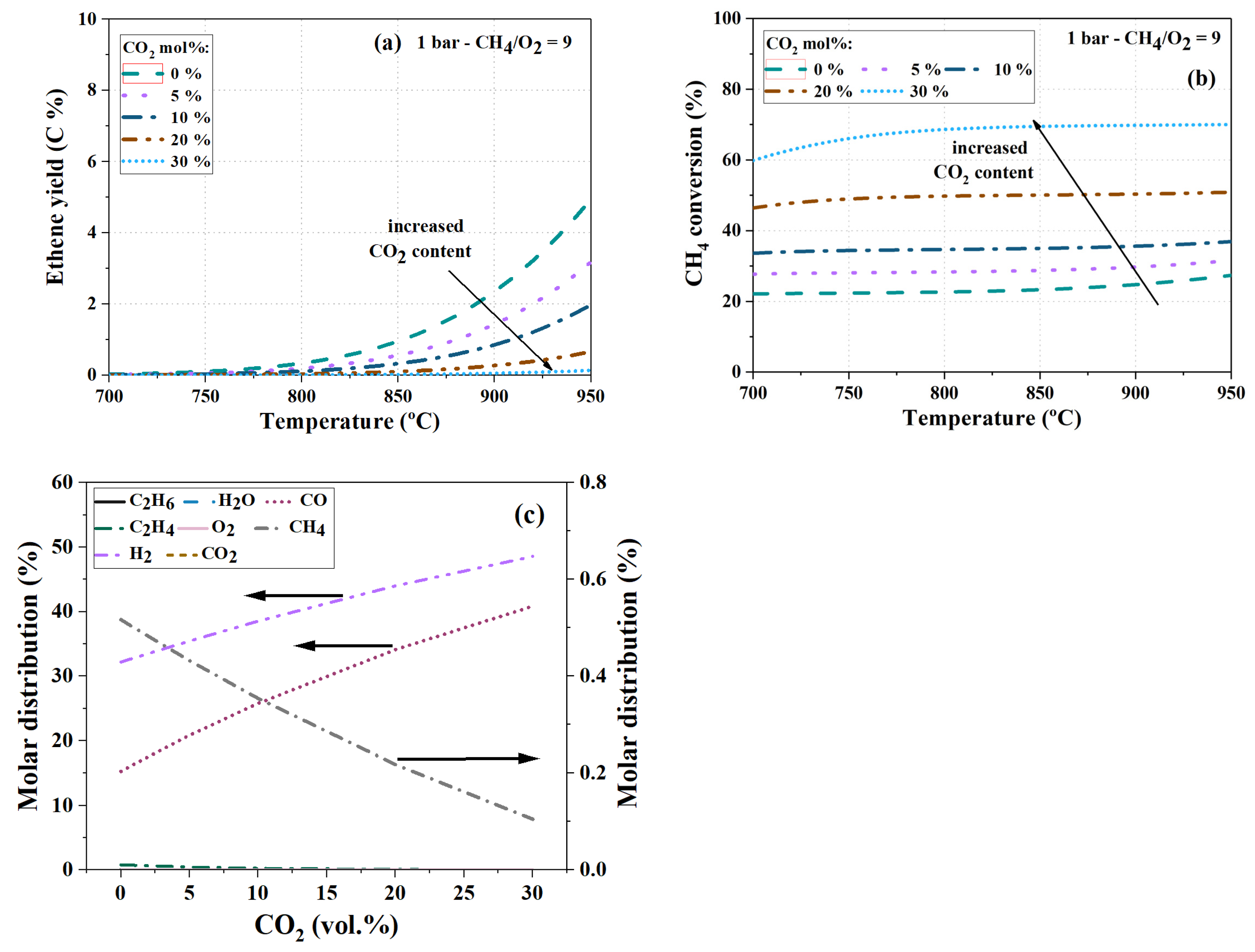
4.4. Effect of Co-Feeding Ethane and CO2
4.4.1. Co-Feeding of Ethane
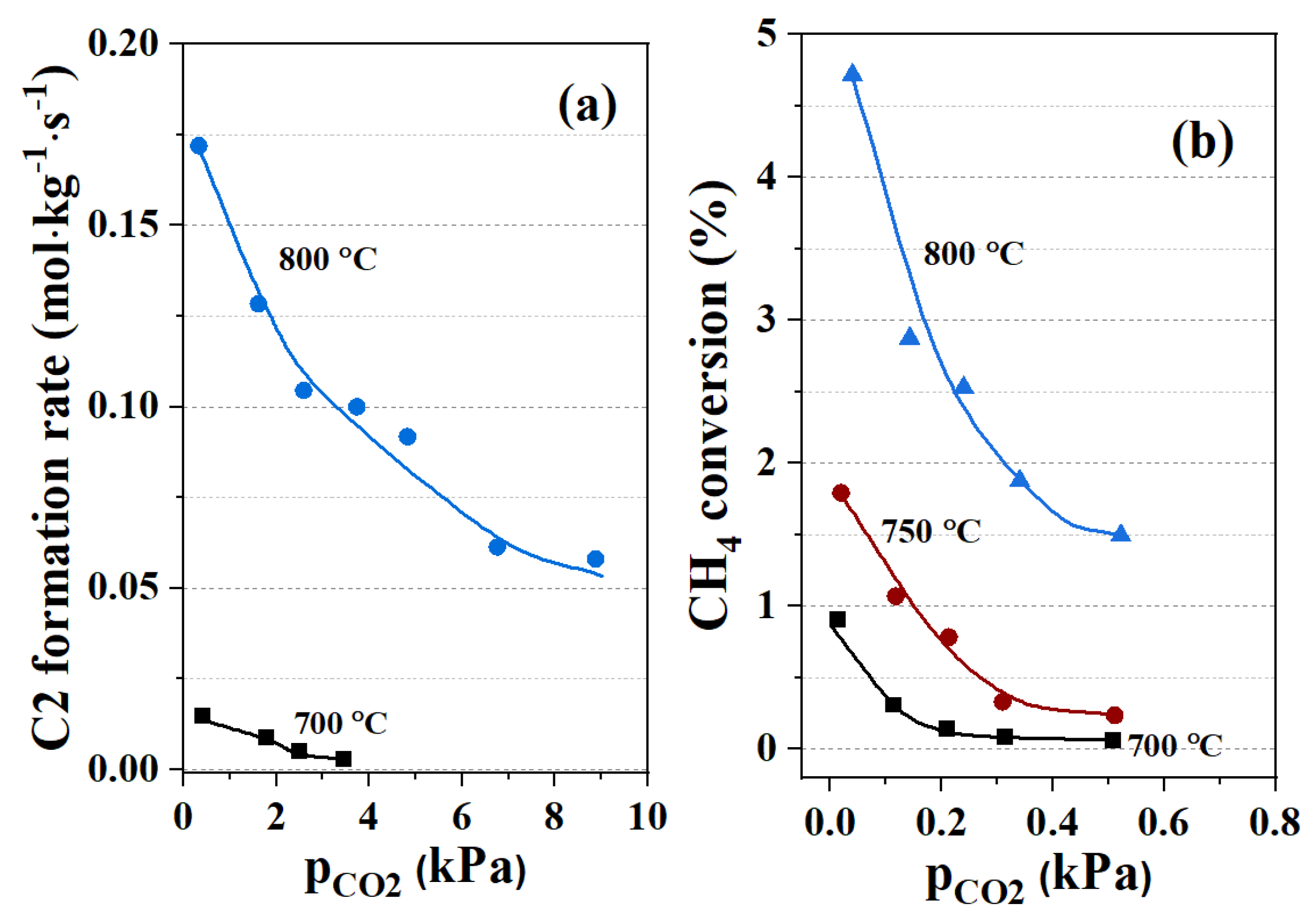
4.4.2. Co-Feeding of CO2
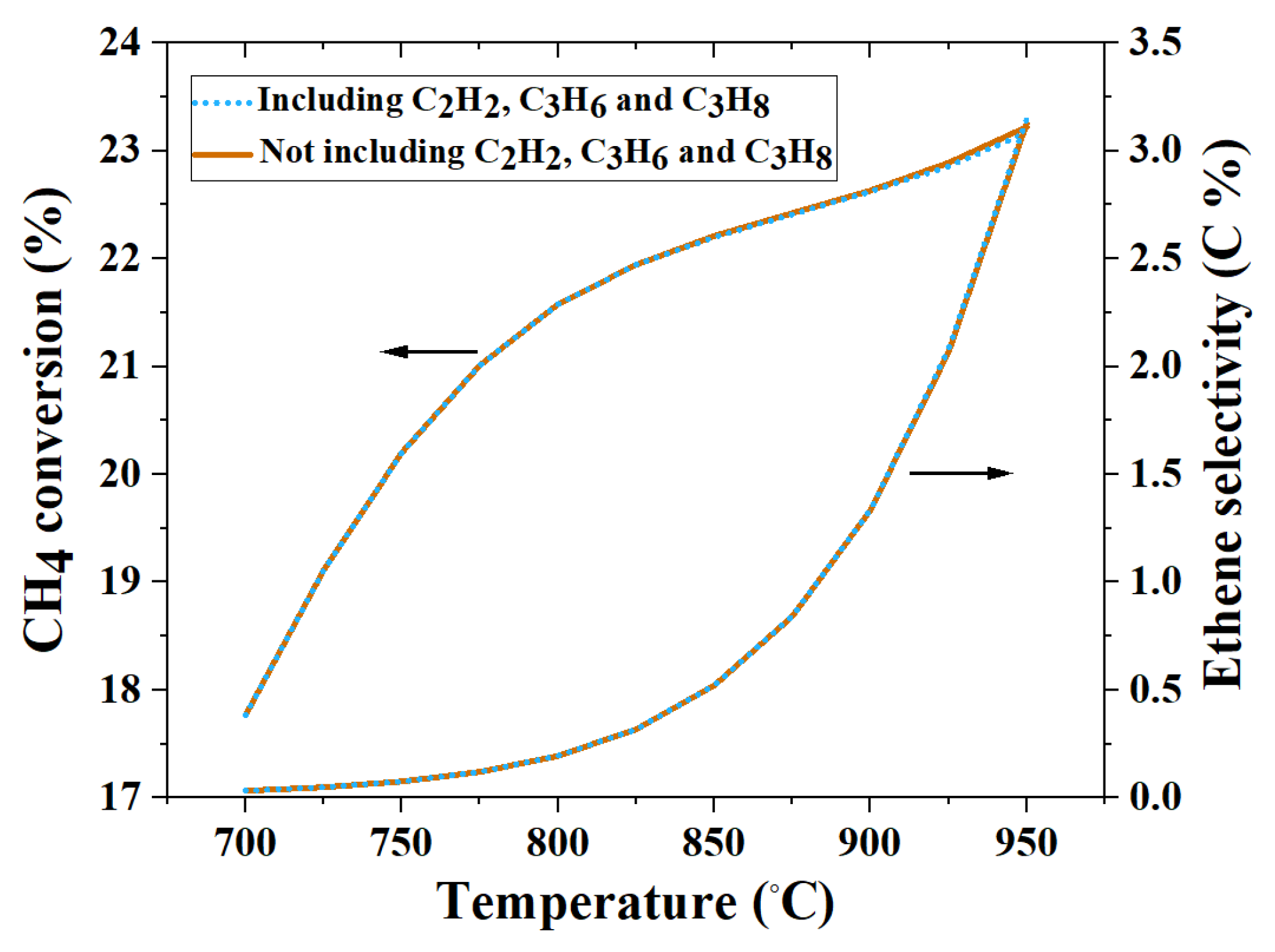
4.5. The Importance of Considering Additional Products on Equilibrium Compositions
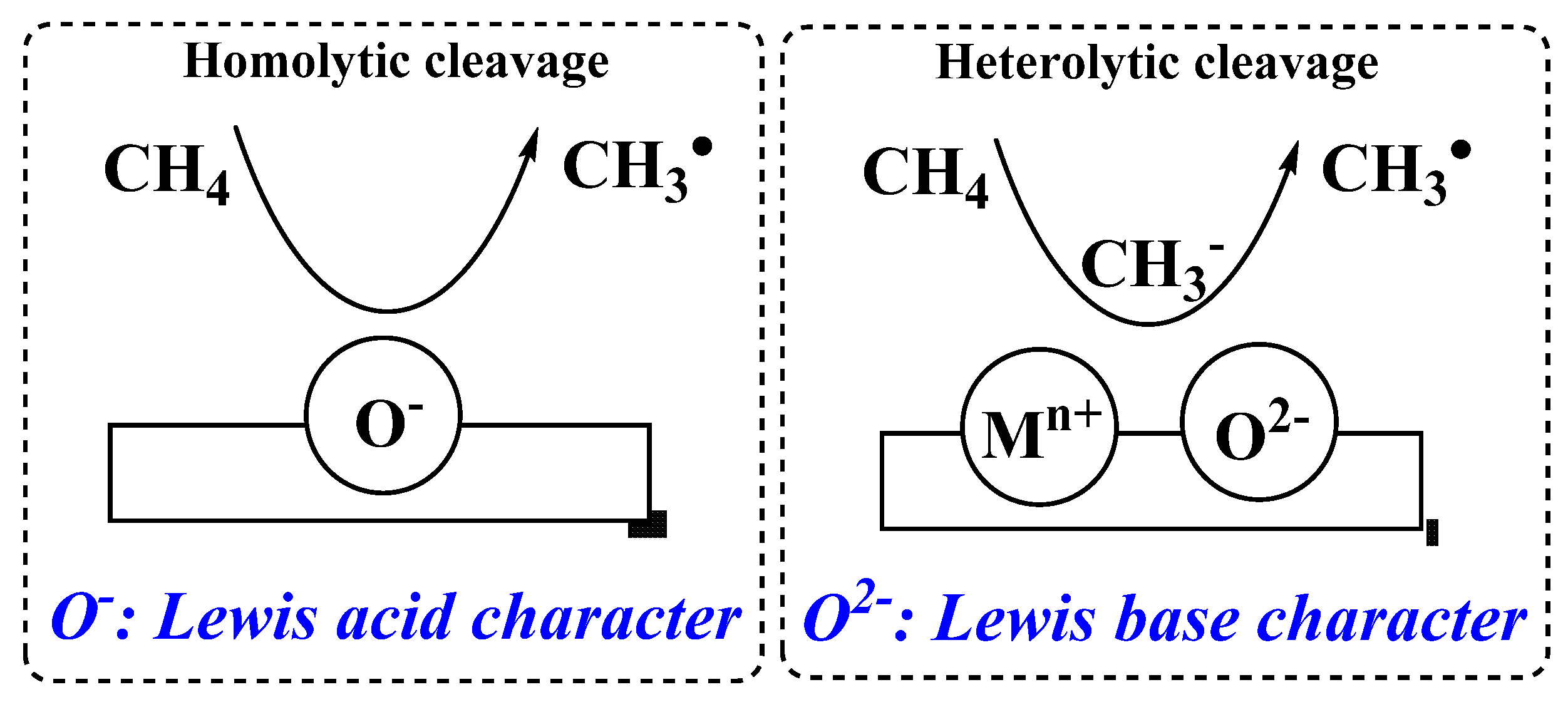
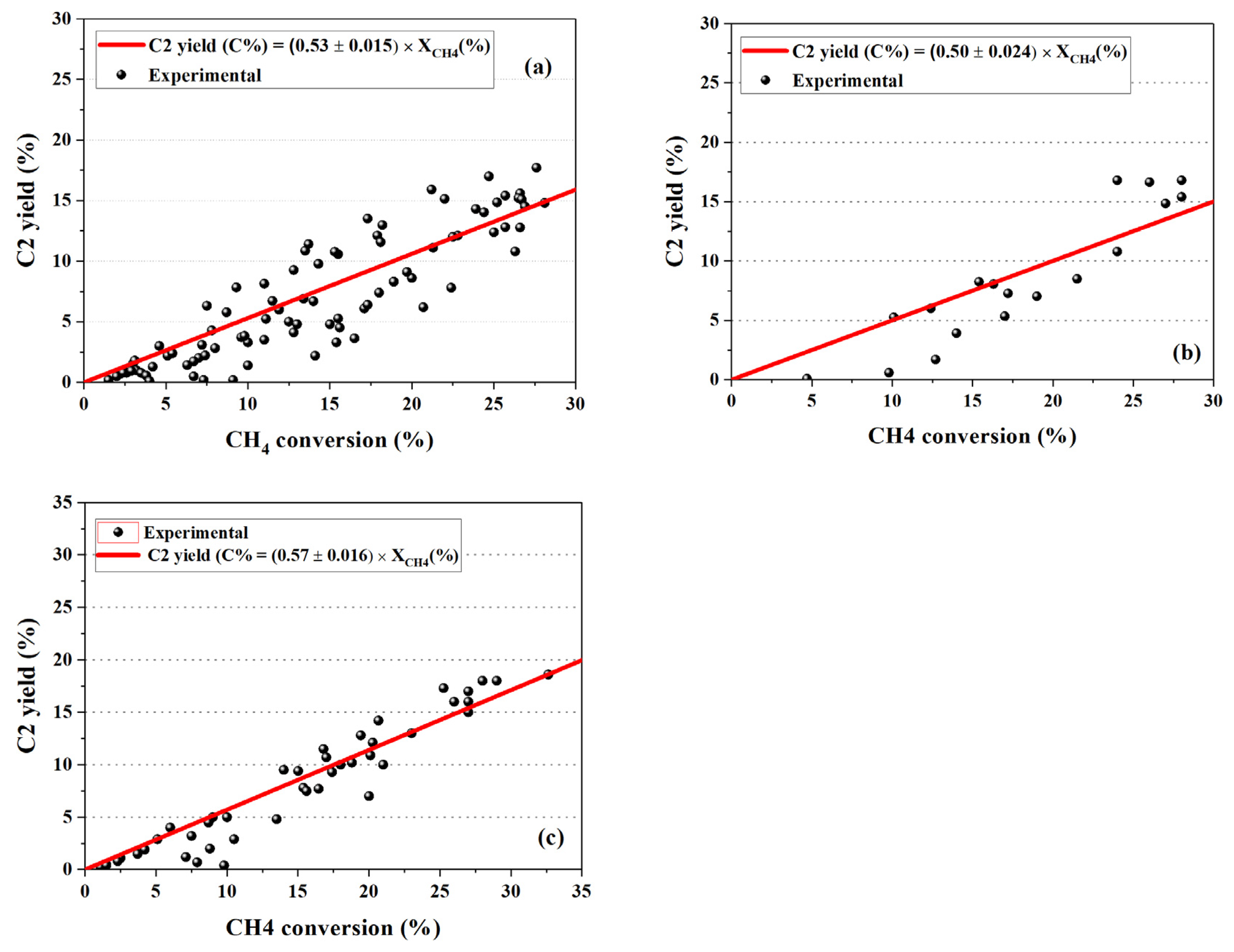
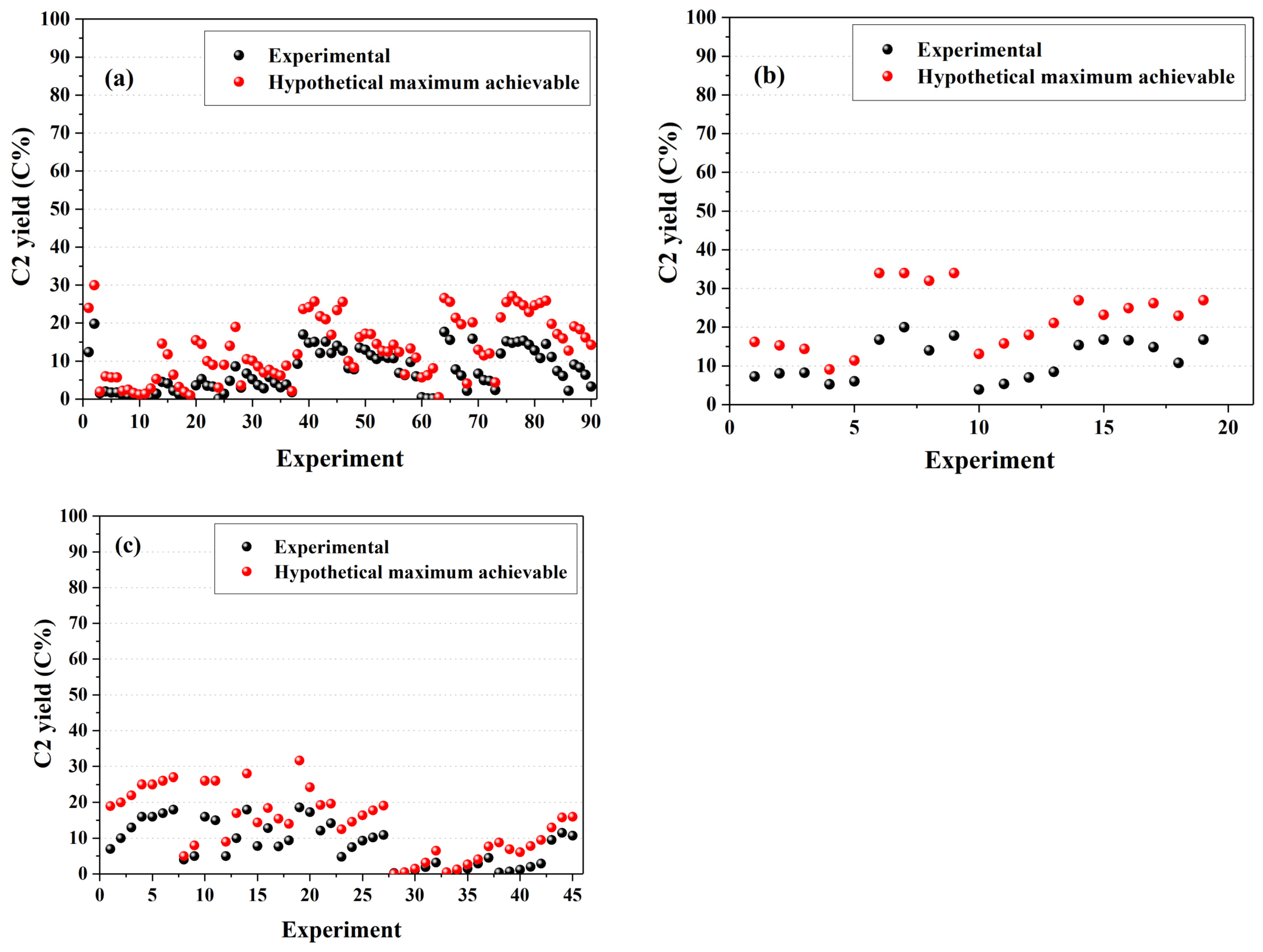
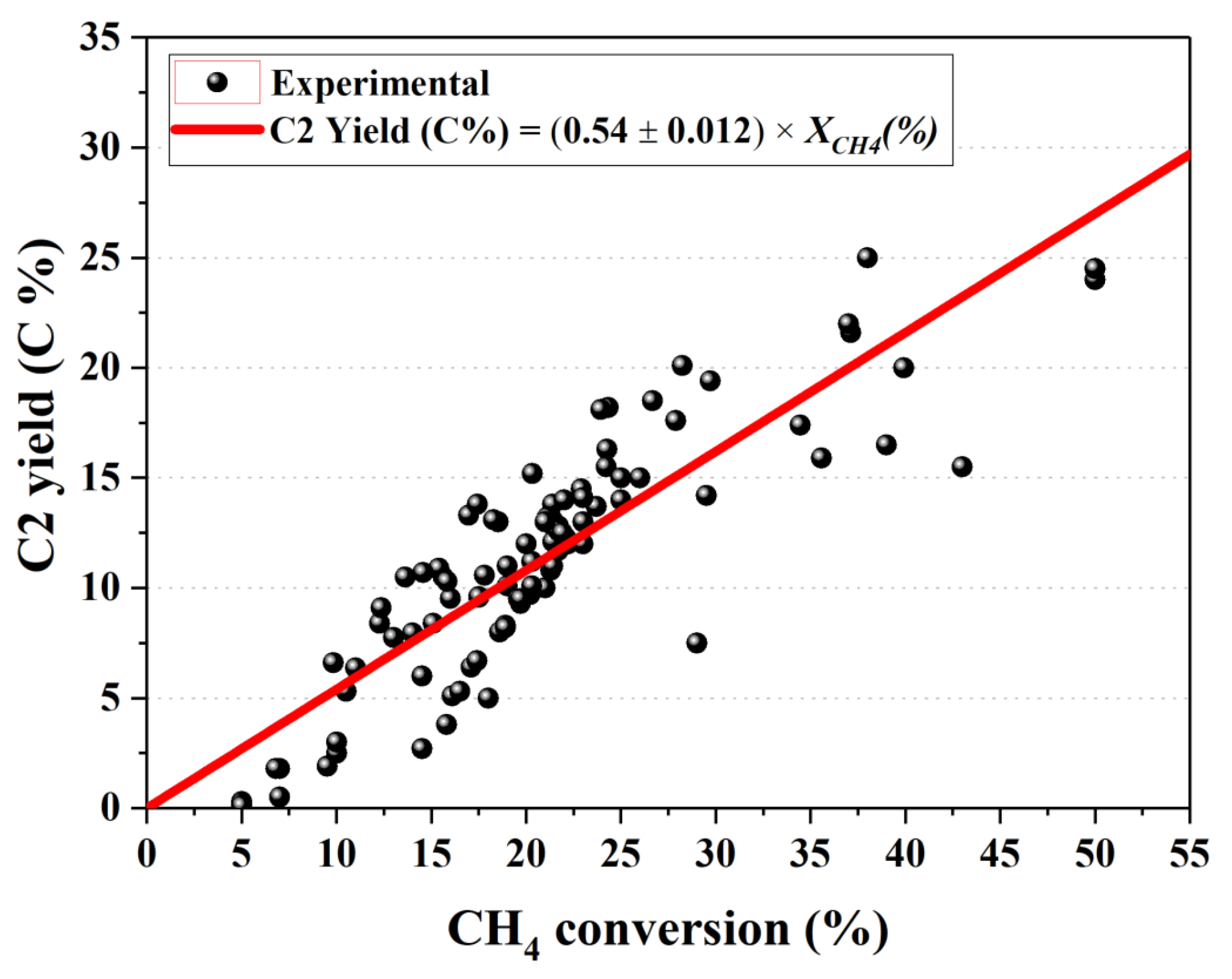
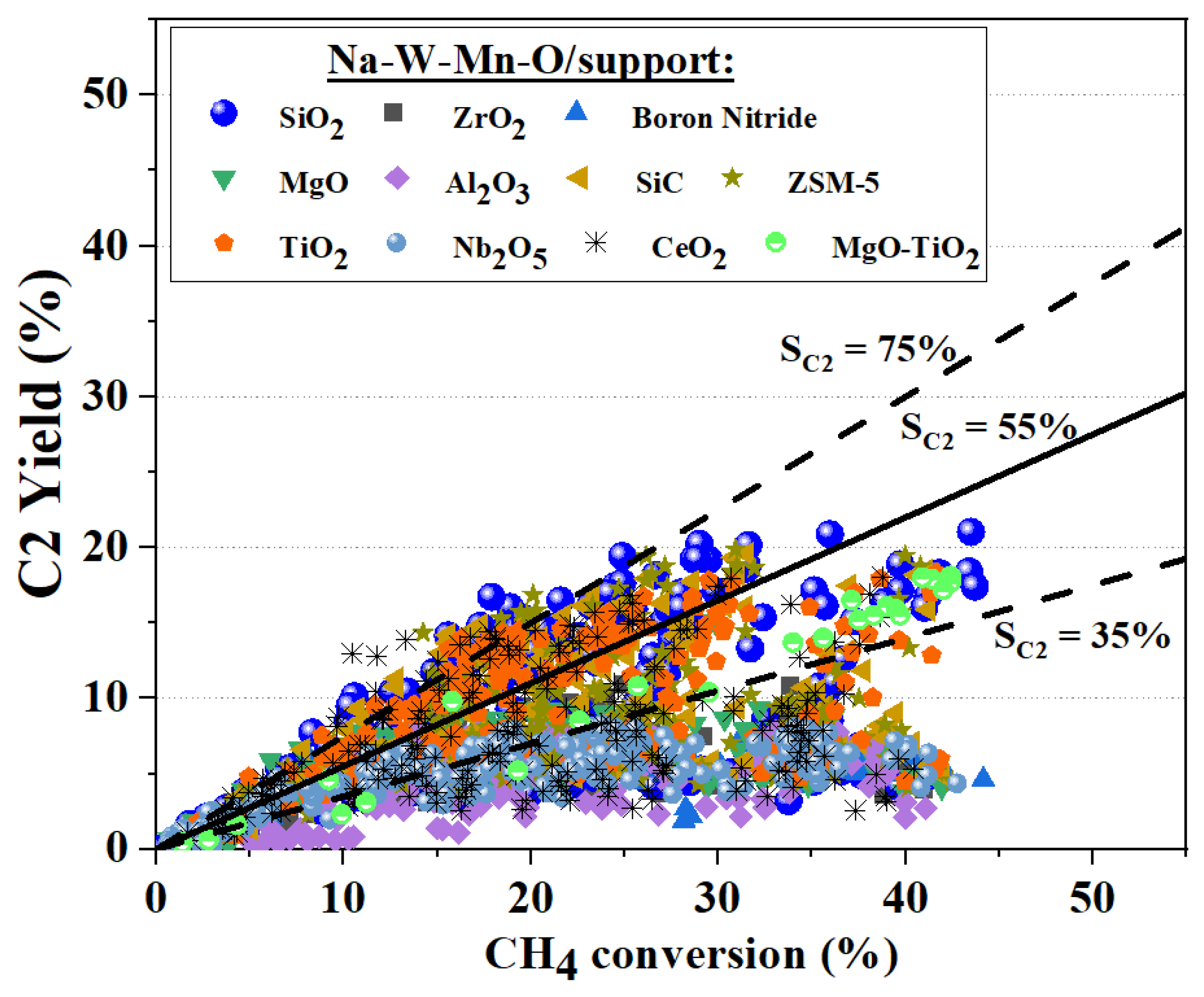
5. Active OCM Catalysts
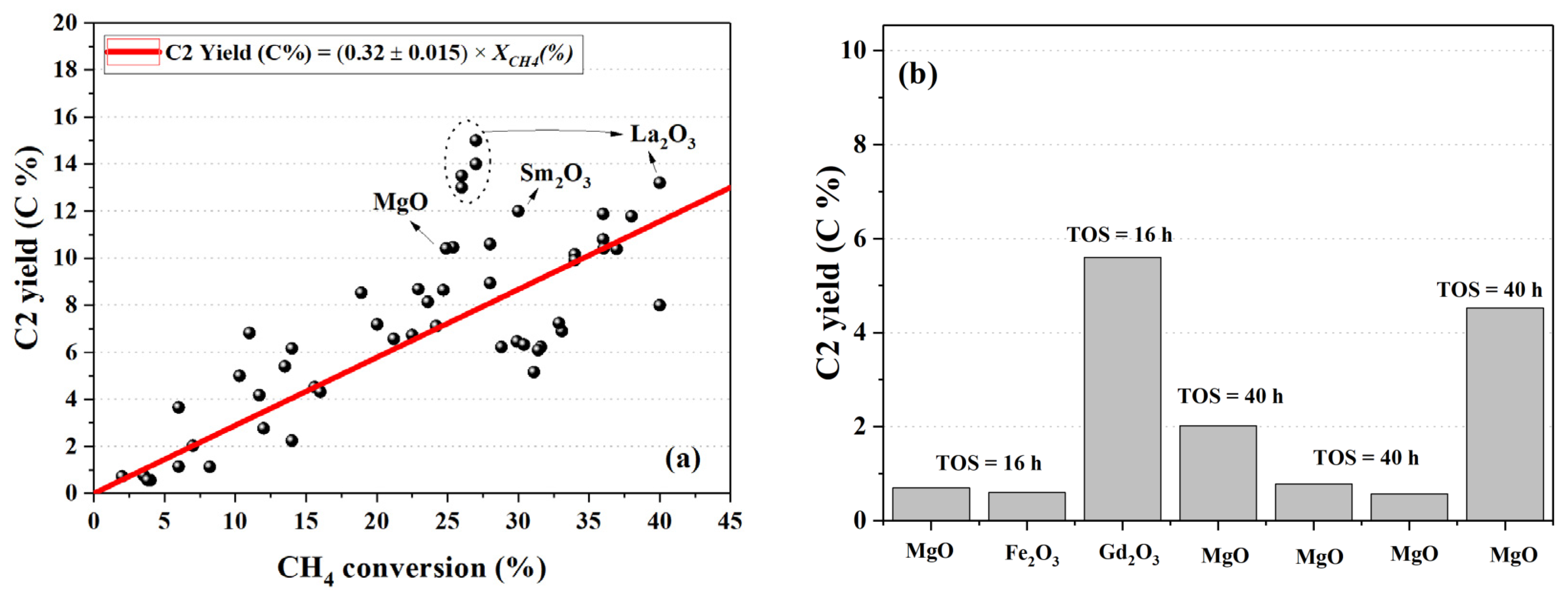
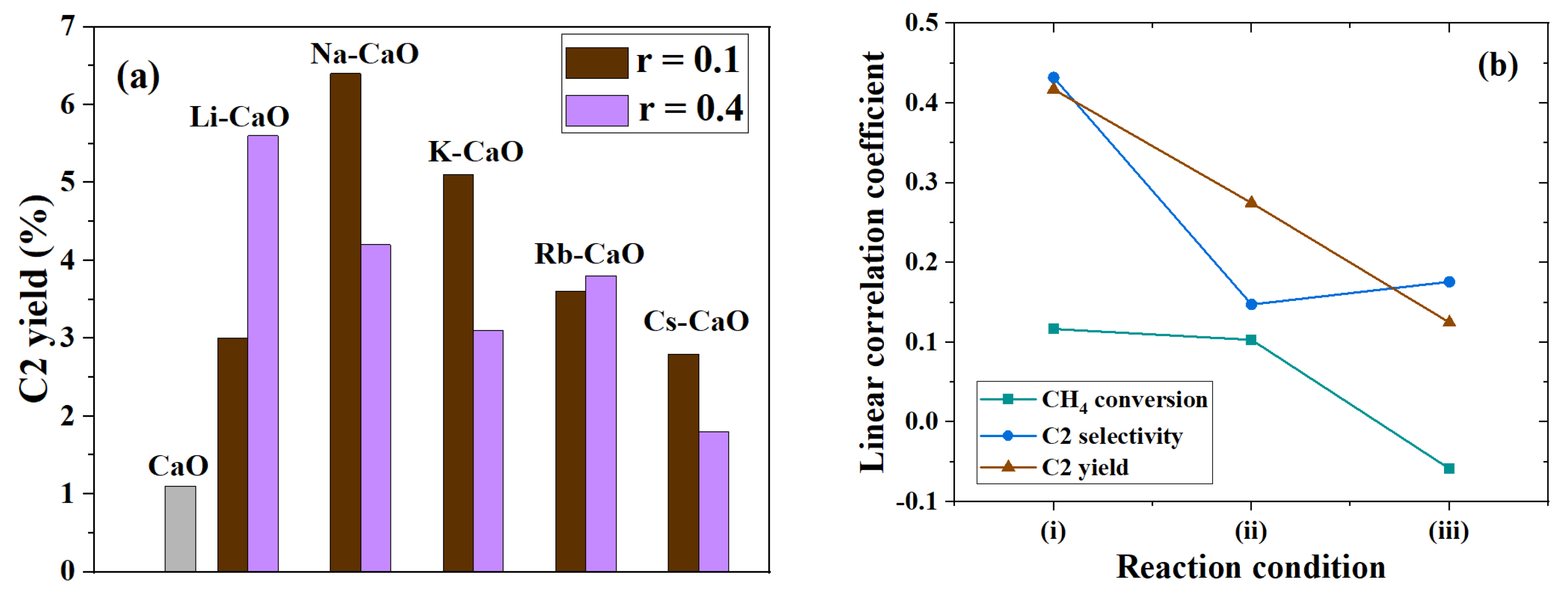
5.1. Monometallic Oxides
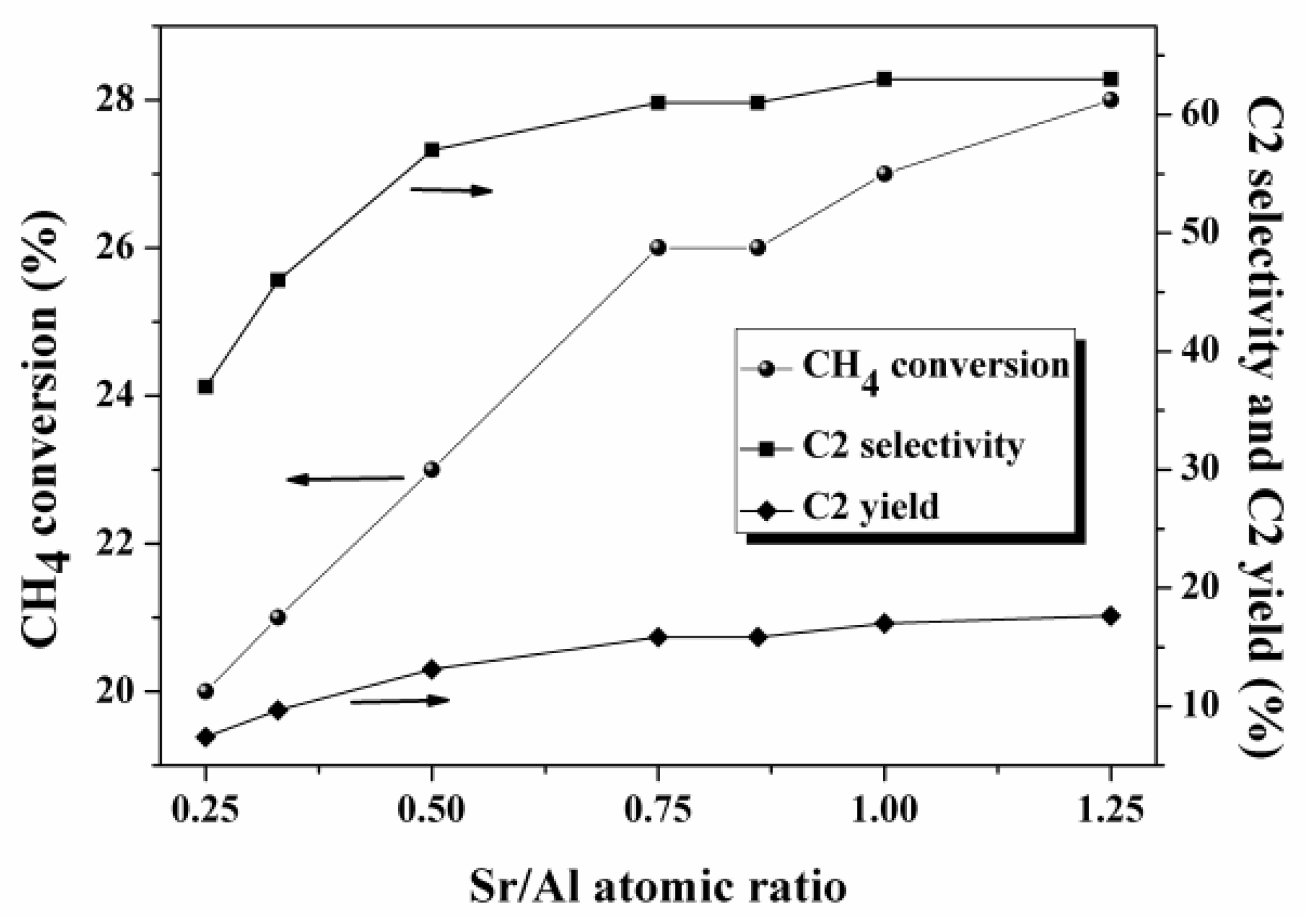
5.2. Bimetallic Oxides
5.3. Trimetallic Oxides
5.4. Multimetallic Oxides
6. Environmental Concerns: Can OCM Be Sustainable?
7. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Da Ros, S.; Jones, M.D.; Mattia, D.; Pinto, J.C.; Schwaab, M.; Noronha, F.B.; Kondrat, S.A.; Clarke, T.C.; Taylor, S.H. Ethanol to 1,3-Butadiene Conversion by Using ZrZn-Containing MgO/SiO2 Systems Prepared by Co-Precipitation and Effect of Catalyst Ccidity Modification. ChemCatChem 2016, 8, 2376–2386. [Google Scholar] [CrossRef] [Green Version]
- Da Ros, S.; Jones, M.D.; Mattia, D.; Schwaab, M.; Noronha, F.B.; Pinto, J.C. Modelling the Effects of Reaction Temperature and Flow Rate on the Conversion of Ethanol to 1,3-Butadiene. Appl. Catal. A Gen. 2017, 530, 37–47. [Google Scholar] [CrossRef] [Green Version]
- EIA. Petroleum & Other Liquids, WTI Spot Price. Available online: www.eia.gov/dnav/pet/hist/rwtcA.htm (accessed on 4 August 2021).
- EIA. Henry Hub Natural Gas Spot Price (Dollars per Million Btu). Available online: www.eia.gov/dnav/ng/hist/rngwhhda.htm (accessed on 4 August 2021).
- EIA. Natural Gas: Dry Shale Gas Production Estimates by Play. Available online: https://www.eia.gov/naturalgas/data.php#production (accessed on 4 August 2021).
- BP British Petrolium. BP Statistical Review of World Energy 2019 Report. Available online: https://www.bp.com/content/dam/bp/business-sites/en/global/corporate/pdfs/energy-economics/statistical-review/bp-stats-review-2019-full-report.pdf (accessed on 4 August 2021).
- Ohyama, J.; Nishimura, S.; Takahashi, K. Data Driven Determination of Reaction Conditions in Oxidative Coupling of Methane via Machine Learning. ChemCatChem 2019, 11, 4307–4313. [Google Scholar] [CrossRef]
- Miyazato, I.; Nishimura, S.; Takahashi, L.; Ohyama, J.; Takahashi, K. Data-Driven Identification of the Reaction Network in Oxidative Coupling of the Methane Reaction via Experimental Data. J. Phys. Chem. Lett. 2020, 11, 787–795. [Google Scholar] [CrossRef]
- Ghareghashi, A.; Sarrafi, A.; Ghader, S. The Effect of Heat Transfer on Products of a Thermally Coupled Shell and Tube Reactor Consisting of Two Processes: Steam Reforming of Methane and Oxidative Coupling of Methane. Chem. Eng. Process. Process Intensif. 2018, 133, 263–277. [Google Scholar] [CrossRef]
- Li, D.; Baslyman, W.S.; Sarathy, S.M.; Takanabe, K. Impact of OH Radical Generator Involvement in the Gas-Phase Radical Reaction Network on the Oxidative Coupling of Methane—A Simulation Study. Energy Technol. 2019, 8, 1900563. [Google Scholar] [CrossRef]
- Yancy-Caballero, D.M.; Biegler, L.T.; Guirardello, R. Large-Scale DAE-Constrained Optimization Applied to a Modified Spouted Bed Reactor for Ethylene Production from Methane. Comput. Chem. Eng. 2018, 113, 162–183. [Google Scholar] [CrossRef]
- Mohammadi, Y.; Penlidis, A. “Optimulation” in Chemical Reaction Engineering: Oxidative Coupling of Methane as a Case Study. Ind. Eng. Chem. Res. 2018, 57, 8664–8678. [Google Scholar] [CrossRef]
- Wiyaratn, W.; Manundawee, S.; Arpornwichanop, A.; Assabumrungrat, S.; Watanapa, A.; Worawimut, C. Two-Dimensional Mathematical Modeling of the Oxidative Coupling of Methane in a Membrane Reactor. Eng. J. 2016, 20, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Manundawee, S.; Arpornwichanop, A.; Assabumrungrat, S.; Wiyaratn, W. Two-Dimensional Modeling of the Oxidative Coupling of Methane in a Fixed Bed Reactor: A Comparison among Different Catalysts. Eng. J. 2017, 21, 77–99. [Google Scholar] [CrossRef] [Green Version]
- Lomonosov, V.; Gordienko, Y.; Ponomareva, E.; Sinev, M. Kinetic Conjugation Effects in Oxidation of C1-C2 Hydrocarbons: Experiment and Modeling. Chem. Eng. J. 2019, 370, 1210–1217. [Google Scholar] [CrossRef]
- Alexiadis, V.I.; Serres, T.; Marin, G.B.; Mirodatos, C.; Thybaut, J.W.; Schuurman, Y. Analysis of Volume-to-surface Ratio Effects on Methane Oxidative Coupling Using Microkinetic Modeling. AIChE J. 2018, 64, 2603–2611. [Google Scholar] [CrossRef]
- Alexiadis, V.I.; Chaar, M.; van Veen, A.; Muhler, M.; Thybaut, J.W.; Marin, G.B. Quantitative Screening of an Extended Oxidative Coupling of Methane Catalyst Library. Appl. Catal. B Environ. 2016, 199, 252–259. [Google Scholar] [CrossRef]
- Obradović, A.; Thybaut, J.W.; Marin, G.B. Oxidative Coupling of Methane: Opportunities for Microkinetic Model-Assisted Process Implementations. Chem. Eng. Technol. 2016, 39, 1996–2010. [Google Scholar] [CrossRef]
- Karakaya, C.; Zhu, H.; Loebick, C.; Weissman, J.G.; Kee, R.J. A Detailed Reaction Mechanism for Oxidative Coupling of Methane over Mn/Na2WO4/SiO2 Catalyst for Non-Isothermal Conditions. Catal. Today 2018, 312, 10–22. [Google Scholar] [CrossRef]
- Pirro, L.; Obradović, A.; Vandegehuchte, B.D.; Marin, G.B.; Thybaut, J.W. Model-Based Catalyst Selection for the Oxidative Coupling of Methane in an Adiabatic Fixed-Bed Reactor. Ind. Eng. Chem. Res. 2018, 57, 16295–16307. [Google Scholar] [CrossRef]
- Sun, Z.; Kota, A.; Sarsani, S.; West, D.H.; Balakotaiah, V. Bifurcation Analysis of Methane Oxidative Coupling without Catalyst. Chem. Eng. J. 2018, 343, 770–788. [Google Scholar] [CrossRef]
- Liu, Z.; Ho Li, J.P.; Vovk, E.; Zhu, Y.; Li, S.; Wang, S.; Van Bavel, A.P.; Yang, Y. Online Kinetics Study of Oxidative Coupling of Methane over La2O3 for Methane Activation: What Is behind the Distinguished Light-off Temperatures? ACS Catal. 2018, 8, 11761–11772. [Google Scholar] [CrossRef]
- Chaudhari, V.; Dutta, K.; Li, C.J.; Kopyscinski, J. Mechanistic Insights of Methane Conversion to Ethylene over Gallium Oxide and Gallium Nitride Using Density Functional Theory. Mol. Catal. 2020, 482, 110606. [Google Scholar] [CrossRef]
- Pirro, L.; Mendes, P.S.F.; Paret, S.; Vandegehuchte, B.D.; Marin, G.B.; Thybaut, J.W. Descriptor-Property Relationships in Heterogeneous Catalysis: Exploiting Synergies between Statistics and Fundamental Kinetic Modelling. Catal. Sci. Technol. 2019, 9, 3109–3125. [Google Scholar] [CrossRef]
- Luo, L.; You, R.; Liu, Y.; Yang, J.; Zhu, Y.; Wen, W.; Pan, Y.; Qi, F.; Huang, W. Gas-Phase Reaction Network of Li/MgO-Catalyzed Oxidative Coupling of Methane and Oxidative Dehydrogenation of Ethane. ACS Catal. 2019, 9, 2514–2520. [Google Scholar] [CrossRef]
- Parishan, S.; Nowicka, E.; Fleischer, V.; Schulz, C.; Colmenares, M.G.; Rosowski, F.; Schomäcker, R. Investigation into Consecutive Reactions of Ethane and Ethene Under the OCM Reaction Conditions over MnxOy–Na2WO4/SiO2 Catalyst. Catal. Lett. 2018, 148, 1659–1675. [Google Scholar] [CrossRef]
- Noon, D.; Zohour, B.; Bae, A.; Seubsai, A.; Senkan, S. Effects of Ir-Doping on the Transition from Oxidative Coupling to Partial Oxidation of Methane in La2O3-CeO2 Nanofiber Catalysts: Spatial Concentration and Temperature Profiles. RSC Adv. 2017, 7, 26783–26789. [Google Scholar] [CrossRef] [Green Version]
- Schmack, R.; Friedrich, A.; Kondratenko, E.V.; Kraehnert, R.; Polte, J.; Werwatz, A. A Meta-Analysis of Catalytic Literature Data Reveals Property-Performance Correlations for the OCM Reaction. Nat. Commun. 2019, 10, 441. [Google Scholar] [CrossRef]
- Lomonosov, V.I.; Sinev, M.Y. Oxidative Coupling of Methane: Mechanism and Kinetics. Kinet. Catal. 2016, 57, 647–676. [Google Scholar] [CrossRef]
- Penteado, A.T.; Kim, M.; Godini, H.R.; Esche, E.; Repke, J.U. Techno-Economic Evaluation of a Biogas-Based Oxidative Coupling of Methane Process for Ethylene Production. Front. Chem. Sci. Eng. 2018, 12, 598–618. [Google Scholar] [CrossRef]
- Ortiz-Espinoza, A.P.; Noureldin, M.M.B.; El-Halwagi, M.M.; Jiménez-Gutiérrez, A. Design, Simulation and Techno-Economic Analysis of Two Processes for the Conversion of Shale Gas to Ethylene. Comput. Chem. Eng. 2017, 107, 237–246. [Google Scholar] [CrossRef]
- Cruellas, A.; Heezius, J.; Spallina, V.; van Sint Annaland, M.; Medrano, J.A.; Gallucci, F. Oxidative Coupling of Methane in Membrane Reactors; A Techno-Economic Assessment. Processes 2020, 8, 247. [Google Scholar] [CrossRef] [Green Version]
- Godini, H.R.; Azadi, M.; Penteado, A.; Khadivi, M.; Wozny, G.; Repke, J.U. A Multi-Perspectives Analysis of Methane Oxidative Coupling Process Based on Miniplant-Scale Experimental Data. Chem. Eng. Res. Des. 2019, 151, 56–69. [Google Scholar] [CrossRef]
- Cruellas, A.; Bakker, J.J.; van Sint Annaland, M.; Medrano, J.A.; Gallucci, F. Techno-Economic Analysis of Oxidative Coupling of Methane: Current State of the Art and Future Perspectives. Energy Convers. Manag. 2019, 198, 111789. [Google Scholar] [CrossRef]
- Spallina, V.; Velarde, I.C.; Jimenez, J.A.M.; Godini, H.R.; Gallucci, F.; Van Sint Annaland, M. Techno-Economic Assessment of Different Routes for Olefins Production through the Oxidative Coupling of Methane (OCM): Advances in Benchmark Technologies. Energy Convers. Manag. 2017, 154, 244–261. [Google Scholar] [CrossRef]
- Dutta, K.; Chaudhari, V.; Li, C.J.; Kopyscinski, J. Methane Conversion to Ethylene over GaN Catalysts. Effect of Catalyst Nitridation. Appl. Catal. A Gen. 2020, 595, 117430. [Google Scholar] [CrossRef]
- Kirik, N.P.; Anshits, N.N.; Rabchevskii, E.V.; Solov’ev, L.A.; Anshits, A.G. Effect of HF Modification on the Catalytic Properties of Ferrospheres in the Oxidative Coupling of Methane. Kinet. Catal. 2019, 60, 196–204. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Liu, Y.; Fang, X.; Xu, X.; Liu, W.; Zheng, R.; Wang, X. Optimizing the Reaction Performance of La2Ce2O7-Based Catalysts for Oxidative Coupling of Methane (OCM) at Lower Temperature by Lattice Doping with Ca Cations. Eur. J. Inorg. Chem. 2019, 2019, 183–194. [Google Scholar] [CrossRef]
- Fei, C.; Jian, Y.; Liang, Y.; Jun, Z.; Huahua, Z.; Huanling, S.; Lingjun, C. Influence of the Composition/Texture of Solid Acid WO3/TiO2-Supported Lithium-Manganese Catalysts on the Oxidative Coupling of Methane. Wuli Huaxue Xuebao Acta Phys. Chim. Sin. 2019, 35, 1027–1036. [Google Scholar] [CrossRef]
- Cheng, F.; Yang, J.; Yan, L.; Zhao, J.; Zhao, H.; Song, H.; Chou, L. Impact of Chloride Ions on the Oxidative Coupling of Methane over Li/SnO2 Catalyst. React. Kinet. Mech. Catal. 2018, 125, 675–688. [Google Scholar] [CrossRef]
- Haneda, M.; Katsuragawa, Y.; Nakamura, Y.; Towata, A. Promoting Effect of Cerium Oxide on the Catalytic Performance of Yttrium Oxide for Oxidative Coupling of Methane. Front. Chem. 2018, 6, 581. [Google Scholar] [CrossRef]
- Peng, L.; Xu, J.; Fang, X.; Liu, W.; Xu, X.; Liu, L.; Li, Z.; Peng, H.; Zheng, R.; Wang, X. SnO2 Based Catalysts with Low-Temperature Performance for Oxidative Coupling of Methane: Insight into the Promotional Effects of Alkali-Metal Oxides. Eur. J. Inorg. Chem. 2018, 2018, 1787–1799. [Google Scholar] [CrossRef]
- Oh, S.C.; Xu, J.; Tran, D.T.; Liu, B.; Liu, D. Effects of Controlled Crystalline Surface of Hydroxyapatite on Methane Oxidation Reactions. ACS Catal. 2018, 8, 4493–4507. [Google Scholar] [CrossRef]
- Li, Z.; Wang, S.; Hong, W.; Zou, S.; Xiao, L.; Fan, J. Intelligent Optimization of Na−Mn−W/SiO2 Catalysts for the Oxidative Coupling of Methane. ChemNanoMat 2018, 4, 487–495. [Google Scholar] [CrossRef]
- Nadezhda, P.K.; Natalia, N.A.; Evgenii, V.R.; Leonid, A.S. The Activity of the HF-Modified Ferrospheres in Oxidative Coupling of Methane. J. Sib. Fed. Univ. Chem. 2018, 11, 347–360. [Google Scholar] [CrossRef] [Green Version]
- Nipan, G.D. Specific Phase Transformations of K/W/Mn/SiO2 Composite Catalyst. Inorg. Mater. 2018, 54, 96–101. [Google Scholar] [CrossRef]
- Ogihara, H.; Imai, N.; Yoshida-Hirahara, M.; Kurokawa, H. Direct Dehydrogenative Conversion of Methane to Hydrogen, Nanocarbons, Ethane, and Ethylene on Pd/SiO2 Catalysts. Chem. Lett. 2020, 49, 236–239. [Google Scholar] [CrossRef]
- Hayek, N.S.; Lucas, N.S.; Warwar Damouny, C.; Gazit, O.M. Critical Surface Parameters for the Oxidative Coupling of Methane over the Mn-Na-W/SiO2 Catalyst. ACS Appl. Mater. Interfaces 2017, 9, 40404–40411. [Google Scholar] [CrossRef] [PubMed]
- Nipan, G.D. Melt-Assisted Phase Transformations of A/W/Mn/SiO2 (A = Li, Na, K, Rb, Cs) Composite Catalysts. Inorg. Mater. 2017, 53, 553–559. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, G.; Wang, Y.; Lu, Y. MnTiO3-Driven Low-Temperature Oxidative Coupling of Methane over TiO2-Doped Mn2O3-Na2WO4/SiO2 Catalyst. Sci. Adv. 2017, 3, e1603180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nipan, G.D.; Buzanov, G.A.; Zhizhin, K.Y.; Kuznetsov, N.T. Phase States of Li(Na,K,Rb,Cs)/W/Mn/SiO2composite Catalysts for Oxidative Coupling of Methane. Russ. J. Inorg. Chem. 2016, 61, 1689–1707. [Google Scholar] [CrossRef]
- Elkins, T.W.; Roberts, S.J.; Hagelin-Weaver, H.E. Effects of Alkali and Alkaline-Earth Metal Dopants on Magnesium Oxide Supported Rare-Earth Oxide Catalysts in the Oxidative Coupling of Methane. Appl. Catal. A Gen. 2016, 528, 175–190. [Google Scholar] [CrossRef]
- Colmenares, M.G.; Simon, U.; Yildiz, M.; Arndt, S.; Schomaecker, R.; Thomas, A.; Rosowski, F.; Gurlo, A.; Goerke, O. Oxidative Coupling of Methane on the Na2WO4-MnxOy Catalyst: COK-12 as an Inexpensive Alternative to SBA-15. Catal. Commun. 2016, 85, 75–78. [Google Scholar] [CrossRef]
- Yildiz, M.; Aksu, Y.; Simon, U.; Otremba, T.; Kailasam, K.; Göbel, C.; Girgsdies, F.; Görke, O.; Rosowski, F.; Thomas, A.; et al. Silica Material Variation for the MnxOy-Na2WO4/SiO2. Appl. Catal. A Gen. 2016, 525, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Anshits, A.G.; Bayukov, O.A.; Kondratenko, E.V.; Anshits, N.N.; Pletnev, O.N.; Rabchevskii, E.V.; Solovyov, L.A. Catalytic Properties and Nature of Active Centers of Ferrospheres in Oxidative Coupling of Methane. Appl. Catal. A Gen. 2016, 524, 192–199. [Google Scholar] [CrossRef]
- Sollier, B.M.; Gómez, L.E.; Boix, A.V.; Miró, E.E. Oxidative Coupling of Methane on Cordierite Monoliths Coated with Sr/La2O3 Catalysts. Influence of Honeycomb Structure and Catalyst-Cordierite Chemical Interactions on the Catalytic Behavior. Appl. Catal. A Gen. 2018, 550, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Igenegbai, V.O.; Almallahi, R.; Meyer, R.J.; Linic, S. Oxidative Coupling of Methane over Hybrid Membrane/Catalyst Active Centers: Chemical Requirements for Prolonged Lifetime. ACS Energy Lett. 2019, 4, 1465–1470. [Google Scholar] [CrossRef]
- Pirro, L.; Mendes, P.S.F.; Vandegehuchte, B.D.; Marin, G.B.; Thybaut, J.W. Catalyst Screening for the Oxidative Coupling of Methane: From Isothermal to Adiabatic Operation: Via Microkinetic Simulations. React. Chem. Eng. 2020, 5, 584–596. [Google Scholar] [CrossRef]
- Igenegbai, V.O.; Meyer, R.J.; Linic, S. In Search of Membrane-Catalyst Materials for Oxidative Coupling of Methane: Performance and Phase Stability Studies of Gadolinium-Doped Barium Cerate and the Impact of Zr Doping. Appl. Catal. B Environ. 2018, 230, 29–35. [Google Scholar] [CrossRef]
- Garcia-Fayos, J.; Lobera, M.P.; Balaguer, M.; Serra, J.M. Catalyst Screening for Oxidative Coupling of Methane Integrated in Membrane Reactors. Front. Mater. 2018, 5, 1–11. [Google Scholar] [CrossRef]
- Oh, S.C.; Wu, Y.; Tran, D.T.; Lee, I.C.; Lei, Y.; Liu, D. Influences of Cation and Anion Substitutions on Oxidative Coupling of Methane over Hydroxyapatite Catalysts. Fuel 2016, 167, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Farrell, B.L.; Linic, S. Oxidative Coupling of Methane over Mixed Oxide Catalysts Designed for Solid Oxide Membrane Reactors. Catal. Sci. Technol. 2016, 6, 4370–4376. [Google Scholar] [CrossRef]
- Zhao, M.; Ke, S.; Wu, H.; Xia, W.; Wan, H. Flower-like Sr-La2O3 Microspheres with Hierarchically Porous Structures for Oxidative Coupling of Methane. Ind. Eng. Chem. Res. 2019, 58, 22847–22856. [Google Scholar] [CrossRef]
- Seubsai, A.; Tiencharoenwong, P.; Kidamorn, P.; Niamnuy, C. Synthesis of Light Hydrocarbons via Oxidative Coupling of Methane over Silica-Supported Na2WO4-Tio2 Catalyst. Eng. J. 2019, 23, 169–182. [Google Scholar] [CrossRef]
- Yıldız, M. Mesoporous TiO2-Rutile Supported MnxOy-Na2WO4: Preparation, Characterization and Catalytic Performance in the Oxidative Coupling of Methane. J. Ind. Eng. Chem. 2019, 76, 488–499. [Google Scholar] [CrossRef]
- Kwon, G.; Shin, D.; Jeong, H.; Sahoo, S.K.; Lee, J.; Kim, G.; Choi, J.; Kim, D.H.; Han, J.W.; Lee, H. Oxidative Methane Conversion to Ethane on Highly Oxidized Pd/CeO2 Catalysts Below 400 °C. ChemSusChem 2020, 13, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, H.; Öksüzömer, M.A.F.; Ali Gürkaynak, M. Studies on Oxidative Coupling of Methane Using Sm2O3-Based Catalysts. Chem. Eng. Commun. 2019, 206, 48–60. [Google Scholar] [CrossRef]
- Matsumoto, T.; Saito, M.; Ishikawa, S.; Fujii, K.; Yashima, M.; Ueda, W.; Motohashi, T. High Catalytic Activity of Crystalline Lithium Calcium Silicate for Oxidative Coupling of Methane Originated from Crystallographic Joint Effects of Multiple Cations. ChemCatChem 2020, 12, 1968–1972. [Google Scholar] [CrossRef]
- Cheng, F.; Yang, J.; Yan, L.; Zhao, J.; Zhao, H.; Song, H.; Chou, L.J. Enhancement of La2O3 to Li-Mn/WO3/TiO2 for Oxidative Coupling of Methane. J. Rare Earths 2020, 38, 167–174. [Google Scholar] [CrossRef]
- Bosch, C.E.; Poulston, S.; Collier, P.; Thybaut, J.W.; Marin, G.B. Exploring Microemulsion-Prepared Lanthanum Catalysts for Natural Gas Valorisation. Johnson Matthey Technol. Rev. 2019, 63, 265–276. [Google Scholar] [CrossRef]
- Lee, B.J.; Hur, Y.G.; Kim, D.H.; Lee, S.H.; Lee, K.Y. Non-Oxidative Aromatization and Ethylene Formation over Ga/HZSM-5 Catalysts Using a Mixed Feed of Methane and Ethane. Fuel 2019, 253, 449–459. [Google Scholar] [CrossRef]
- Jones, A.S.; Alfonso, N.; Hagelin-Weaver, H.E. Transition-Metal Doped, Magnesium Oxide-Supported Terbium Oxides as Catalysts for the Oxidative Coupling of Methane. Polyhedron 2019, 170, 602–611. [Google Scholar] [CrossRef]
- Chukeaw, T.; Sringam, S.; Chareonpanich, M.; Seubsai, A. Screening of Single and Binary Catalysts for Oxidative Coupling of Methane to Value-Added Chemicals. Mol. Catal. 2019, 470, 40–47. [Google Scholar] [CrossRef]
- Kolesnichenko, N.V.; Ezhova, N.N.; Snatenkova, Y.M. Lower Olefins from Methane: Recent Advances. Russ. Chem. Rev. 2020, 89, 191–224. [Google Scholar] [CrossRef]
- Gao, Y.; Neal, L.; Ding, D.; Wu, W.; Baroi, C.; Gaffney, A.M.; Li, F. Recent Advances in Intensified Ethylene Production—A Review. ACS Catal. 2019, 9, 8592–8621. [Google Scholar] [CrossRef]
- Scapinello, M.; Delikonstantis, E.; Stefanidis, G.D. The Panorama of Plasma-Assisted Non-Oxidative Methane Reforming. Chem. Eng. Process. Process Intensif. 2017, 117, 120–140. [Google Scholar] [CrossRef]
- García, L.; Poveda, Y.A.; Rodríguez, G.; Esche, E.; Godini, H.R.; Wozny, G.; Repke, J.U.; Orjuela, Á. Adsorption Separation of Oxidative Coupling of Methane Effluent Gases. Mini-Plant Scale Experiments and Modeling. J. Nat. Gas Sci. Eng. 2019, 61, 106–118. [Google Scholar] [CrossRef]
- Bachman, J.E.; Reed, D.A.; Kapelewski, M.T.; Chachra, G.; Jonnavittula, D.; Radaelli, G.; Long, J.R. Enabling Alternative Ethylene Production through Its Selective Adsorption in the Metal-Organic Framework Mn2(: M -Dobdc). Energy Environ. Sci. 2018, 11, 2423–2431. [Google Scholar] [CrossRef]
- Penteado, A.; Esche, E.; Salerno, D.; Godini, H.R.; Wozny, G. Design and Assessment of a Membrane and Absorption Based Carbon Dioxide Removal Process for Oxidative Coupling of Methane. Ind. Eng. Chem. Res. 2016, 55, 7473–7483. [Google Scholar] [CrossRef]
- Parishan, S.; Littlewood, P.; Arinchtein, A.; Fleischer, V.; Schomäcker, R. Chemical Looping as a Reactor Concept for the Oxidative Coupling of Methane over the MnxOy-Na2WO4/SiO2 Catalyst, Benefits and Limitation. Catal. Today 2018, 311, 40–47. [Google Scholar] [CrossRef]
- Fleischer, V.; Simon, U.; Parishan, S.; Colmenares, M.G.; Görke, O.; Gurlo, A.; Riedel, W.; Thum, L.; Schmidt, J.; Risse, T.; et al. Investigation of the Role of the Na2WO4/Mn/SiO2 Catalyst Composition in the Oxidative Coupling of Methane by Chemical Looping Experiments. J. Catal. 2018, 360, 102–117. [Google Scholar] [CrossRef]
- Chung, E.Y.; Wang, W.K.; Nadgouda, S.G.; Baser, D.S.; Sofranko, J.A.; Fan, L.S. Catalytic Oxygen Carriers and Process Systems for Oxidative Coupling of Methane Using the Chemical Looping Technology. Ind. Eng. Chem. Res. 2016, 55, 12750–12764. [Google Scholar] [CrossRef]
- Fleischer, V.; Littlewood, P.; Parishan, S.; Schomäcker, R. Chemical Looping as Reactor Concept for the Oxidative Coupling of Methane over a Na2WO4/Mn/SiO2 Catalyst. Chem. Eng. J. 2016, 306, 646–654. [Google Scholar] [CrossRef]
- Bayanak, M.; Azimi, A. Investigation of Activity and Selectivity of Redox Catalysts in Oxidative Coupling of Methane in Fluidized Bed Reactor. J. Fundam. Appl. Sci. 2016, 8, 397. [Google Scholar] [CrossRef] [Green Version]
- Aseem, A.; Harold, M.P. C2 Yield Enhancement during Oxidative Coupling of Methane in a Nonpermselective Porous Membrane Reactor. Chem. Eng. Sci. 2018, 175, 199–207. [Google Scholar] [CrossRef]
- Liang, W.; Sarsani, S.; West, D.; Mamedov, A.; Lengyel, I.; Perez, H.; Lowrey, J. Performance Improvement for a Fixed-Bed Reactor with Layered Loading Catalysts of Different Catalytic Properties for Oxidative Coupling of Methane. Catal. Today 2018, 299, 60–66. [Google Scholar] [CrossRef]
- Han, Q.; Tanaka, A.; Matsumoto, M.; Endo, A.; Kubota, Y.; Inagaki, S. Conversion of Methane to C2 and C3 Hydrocarbons over TiO2/ZSM-5 Core-Shell Particles in an Electric Field. RSC Adv. 2019, 9, 34793–34803. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Ogo, S.; Takeno, Y.; Takise, K.; Seo, J.G.; Sekine, Y. Electric Field and Mobile Oxygen Promote Low-Temperature Oxidative Coupling of Methane over La1-XCaxAlO3-δ Perovskite Catalysts. ACS Omega 2019, 4, 10438–10443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cho, Y.; Yamaguchi, A.; Peng, X.; Miyauchi, M.; Abe, H.; Fujita, T. CO2 Oxidative Coupling of Methane Using an Earth-Abundant CaO-Based Catalyst. Sci. Rep. 2019, 9, 15454. [Google Scholar] [CrossRef] [Green Version]
- Hayek, N.S.; Khlief, G.J.; Horani, F.; Gazit, O.M. Effect of Reaction Conditions on the Oxidative Coupling of Methane over Doped MnOx-Na2WO4/SiO2 Catalyst. J. Catal. 2019, 376, 25–31. [Google Scholar] [CrossRef]
- Valadkhani, A.; Shahrokhi, M.; Pishvaie, M.R.; Zarrinpashneh, S. Simulation and Experimental Studies of Methane Oxidative Coupling Reaction in a Bench Scale Fixed Bed Reactor. Energy Sources Part A Recover. Util. Environ. Eff. 2013, 35, 1418–1426. [Google Scholar] [CrossRef]
- Stansch, Z.; Mleczko, M.; Baerns, L. Comprehensive Kinetics of Oxidative Coupling of Methane over the La2O3/CaO Catalyst. Ind. Eng. Chem. Res. 1997, 36, 2568–2579. [Google Scholar] [CrossRef]
- Couwenberg, P.M.; Chen, Q.; Marin, G.B. Kinetics of a Gas-Phase Chain Reaction Catalyzed by a Solid: The Oxidative Coupling of Methane over Li/MgO-Based Catalysts. Ind. Eng. Chem. Res. 1996, 35, 3999–4011. [Google Scholar] [CrossRef] [Green Version]
- Simon, Y.; Baronnet, F.; Côme, G.M.; Marquaire, P.M. Detailed Mechanism of the Oxidative Coupling of Methane. Stud. Surf. Sci. Catal. 2004, 147, 571–576. [Google Scholar] [CrossRef]
- Sinev, M.Y.; Fattakhova, Z.T.; Lomonosov, V.I.; Gordienko, Y.A. Kinetics of Oxidative Coupling of Methane: Bridging the Gap between Comprehension and Description. J. Nat. Gas Chem. 2009, 18, 273–287. [Google Scholar] [CrossRef]
- Chen, Q.; Hoebink, J.H.B.J.; Marin, G.B. Kinetics of the Oxidative Coupling of Methane at Atmospheric Pressure in the Absence of Catalyst. Ind. Eng. Chem. Res. 1991, 30, 2088–2097. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Couwenberg, P.M.; Marin, G.B. Effect of Pressure on the Oxidative Coupling of Methane in the Absence of Catalyst. AIChE J. 1994, 40, 521–535. [Google Scholar] [CrossRef]
- Zanthoff, H.W.; Baerns, M. Oxidative Coupling of Methane in the Gas Phase. Kinetic Simulation and Experimental Verification. Ind. Eng. Chem. Res. 1990, 29, 2–10. [Google Scholar] [CrossRef]
- Chen, Q.; Couwenberg, P.M.; Marin, G.B. The Oxidative Coupling of Methane with Cofeeding of Ethane. Catal. Today 1994, 21, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Ekstrom, A.; Regtop, R.; Bhargava, S. Effect of Pressure on the Oxidative Coupling Reaction of Methane. Appl. Catal. 1990, 62, 253–269. [Google Scholar] [CrossRef]
- Beck, B.; Fleischer, V.; Arndt, S.; Hevia, M.G.; Urakawa, A.; Hugo, P.; Schomäcker, R. Oxidative Coupling of Methane—A Complex Surface/Gas Phase Mechanism with Strong Impact on the Reaction Engineering. Catal. Today 2014, 228, 212–218. [Google Scholar] [CrossRef]
- Al-Zahrani, S.; Song, Q.; Lobban, L.L. Effects of CO2 during Oxidative Coupling of Methane over Li/MgO: Mechanisms and Models. Ind. Eng. Chem. Res. 1994, 33, 251–258. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Rane, V.H.; Pandit, M.Y. Comparison of Alkali Metal Promoted MgO Catalysts for Their Surface Acidity/Basicity and Catalytic Activity/Selectivity in the Oxidative Coupling of Methane. J. Chem. Technol. Biotechnol. 1997, 68, 177–186. [Google Scholar] [CrossRef]
- Sun, J.; Thybaut, J.W.; Marin, G.B. Microkinetics of Methane Oxidative Coupling. Catal. Today 2008, 137, 90–102. [Google Scholar] [CrossRef]
- Kechagiopoulos, P.N.; Thybaut, J.W.; Marin, G.B. Oxidative Coupling of Methane: A Microkinetic Model Accounting for Intraparticle Surface-Intermediates Concentration Profiles. Ind. Eng. Chem. Res. 2014, 53, 1825–1840. [Google Scholar] [CrossRef]
- Nibbelke, R.H.; Scheerová, J.; De Croon, M.H.J.M.; Marin, G.B. The Oxidative Coupling of Methane over MgO-Based Catalysts: A Steady-State Isotope Transient Kinetic Analysis. J. Catal. 1995, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Buyevskaya, M.O.V.; Rothaemel, M.; Zanthoff, H.W.; Baerns, M. Transient Studies on Reactions Steps in the Oxidative Coupling of Methane over Catalytic Surfaces of MgO and Sm2O3. J. Catal. 1994, 146, 346–357. [Google Scholar] [CrossRef]
- Luo, L.; Tang, X.; Wang, W.; Wang, Y.; Sun, S.; Qi, F.; Huang, W. Methyl Radicals in Oxidative Coupling of Methane Directly Confirmed by Synchrotron VUV Photoionization Mass Spectroscopy. Sci. Rep. 2013, 3, 1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef] [PubMed]
- Kuś, S.L.S.; Otremba, M.; Taniewski, M. The Catalytic Performance in Oxidative Coupling of Methane and the Surface Basicity of La2O3, Nd2O3, ZrO2 and Nb2O5. Fuel 2003, 82, 1331–1338. [Google Scholar] [CrossRef]
- Arndt, S.; Simon, U.; Heitz, S.; Berthold, A.; Beck, B.; Görke, O.; Epping, J.D.; Otremba, T.; Aksu, Y.; Irran, E.; et al. Li-Doped MgO from Different Preparative Routes for the Oxidative Coupling of Methane. Top. Catal. 2011, 54, 1266–1285. [Google Scholar] [CrossRef]
- Pannek, U.; Mleczko, L. Comprehensive Model of Oxidative Coupling of Methane in a Fluidized-Bed Reactor. Chem. Eng. Sci. 1996, 51, 3575–3590. [Google Scholar] [CrossRef]
- Jašo, S.; Arellano-Garcia, H.; Wozny, G. Oxidative Coupling of Methane in a Fluidized Bed Reactor: Influence of Feeding Policy, Hydronamics and Rector Geometry. Chem. Eng. J. 2011, 171, 255–271. [Google Scholar] [CrossRef]
- Salehi, M.S.; Askarishahi, M.; Godini, H.R.; Schomäcker, R.; Wozny, G. CFD Simulation of Oxidative Coupling of Methane in Fluidized-Bed Reactors: A Detailed Analysis of Flow-Reaction Characteristics and Operating Conditions. Ind. Eng. Chem. Res. 2016, 55, 1149–1163. [Google Scholar] [CrossRef]
- Holst, N.; Jašo, S.; Godini, H.R.; Glöser, S.; Arellano-Garcia, H.; Wozny, G.; Steinbach, J. Two-Dimensional Model for Oxidative Coupling of Methane in a Packed-Bed Membrane Reactor. Chem. Eng. Technol. 2012, 35, 294–301. [Google Scholar] [CrossRef]
- Godini, H.R.; Xiao, S.; Kim, M.; Holst, N.; Jašo, S.; Görke, O.; Steinbach, J.; Wozny, G. Experimental and Model-Based Analysis of Membrane Reactor Performance for Methane Oxidative Coupling: Effect of Radial Heat and Mass Transfer. J. Ind. Eng. Chem. 2014, 20, 1993–2002. [Google Scholar] [CrossRef]
- Onoja, O.P.; Wang, X.; Kechagiopoulos, P.N. Influencing Selectivity in the Oxidative Coupling of Methane by Modulating Oxygen Permeation in a Variable Thickness Membrane Reactor. Chem. Eng. Process. Process Intensif. 2019, 135, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Couwenberg, P.M.; Chen, Q.; Marin, G.B. Irreducible Mass-Transport Limitations during a Heterogeneously Catalyzed Gas-Phase Chain Reaction: Oxidative Coupling of Methane. Ind. Eng. Chem. Res. 1996, 35, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Yaghobi, N.; Ghoreishy, M.H.R. Oxidative Coupling of Methane in a Fixed Bed Reactor over Perovskite Catalyst: A Simulation Study Using Experimental Kinetic Model. J. Nat. Gas Chem. 2008, 17, 8–16. [Google Scholar] [CrossRef]
- Sun, Z.; West, D.H.; Balakotaiah, V. Bifurcation Analysis of Catalytic Partial Oxidations in Laboratory-Scale Packed-Bed Reactors with Heat Exchange. Chem. Eng. J. 2019, 377. [Google Scholar] [CrossRef]
- Sun, Z.; West, D.H.; Gautam, P.; Balakotaiah, V. Scale-up Analysis of Autothermal Operation of Methane Oxidative Coupling with La2O3/CaO Catalyst. AIChE J. 2020, 66, e16949. [Google Scholar] [CrossRef]
- Zhang, Z.; Ji, S. Numerical Simulation of Particle/Monolithic Two-Stage Catalyst Bed Reactor with Beds-Interspace Distributed Dioxygen Feeding for Oxidative Coupling of Methane. Comput. Chem. Eng. 2016, 90, 247–259. [Google Scholar] [CrossRef]
- Vandewalle, L.A.; Van de Vijver, R.; Van Geem, K.M.; Marin, G.B. The Role of Mass and Heat Transfer in the Design of Novel Reactors for Oxidative Coupling of Methane. Chem. Eng. Sci. 2019, 198, 268–289. [Google Scholar] [CrossRef]
- Cruellas, A.; Melchiori, T.; Gallucci, F.; van Sint Annaland, M. Advanced Reactor Concepts for Oxidative Coupling of Methane. Catal. Rev. Sci. Eng. 2017, 59, 234–294. [Google Scholar] [CrossRef] [Green Version]
- Froment, G.; Bischoff, K.; De Wilde, J. Chemical Reactor Analysis and Design, 2nd ed.; Wiley Subscription Services, Inc., A Wiley Company: New York, NY, USA, 2011. [Google Scholar]
- Alexiadis, V.I.; Thybaut, J.W.; Kechagiopoulos, P.N.; Chaar, M.; Van Veen, A.C.; Muhler, M.; Marin, G.B. Oxidative Coupling of Methane: Catalytic Behaviour Assessment via Comprehensive Microkinetic Modelling. Appl. Catal. B Environ. 2014, 150–151, 496–505. [Google Scholar] [CrossRef]
- Sarsani, S.; West, D.; Liang, W.; Balakotaiah, V. Autothermal Oxidative Coupling of Methane with Ambient Feed Temperature. Chem. Eng. J. 2017, 328, 484–496. [Google Scholar] [CrossRef]
- Vandewalle, L.A.; Lengyel, I.; West, D.H.; Van Geem, K.M.; Marin, G.B. Catalyst Ignition and Extinction: A Microkinetics-Based Bifurcation Study of Adiabatic Reactors for Oxidative Coupling of Methane. Chem. Eng. Sci. 2019, 199, 635–651. [Google Scholar] [CrossRef]
- Dautzenberg, F.M.; Schlatter, J.C. Catalyst and Reactor Requirements for the Oxidative Coupling of Methane. Catal. Today 1992, 13, 503–5r09. [Google Scholar] [CrossRef]
- Kato, K.; Wen, C.Y. Bubble Assemblage Model for Fluidized Bed Catalytic Reactors. Chem. Eng. Sci. 1969, 24, 1351–1369. [Google Scholar] [CrossRef]
- Mleczko, L.; Schweer, D.; Durjanova, Z.; Andorf, R.; Baerns, M. Reaction Engineering Approaches to the Oxidative Coupling of Methane to C2+ Hydrocarbons. Stud. Surf Sci. Catal. 1992, 81, 155–164. [Google Scholar]
- Uglietti, R.; Bracconi, M.; Maestri, M. Coupling CFD-DEM and Microkinetic Modeling of Surface Chemistry for the Simulation of Catalytic Fluidized Systems. React. Chem. Eng. 2018, 3, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Cruellas, A.; Melchiori, T.; Gallucci, F.; van Sint Annaland, M. Oxidative Coupling of Methane: A Comparison of Different Reactor Configurations. Energy Technol. 2019, 8, 1900148. [Google Scholar] [CrossRef]
- Pak, S.; Lunsford, J.H. Thermal Effects during the Oxidative Coupling of Methane over Mn/Na2WO4/SiO2 and Mn/Na2WO4/MgO Catalysts. Appl. Catal. A Gen. 1998, 168, 131–137. [Google Scholar] [CrossRef]
- Noon, D.; Zohour, B.; Senkan, S. Oxidative Coupling of Methane with La2O3-CeO2 Nanofiber Fabrics: A Reaction Engineering Study. J. Nat. Gas Sci. Eng. 2014, 18, 406–411. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jeon, W.; Choi, J.W.; Suh, Y.W.; Ha, J.M.; Suh, D.J.; Park, Y.K. Scaled-up Production of C2 Hydrocarbons by the Oxidative Coupling of Methane over Pelletized Na2WO4/Mn/SiO2 Catalysts: Observing Hot Spots for the Selective Process. Fuel 2013, 106, 851–857. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Yang, D.; Gao, R.; Wang, Z.; Yang, J. Scale up and Stability Test for Oxidative Coupling of Methane over Na2WO4-Mn/SiO2 Catalyst in a 200 Ml Fixed-Bed Reactor. J. Nat. Gas Chem. 2008, 17, 59–63. [Google Scholar] [CrossRef]
- Geerts, J.W.M.H.; Chen, Q.; van Kasteren, J.M.N.; van der Wiele, K. Thermodynamics and Kinetic Modeling of the Homogeneous Gas Phase Reactions of the Oxidative Coupling of Methane. Catal. Today 1990, 6, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Farrell, B.L.; Igenegbai, V.O.; Linic, S. A Viewpoint on Direct Methane Conversion to Ethane and Ethylene Using Oxidative Coupling on Solid Catalysts. ACS Catal. 2016, 6, 4340–4346. [Google Scholar] [CrossRef] [Green Version]
- Thybaut, J.W.; Marin, G.B.; Mirodatos, C.; Schuurman, Y.; Van Veen, A.C.; Sadykov, V.A.; Pennemann, H.; Bellinghausen, R.; Mleczko, L. A Novel Technology for Natural Gas Conversion by Means of Integrated Oxidative Coupling and Dry Reforming of Methane. Chem. Ingenieur Tech. 2014, 86, 1855–1870. [Google Scholar] [CrossRef]
- Koretsky, M.D. Engineering and Chemical Thermodynamics, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Su, Y.S.; Ying, J.Y.; Green, W.H. Upper Bound on the Yield for Oxidative Coupling of Methane. J. Catal. 2003, 218, 321–333. [Google Scholar] [CrossRef]
- Scheid, A.J.; Barbosa-Coutinho, E.; Schwaab, M.; Salau, N.P.G. GPREC—Gas-Phase Reaction Equilibrium Calculator. Computer program in Fortran/Matlab, registered in the Brazilian National Institute of Intellectual Property (INPI), under No BR 51 2017 001692 5.
- Barnes, C.; Koretsky, M.D. ThermoSolver; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Machocki, A.; Denis, A. Simultaneous Oxidative Coupling of Methane and Oxidative Dehydrogenation of Ethane on the Na+/CaO Catalyst. Chem. Eng. J. 2002, 90, 165–172. [Google Scholar] [CrossRef]
- Snytnikov, V.N.; Mishchenko, T.I.; Snytnikov, V.N.; Malykhin, S.E.; Avdeev, V.I.; Parmon, V.N. Autocatalytic Gas-Phase Dehydrogenation of Ethane. Res. Chem. Intermed. 2012, 38, 1133–1147. [Google Scholar] [CrossRef]
- Korf, S.J.; Roos, J.A.; De Bruijn, N.A.; Van Ommen, J.G.; Ross, J.R.H. Influence of CO2 on the Oxidative Coupling of Methane over a Lithium Promoted Magnesium Oxide Catalyst. J. Chem. Soc. Chem. Commun. 1987, 19, 1433–1434. [Google Scholar] [CrossRef]
- Thum, L.; Rudolph, M.; Schomäcker, R.; Wang, Y.; Tarasov, A.; Trunschke, A.; Schlögl, R. Oxygen Activation in Oxidative Coupling of Methane on Calcium Oxide. J. Phys. Chem. C 2019, 123, 8018–8026. [Google Scholar] [CrossRef] [Green Version]
- Sarsani, V.S.R.; West, D.; Lengyel, I. Method for Producing Hydrocarbons by Oxidative Coupling of Methane with a Heavy Diluent. U.S. Patent 2017/0057889 A1, 2 March 2017. [Google Scholar]
- Cai, X.; Hu, Y.H. Advances in Catalytic Conversion of Methane and Carbon Dioxide to Highly Valuable Products. Energy Sci. Eng. 2019, 7, 4–29. [Google Scholar] [CrossRef] [Green Version]
- Istadi Amin, N.A.S. Co-Generation of C2 Hydrocarbons and Synthesis Gases from Methane and Carbon Dioxide: A Thermodynamic Analysis. J. Nat. Gas Chem. 2005, 14, 140–150. [Google Scholar]
- Rafique, H.A.; Vuddagiri, S.; Radaelli, G.; Scher, E.C.; McCormick, J.; Cizeron, J. Oxidative Coupling of Methane Implementation for Olefin Production. U.S. Patent 9352295B2, 31 May 2016. [Google Scholar]
- Iwamoto, M.; Lunsford, J.H. Surface Reactions of Oxygen Ions. 5. Oxidation of Alkanes and Alkenes by O2− on MgO. J. Phys. Chem. 1980, 84, 3079–3084. [Google Scholar] [CrossRef]
- Kumar, G.; Lau, S.L.J.; Krcha, M.D.; Janik, M.J. Correlation of Methane Activation and Oxide Catalyst Reducibility and Its Implications for Oxidative Coupling. ACS Catal. 2016, 6, 1812–1821. [Google Scholar] [CrossRef]
- Zavyalova, U.; Holena, M.; Schlögl, R.; Baerns, M. Statistical Analysis of Past Catalytic Data on Oxidative Methane Coupling for New Insights into the Composition of High-Performance Catalysts. ChemCatChem 2011, 3, 1935–1947. [Google Scholar] [CrossRef]
- Gambo, Y.; Jalil, A.A.; Triwahyono, S.; Abdulrasheed, A.A. Recent Advances and Future Prospect in Catalysts for Oxidative Coupling of Methane to Ethylene: A Review. J. Ind. Eng. Chem. 2018, 59, 218–229. [Google Scholar] [CrossRef]
- Schucker, R.C.; Derrickson, K.J.; Ali, A.K.; Caton, N.J. Identification of the Optimum Catalyst and Operating Conditions for Oxidative Coupling of Methane: Activity and Selectivity of Alkaline Earth-Doped Lanthanides. Ind. Eng. Chem. Res. 2020, 59, 18434–18446. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Wang, R. Formation of Hydrogen in Oxidative Coupling of Methane over BaCO3 and MgO Catalysts. J. Nat. Gas Chem. 2008, 17, 238–241. [Google Scholar] [CrossRef]
- Rane, V.H.; Chaudhari, S.T.; Choudhary, V.R. Oxidative Coupling of Methane over La-Promoted CaO Catalysts: Influence of Precursors and Catalyst Preparation Method. J. Nat. Gas Chem. 2010, 19, 25–30. [Google Scholar] [CrossRef]
- Baidya, T.; Van Vegten, N.; Verel, R.; Jiang, Y.; Yulikov, M.; Kohn, T.; Jeschke, G.; Baiker, A. SrO·Al2O3 Mixed Oxides: A Promising Class of Catalysts for Oxidative Coupling of Methane. J. Catal. 2011, 281, 241–253. [Google Scholar] [CrossRef]
- Yunarti, R.T.; Lee, M.; Hwang, Y.J.; Choi, J.W.; Suh, D.J.; Lee, J.; Kim, I.W.; Ha, J.M. Transition Metal-Doped TiO2 Nanowire Catalysts for the Oxidative Coupling of Methane. Catal. Commun. 2014, 50, 54–58. [Google Scholar] [CrossRef]
- Simon, U.; Arndt, S.; Otremba, T.; Schlingmann, T.; Görke, O.; Dinse, K.P.; Schomäcker, R.; Schubert, H. Li/MgO with Spin Sensors as Catalyst for the Oxidative Coupling of Methane. Catal. Commun. 2012, 18, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Scher, E.C.; Zurcher, F.R.; Cizeron, J.M.; Schammel, W.P.; Tkachenko, A.; Karshtedt, D.; Nyce, G. Nanowire Catalysts. U.S. Patent Application WO2011/149996A2, 1 December 2011. [Google Scholar]
- Rane, V.H.; Chaudhari, S.T.; Choudhary, V.R. Influence of Alkali Metal Doping on Surface Properties and Catalytic Activity/Selectivity of CaO Catalysts in Oxidative Coupling of Methane. J. Nat. Gas Chem. 2008, 17, 313–320. [Google Scholar] [CrossRef]
- Arndt, S.; Laugel, G.; Levchenko, S.; Horn, R.; Baerns, M.; Scheffler, M.; Schlögl, R.; Schomäcker, R. A Critical Assessment of Li/MgO-Based Catalysts for the Oxidative Coupling of Methane. Catal. Rev. Sci. Eng. 2011, 53, 424–514. [Google Scholar] [CrossRef]
- Song, J.; Sun, Y.; Ba, R.; Huang, S.; Zhao, Y.; Zhang, J.; Sun, Y.; Zhu, Y. Monodisperse Sr-La2O3 Hybrid Nanofibers for Oxidative Coupling of Methane to Synthesize C2 Hydrocarbons. Nanoscale 2015, 7, 2260–2264. [Google Scholar] [CrossRef] [PubMed]
- Galadima, A.; Muraza, O. Revisiting the Oxidative Coupling of Methane to Ethylene in the Golden Period of Shale Gas: A Review. J. Ind. Eng. Chem. 2016, 37, 1–13. [Google Scholar] [CrossRef]
- He, Y.; Yang, B.; Cheng, G. On the Oxidative Coupling of Methane with Carbon Dioxide over CeO2/ZnO Nanocatalysts. Catal. Today 2004, 98, 595–600. [Google Scholar] [CrossRef]
- Noon, D.; Seubsai, A.; Senkan, S. Oxidative Coupling of Methane by Nanofiber Catalysts. ChemCatChem 2013, 5, 146–149. [Google Scholar] [CrossRef]
- Kolmakov, A.; Moskovits, M. Chemical Sensing and Catalysis By One-Dimensional Metal-Oxide Nanostructures. Annu. Rev. Mater. Res. 2004, 34, 151–180. [Google Scholar] [CrossRef] [Green Version]
- Chinta, S.; Thorman, J.; Butler, J.; Rives, T. Process for the Oxidative Coupling. U.S. Patent 8912381B2, 29 June 2009. [Google Scholar]
- Tanaka, K.; Sekine, Y.; Inoue, J.; Araki, H.; Matsukata, M.; Kikuchi, E. Oxidative Coupling of Methane over Ba-Incorporate LaInO3 Perovskite Catalyst. J. Jpn. Pet. Inst. 2012, 55, 71–72. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, D.V.; Isupova, L.A.; Gerasimov, E.Y.; Dovlitova, L.S.; Glazneva, T.S.; Prosvirin, I.P. Oxidative Methane Coupling over Mg, Al, Ca, Ba, Pb-Promoted SrTiO3 and Sr2TiO4: Influence of Surface Composition and Microstructure. Appl. Catal. A Gen. 2014, 485, 10–19. [Google Scholar] [CrossRef]
- Ghose, R.; Hwang, H.T.; Varma, A. Catalysts for Oxidative Coupling of Methane and Solution Combustion Method for the Production of the Same. U.S. Patent 9610565 B2, 4 April 2017. [Google Scholar]
- Othman, N.H.; Wu, Z.; Li, K. Micro-Structured Bi1.5Y0.3Sm0.2O3-Alpha Catalysts for Oxidative Couplinf of Methane. AIChE J. 2015, 61, 3451–3458. [Google Scholar] [CrossRef]
- Huang, K.; Chen, F.-Q.; Lu, D.-W. Artificial Neural Network-Aided Design of Multi-Component Catalyst for Methane Oxidative Coupling. Appl. Catal. A Gen. 2001, 219, 61–68. [Google Scholar] [CrossRef]
- Huang, K.; Zhan, X.L.; Chen, F.Q.; Lü, D.W. Catalyst Design for Methane Oxidative Coupling by Using Artificial Neural Network and Hybrid Genetic Algorithm. Chem. Eng. Sci. 2003, 58, 81–87. [Google Scholar] [CrossRef]
- Cizeron, J.M.; Scher, E.C.; Zurcher, F.R.; Schammel, W.P.; Nyce, G.; Rumplecker, A.; McCormick, J.; Alcid, M.; Gamoras, J.; Rosenberg, D.; et al. Catalysts for Petrochemical Catalysis. U.S. Patent 2013/0023709A1, 24 January 2013. [Google Scholar]
- Dedov, A.G.; Loktev, A.S.; Nipan, G.D.; Dorokhov, S.N.; Golikov, S.D.; Spesivtsev, N.A.; Moiseev, I.I. Oxidative Coupling of Methane to Form Ethylene: Effect of the Preparation Method on the Phase Composition and Catalytic Properties of Li-W-Mn-O-SiO2 Composite Materials. Pet. Chem. 2015, 55, 163–168. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Chou, L.; Song, H.; Zhao, J.; Yang, J.; Li, S. Effective and Stable CeO2-W-Mn/SiO2 Catalyst for Methane Oxidation to Ethylene and Ethane. Stud. Surf. Sci. Catal. 2007, 167, 237–242. [Google Scholar] [CrossRef]
- Arndt, S.; Otremba, T.; Simon, U.; Yildiz, M.; Schubert, H.; Schomäcker, R. Mn-Na 2WO 4/SiO2 as Catalyst for the Oxidative Coupling of Methane. What Is Really Known? Appl. Catal. A Gen. 2012, 425, 53–61. [Google Scholar] [CrossRef]
- Werny, M.J.; Wang, Y.; Girgsdies, F.; Schlögl, R.; Trunschke, A. Fluctuating Storage of the Active Phase in a Mn-Na2WO4/SiO2 Catalyst for the Oxidative Coupling of Methane. Angew. Chem. Int. Ed. 2020, 59, 14921–14926. [Google Scholar] [CrossRef]
- Liu, W.C.; Ralston, W.T.; Melaet, G.; Somorjai, G.A. Oxidative Coupling of Methane (OCM): Effect of Noble Metal (M = Pt, Ir, Rh) Doping on the Performance of Mesoporous Silica MCF-17 Supported MnxOy-Na2WO4 Catalysts. Appl. Catal. A Gen. 2017, 545, 17–23. [Google Scholar] [CrossRef]
- Gordienko, Y.; Usmanov, T.; Bychkov, V.; Lomonosov, V.; Fattakhova, Z.; Tulenin, Y.; Shashkin, D.; Sinev, M. Oxygen Availability and Catalytic Performance of NaWMn/SiO2 Mixed Oxide and Its Components in Oxidative Coupling of Methane. Catal. Today 2016, 278, 127–134. [Google Scholar] [CrossRef]
- Simon, U.; Görke, O.; Berthold, A.; Arndt, S.; Schomäcker, R.; Schubert, H. Fluidized Bed Processing of Sodium Tungsten Manganese Catalysts for the Oxidative Coupling of Methane. Chem. Eng. J. 2011, 168, 1352–1359. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Nhat, T.T.P.; Takimoto, K.; Thakur, A.; Nishimura, S.; Ohyama, J.; Miyazato, I.; Takahashi, L.; Fujima, J.; Takahashi, K.; et al. High-Throughput Experimentation and Catalyst Informatics for Oxidative Coupling of Methane. ACS Catal. 2020, 10, 921–932. [Google Scholar] [CrossRef]
- Yildiz, M.; Simon, U.; Otremba, T.; Aksu, Y.; Kailasam, K.; Thomas, A.; Schomäcker, R.; Arndt, S. Support Material Variation for the MnxOy-Na2WO4/SiO2 Catalyst. Catal. Today 2014, 228, 5–14. [Google Scholar] [CrossRef]
- Ha, J.M.; Suh, D.J.; Choi, J.W.; Yoon, Y.H.; Yang, G.S.; Jeon, W. Catalyst for Oxidative Coupling of Methane, Method for Preparing the Same, and Method for Oxidative Coupling Reaction of Methane Using the Same. U.S. Patent 2013178680A1, 11 July 2013. [Google Scholar]
- Ji, S.; Xiao, T.; Li, S.; Chou, L.; Zhang, B.; Xu, C.; Hou, R.; York, A.P.E.; Green, M.L.H. Surface WO4 Tetrahedron: The Essence of the Oxidative Coupling of Methane over M-W-Mn/SiO2 Catalysts. J. Catal. 2003, 220, 47–56. [Google Scholar] [CrossRef]
- Park, L.H.; Jo, Y.R.; Choi, J.W.; Suh, D.J.; Song, K.H.; Ha, J.M. SiO2@MnO: X@Na2WO4@SiO2core-Shell-Derived Catalyst for Oxidative Coupling of Methane. RSC Adv. 2020, 10, 37749–37756. [Google Scholar] [CrossRef]
- Shi, J.; Yao, L.; Hu, C. Effect of CO2 on the Structural Variation of Na2WO4/Mn/SiO2 Catalyst for Oxidative Coupling of Methane to Ethylene. J. Energy Chem. 2015, 24, 394–400. [Google Scholar] [CrossRef]
- Yildiz, M.; Aksu, Y.; Simon, U.; Kailasam, K.; Goerke, O.; Rosowski, F.; Schomacker, R.; Thomas, A.; Arndt, S. Enhanced Catalytic Performance of MnxOy–Na2WO4/SiO2 for the Oxidative Coupling of Methane Using an Ordered Mesoporous Silica Support. Chem. Commun. 2014, 50, 14440–14442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakaya, C.; Kee, R.J. Progress in the Direct Catalytic Conversion of Methane to Fuels and Chemicals. Prog. Energy Combust. Sci. 2016, 55, 60–97. [Google Scholar] [CrossRef] [Green Version]
- Thiruvenkataswamy, P.; Eljack, F.T.; Roy, N.; Mannan, M.S.; El-Halwagi, M.M. Safety and Techno-Economic Analysis of Ethylene Technologies. J. Loss Prev. Process Ind. 2016, 39, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.H.; Tan, C.S. A Review: CO2 Utilization. Aerosol Air Qual. Res. 2014, 14, 480–499. [Google Scholar] [CrossRef] [Green Version]
- Song, C. Global Challenges and Strategies for Control, Conversion and Utilization of CO2 for Sustainable Development Involving Energy, Catalysis, Adsorption and Chemical Processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Chauvy, R.; De Weireld, G. CO2 Utilization Technologies in Europe: A Short Review. Energy Technol. 2020, 8, 2000627. [Google Scholar] [CrossRef]
- Hepburn, C.; Adlen, E.; Beddington, J.; Carter, E.A.; Fuss, S.; Mac Dowell, N.; Minx, J.C.; Smith, P.; Williams, C.K. The Technological and Economic Prospects for CO2 Utilization and Removal. Nature 2019, 575, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radaelli, G.; Rafique, H.A.; Vuddagiri, S.; Scher, E.C.; McCormick, J.; Cizeron, J.; Patel, B.; Lakhapatri, S. Efficient Oxidative Coupling of Methane Processes and Systems. U.S. Patent Application 2016/0272557A1, 22 September 2016. [Google Scholar]




































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | No. |
|---|---|
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 |
| Reaction | No. | Reaction | No. |
|---|---|---|---|
| 1 | 18 | ||
| 2 | 19 | ||
| 3 | 20 | ||
| 4 | 21 | ||
| 5 | 22 | ||
| 6 | 23 | ||
| 7 | 24 | ||
| 8 | 25 | ||
| 9 a | 26 | ||
| 10 | 27 | ||
| 11 | 28 | ||
| 12 | 29 | ||
| 13 | 30 | ||
| 14 | 31 | ||
| 15 | 32 | ||
| 16 | 33 | ||
| 17 |
| Reaction | No. | Reaction | No. |
|---|---|---|---|
| 1 | 6 | ||
| 2 | 7 | ||
| 3 | 8 | ||
| 4 | 9 | ||
| 5 | 10 |
| Reaction | Reaction Rate Equation |
|---|---|
| No | Catalytic Reaction | No | Catalytic Reaction |
|---|---|---|---|
| 1 | 14 | ||
| 2 | 15 | ||
| 3 | 16 | ||
| 4 | 17 | ||
| 5 | 18 | ||
| 6 | 19 | ||
| 7 | 20 | ||
| 8 | 21 | ||
| 9 | 22 | ||
| 10 | 23 | ||
| 11 | 24 | ||
| 12 | 25 | ||
| 13 | 26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Ros, S.; Barbalho Fontoura, T.; Schwaab, M.; Castro de Jesus, N.J.; Pinto, J.C. Oxidative Coupling of Methane for Ethylene Production: Reviewing Kinetic Modelling Approaches, Thermodynamics and Catalysts. Processes 2021, 9, 2196. https://doi.org/10.3390/pr9122196
Da Ros S, Barbalho Fontoura T, Schwaab M, Castro de Jesus NJ, Pinto JC. Oxidative Coupling of Methane for Ethylene Production: Reviewing Kinetic Modelling Approaches, Thermodynamics and Catalysts. Processes. 2021; 9(12):2196. https://doi.org/10.3390/pr9122196
Chicago/Turabian StyleDa Ros, Simoní, Tahyná Barbalho Fontoura, Marcio Schwaab, Normando José Castro de Jesus, and José Carlos Pinto. 2021. "Oxidative Coupling of Methane for Ethylene Production: Reviewing Kinetic Modelling Approaches, Thermodynamics and Catalysts" Processes 9, no. 12: 2196. https://doi.org/10.3390/pr9122196
APA StyleDa Ros, S., Barbalho Fontoura, T., Schwaab, M., Castro de Jesus, N. J., & Pinto, J. C. (2021). Oxidative Coupling of Methane for Ethylene Production: Reviewing Kinetic Modelling Approaches, Thermodynamics and Catalysts. Processes, 9(12), 2196. https://doi.org/10.3390/pr9122196