
Continuous Crystallization Using Ultrasound Assisted Nucleation, Cubic Cooling Profiles and Oscillatory Flow

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
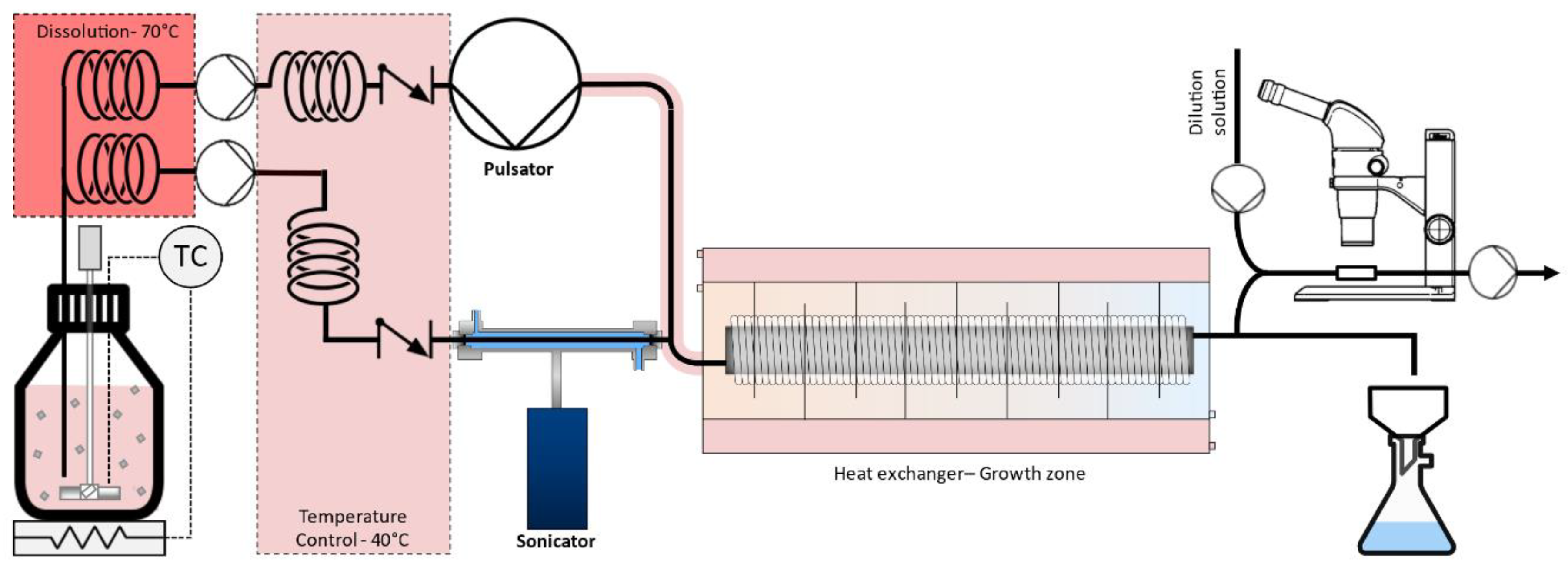
2.2. Experimental Setup
2.3. Experimental Procedure
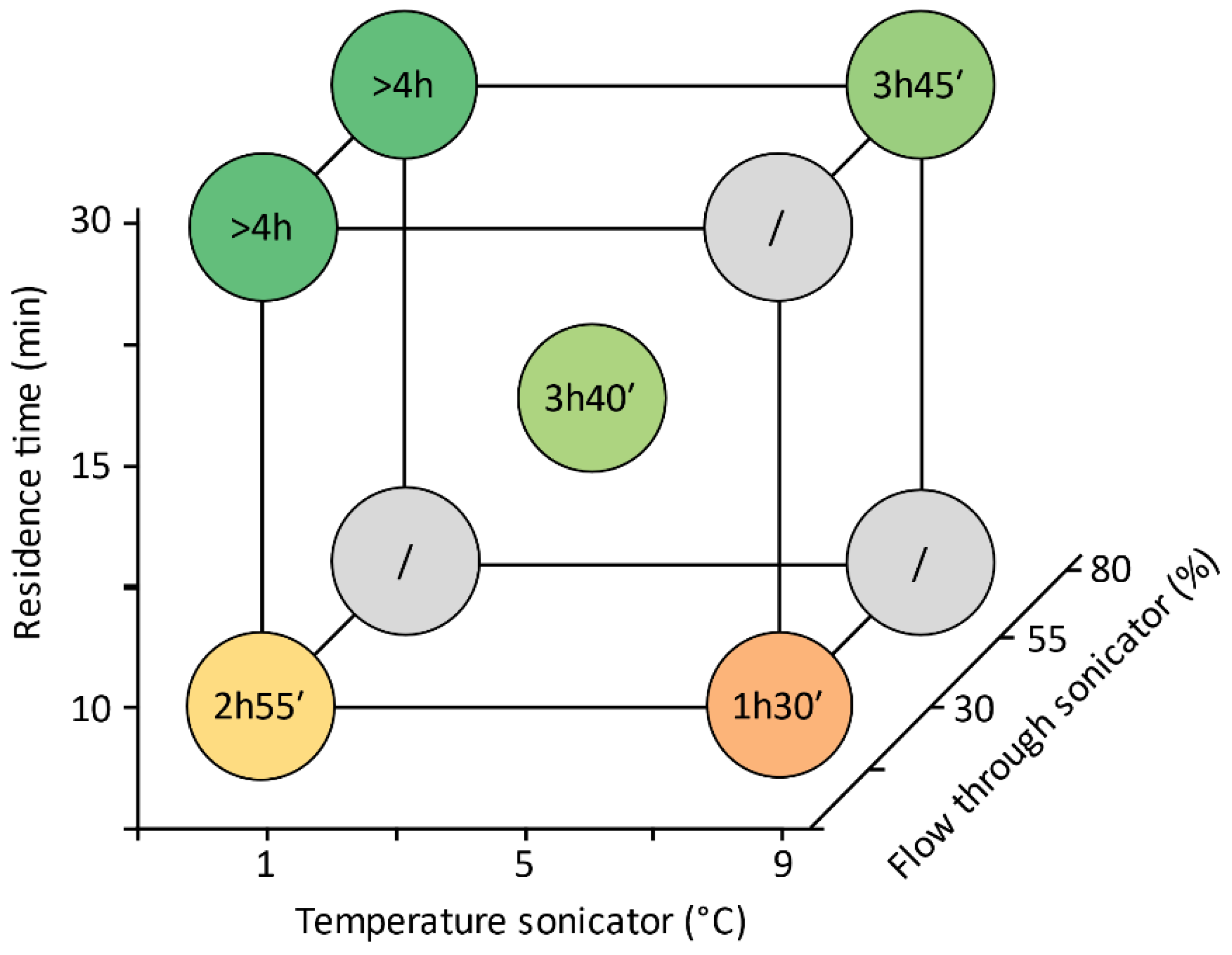
2.4. Experiments
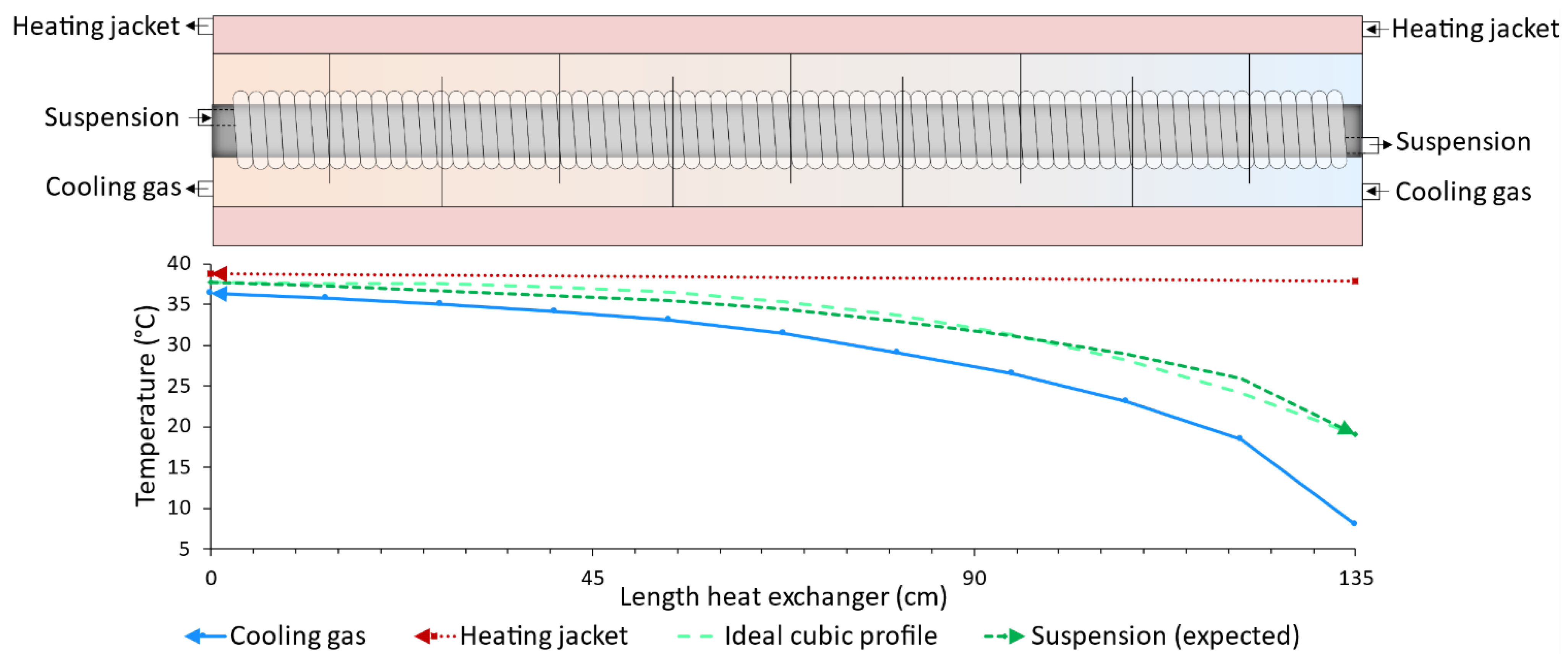
3. Cubic Cooling Profile
4. Results and Discussion
4.1. Operating Times
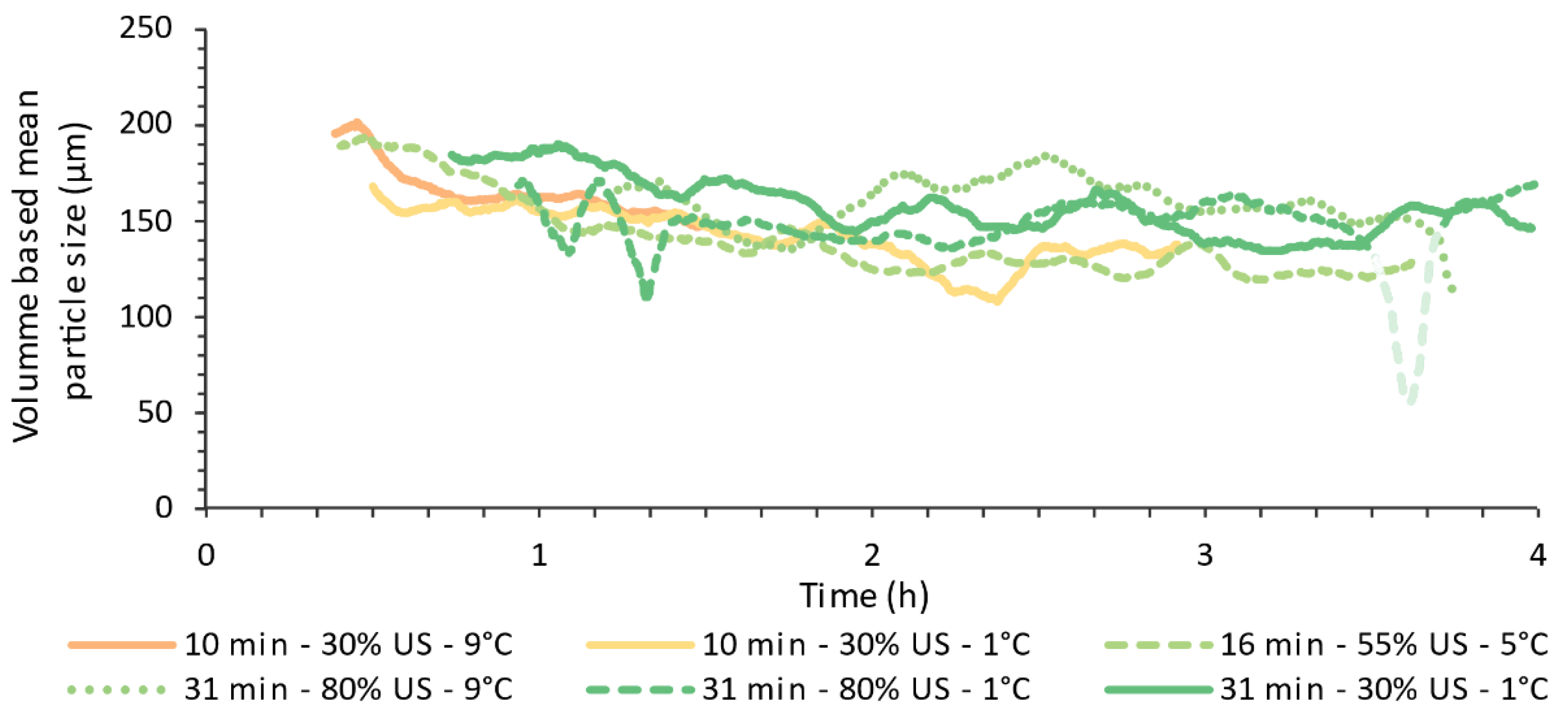
4.2. Online Particle Size Measurements
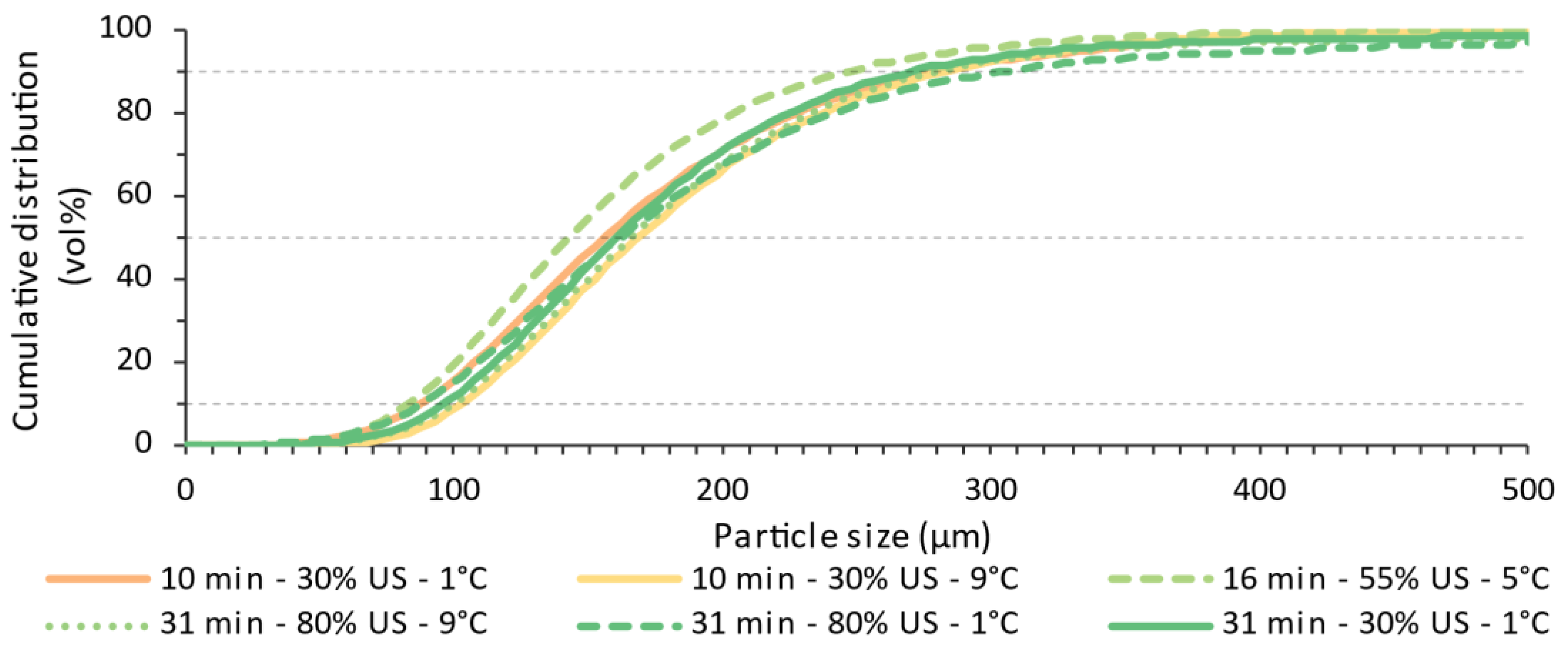
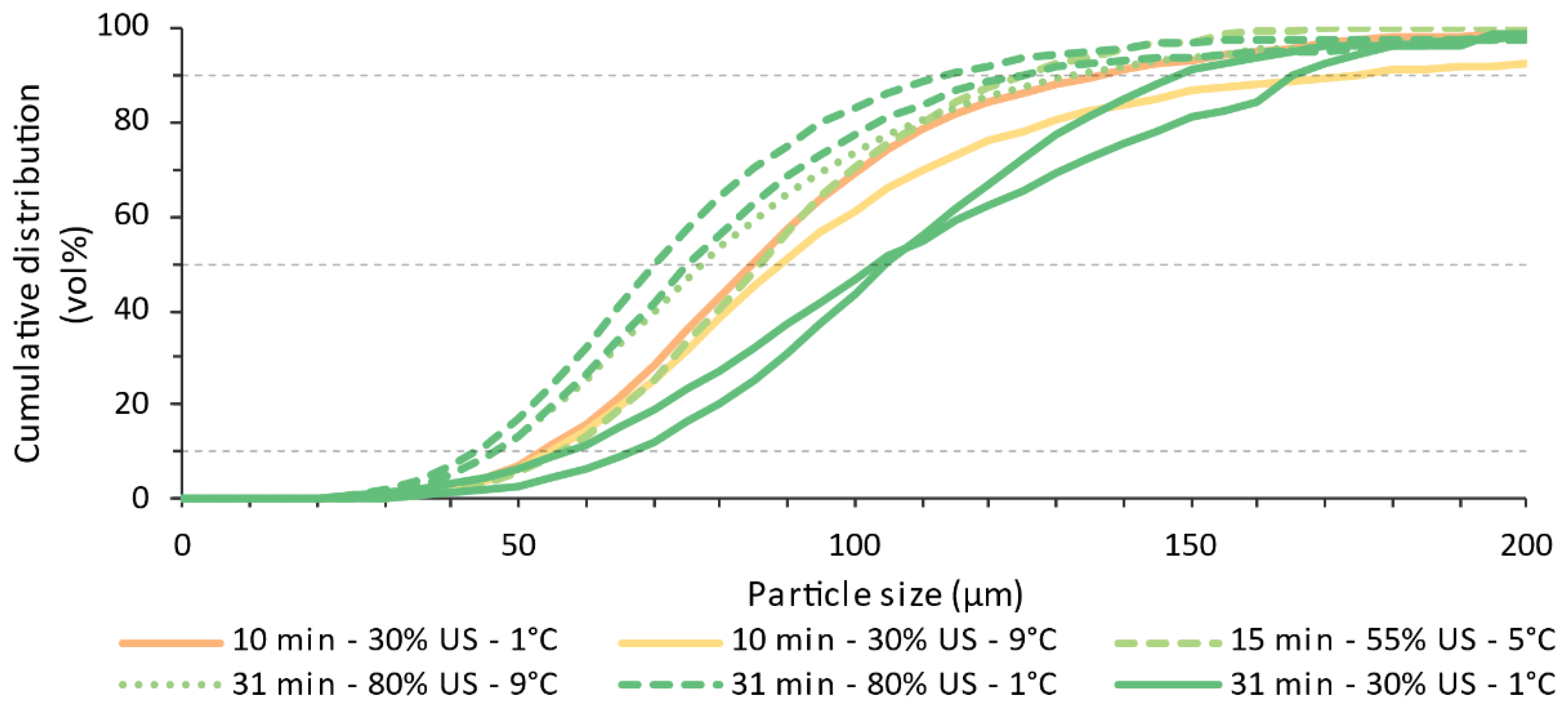
4.3. Offline Particle Sizes Distributions
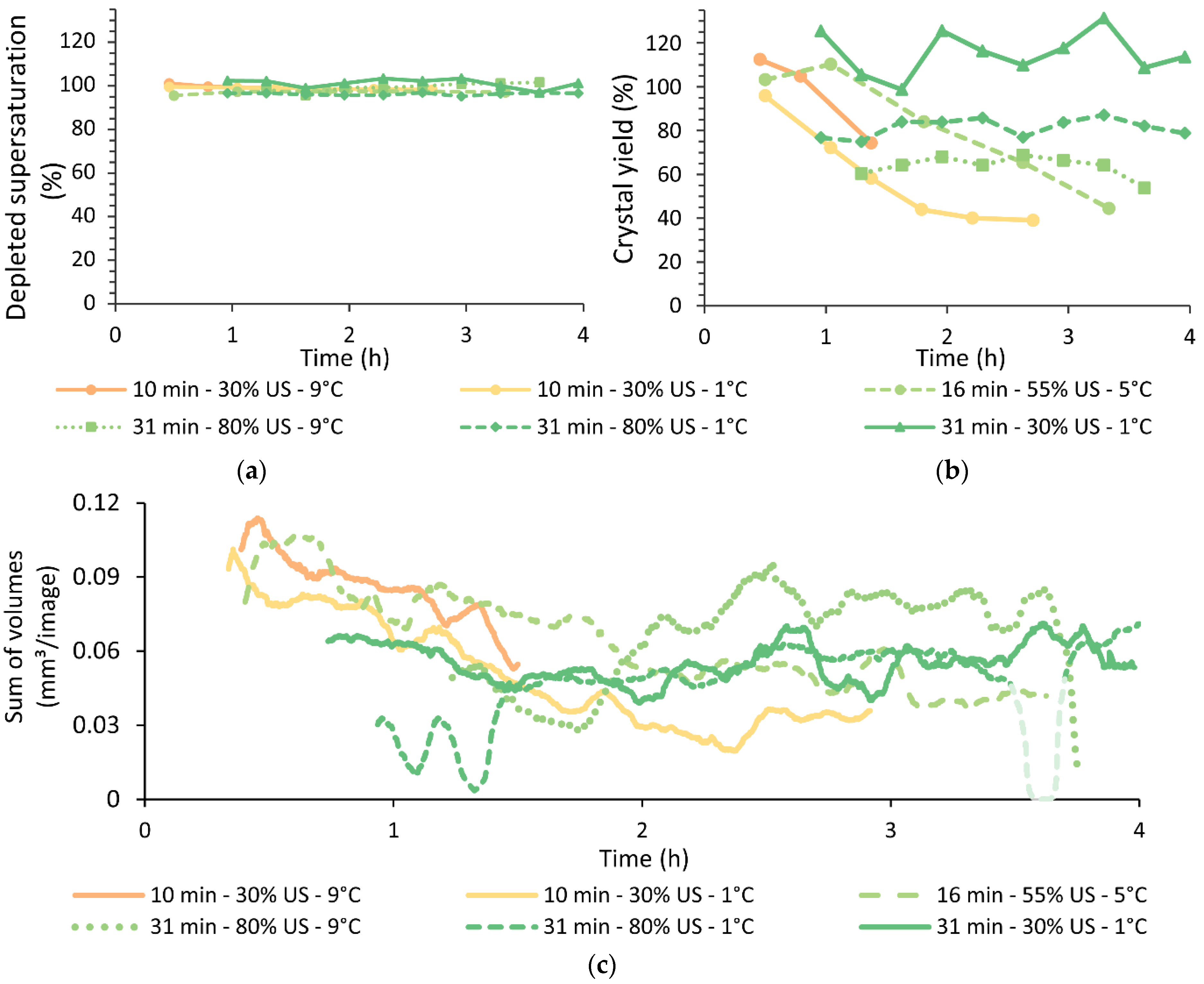
4.4. Yield
4.5. Design Discussion
- A pulsator with a faster discharge stroke and a slower suction stroke: The stroke length of the suction and discharge of the pulsator are currently equal. The speed of the pulsator is currently limited by the suction stroke as it would otherwise cause excessive cavitation that would dampen the discharge stroke. However, a faster and more powerful discharge stroke is expected to reduce fouling and agglomeration and improve mixing.
- Using a single coil for the dissolution step instead of two separate ones for simplicity.
- Splitting up the flow to the sonicator after the sonicator instead of from the beginning so that all flow goes through the pulsator, which will minimize gas buildup through cavitation here.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wood, B.; Girard, K.P.; Polster, C.S.; Croker, D.M. Progress to Date in the Design and Operation of Continuous Crystallization Processes for Pharmaceutical Applications. Org. Process Res. Dev. 2019, 23, 122–144. [Google Scholar] [CrossRef]
- Ferguson, S.; Morris, G.; Hao, H.; Barrett, M.; Glennon, B. Characterization of the anti-solvent batch, plug flow and MSMPR crystallization of benzoic acid. Chem. Eng. Sci. 2013, 104, 44–54. [Google Scholar] [CrossRef]
- Alvarez, A.J.; Myerson, A.S. Continuous plug flow crystallization of pharmaceutical compounds. Cryst. Growth Des. 2010, 10, 2219–2228. [Google Scholar] [CrossRef]
- Eder, R.J.P.; Schrank, S.; Besenhard, M.O.; Roblegg, E.; Gruber-Woelfler, H.; Khinast, J.G. Continuous sonocrystallization of acetylsalicylic acid (ASA): Control of crystal size. Cryst. Growth Des. 2012, 12, 4733–4738. [Google Scholar] [CrossRef]
- Jiang, M.; Papageorgiou, C.D.; Waetzig, J.; Hardy, A.; Langston, M.; Braatz, R.D. Indirect ultrasonication in continuous slug-flow crystallization. Cryst. Growth Des. 2015, 15, 2486–2492. [Google Scholar] [CrossRef]
- Vetter, T.; Burcham, C.L.; Doherty, M.F. Regions of attainable particle sizes in continuous and batch crystallization processes. Chem. Eng. Sci. 2014, 106, 167–180. [Google Scholar] [CrossRef]
- Acevedo, D.; Yang, X.; Liu, Y.C.; O’Connor, T.F.; Koswara, A.; Nagy, Z.K.; Madurawe, R.; Cruz, C.N. Encrustation in Continuous Pharmaceutical Crystallization Processes—A Review. Org. Process Res. Dev. 2019, 23, 1134–1142. [Google Scholar] [CrossRef]
- Furuta, M.; Mukai, K.; Cork, D.; Mae, K. Continuous crystallization using a sonicated tubular system for controlling particle size in an API manufacturing process. Chem. Eng. Process. Process Intensif. 2016, 102, 210–218. [Google Scholar] [CrossRef]
- Ezeanowi, N.; Pajari, H.; Laitinen, A.; Koiranen, T. Monitoring the Dynamics of a Continuous Sonicated Tubular Cooling Crystallizer. Cryst. Growth Des. 2020, 20, 1458–1466. [Google Scholar] [CrossRef]
- Kreimer, M.; Zettl, M.; Aigner, I.; Mannschott, T.; van der Wel, P.; Khinast, J.G.; Krumme, M. Performance characterization of static mixers in precipitating environments. Org. Process Res. Dev. 2019, 23, 1308–1320. [Google Scholar] [CrossRef]
- Rossi, D.; Gavriilidis, A.; Kuhn, S.; Candel, M.A.; Jones, A.G.; Price, C.; Mazzei, L. Adipic acid primary nucleation kinetics from probability distributions in droplet-based systems under stagnant and flow conditions. Cryst. Growth Des. 2015, 15, 1784–1791. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Jamshidi, R.; Saffari, N.; Kuhn, S.; Gavriilidis, A.; Mazzei, L. Continuous-Flow Sonocrystallization in Droplet-Based Microfluidics. Cryst. Growth Des. 2015, 15, 5519–5529. [Google Scholar] [CrossRef]
- Nagasawa, H.; Mae, K. Development of a new microreactor based on annular microsegments for fine particle production. Ind. Eng. Chem. Res. 2006, 45, 2179–2186. [Google Scholar] [CrossRef]
- Termühlen, M.; Etmanski, M.M.; Kryschewski, I.; Kufner, A.C.; Schembecker, G.; Wohlgemuth, K. Continuous slug flow crystallization: Impact of design and operating parameters on product quality. Chem. Eng. Res. Des. 2021, 170, 290–303. [Google Scholar] [CrossRef]
- Vancleef, A.; Seurs, S.; Jordens, J.; Van Gerven, T.; Thomassen, L.C.J.; Braeken, L. Reducing the Induction Time Using Ultrasound and High-Shear Mixing in a Continuous Crystallization Process. Crystals 2018, 8, 326. [Google Scholar] [CrossRef] [Green Version]
- Yazdanpanah, N.; Nagy, Z.K. The Handbook of Continuous Crystallization; The Royal Society of Chemistry: London, UK, 2020; ISBN 9788578110796. [Google Scholar]
- Vancleef, A.; Van Gerven, T.; Thomassen, L.C.J.; Braeken, L. Ultrasound in Continuous Tubular Crystallizers: Parameters affecting the Nucleation Rate. Crystals 2021, 11, 1054. [Google Scholar] [CrossRef]
- Gielen, B. Particle Engineering by Targeted Application of Ultrasound; KU LEUVEN: Leuven, Belgium, 2017. [Google Scholar]
- Mullin, J.W. Crystallization; Elsevier: Amsterdam, The Netherlands, 2001; ISBN 9781626239777. [Google Scholar]
- Han, B.; Ezeanowi, N.C.; Koiranen, T.O.; Häkkinen, A.T.; Louhi-Kultanen, M. Insights into Design Criteria for a Continuous, Sonicated Modular Tubular Cooling Crystallizer. Cryst. Growth Des. 2018, 18, 7286–7295. [Google Scholar] [CrossRef]
- Regensburg, S.I. Cooling Crystallization in an Oscillatory Flow Baffled Crystallizer (OFBC)—Influence of Fluid Dynamics on Crystal Product. Master’s Thesis, Delft University of Technology, Delft, The Netherlands, 2015. [Google Scholar]
- Besenhard, M.O.; Neugebauer, P.; Ho, C.-D.; Khinast, J.G. Crystal Size Control in a Continuous Tubular Crystallizer. Cryst. Growth Des. 2015, 15, 1683–1691. [Google Scholar] [CrossRef]
- Doyle, B.J.; Gutmann, B.; Bittel, M.; Hubler, T.; Macchi, A.; Roberge, D.M. Handling of Solids and Flow Characterization in a Baffleless Oscillatory Flow Coil Reactor. Ind. Eng. Chem. Res. 2020, 59, 4007–4019. [Google Scholar] [CrossRef]
- Gaivoronskii, A.N.; Granzhan, V.A. PHYSICOCHEMICAL STUDIES OF SYSTEMS AND PROCESSES Solubility of Adipic Acid in Organic Solvents and Water. Russ. J. Appl. Chem. 2005, 78, 404–408. [Google Scholar] [CrossRef]
- Vancleef, A.; Maes, D.; Van Gerven, T.; Thomassen, L.C.J.; Braeken, L. Flow-through microscopy and image analysis for crystallization processes. Chem. Eng. Sci. 2021, 248, 117067. [Google Scholar] [CrossRef]
- Vancleef, A.; Vanhemel, J.; Van Gerven, T.; Thomassen, L.; Braeken, L. Anti-Fouling Strategies for Tubular Flow Crystallizers. In Proceedings of the Second International Process Intensification Conference, Leuven, Belgium, 27–29 May 2019. [Google Scholar]
- Nichols, G.; Byard, S.; Bloxham, M.J.; Botterill, J.; Dawson, N.J.; Dennis, A.; Diart, V.; North, N.C.; Sherwood, J.D. A review of the terms agglomerate and aggregate with a recommendation for nomenclature used in powder and particle characterization. J. Pharm. Sci. 2002, 91, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Gielen, B.; Jordens, J.; Thomassen, L.C.J.; Braeken, L.; Van Gerven, T. Agglomeration control during ultrasonic crystallization of an active pharmaceutical ingredient. Crystals 2017, 7, 40. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vancleef, A.; De Beuckeleer, W.; Van Gerven, T.; Thomassen, L.C.J.; Braeken, L. Continuous Crystallization Using Ultrasound Assisted Nucleation, Cubic Cooling Profiles and Oscillatory Flow. Processes 2021, 9, 2268. https://doi.org/10.3390/pr9122268
Vancleef A, De Beuckeleer W, Van Gerven T, Thomassen LCJ, Braeken L. Continuous Crystallization Using Ultrasound Assisted Nucleation, Cubic Cooling Profiles and Oscillatory Flow. Processes. 2021; 9(12):2268. https://doi.org/10.3390/pr9122268
Chicago/Turabian StyleVancleef, Arne, Ward De Beuckeleer, Tom Van Gerven, Leen C. J. Thomassen, and Leen Braeken. 2021. "Continuous Crystallization Using Ultrasound Assisted Nucleation, Cubic Cooling Profiles and Oscillatory Flow" Processes 9, no. 12: 2268. https://doi.org/10.3390/pr9122268
APA StyleVancleef, A., De Beuckeleer, W., Van Gerven, T., Thomassen, L. C. J., & Braeken, L. (2021). Continuous Crystallization Using Ultrasound Assisted Nucleation, Cubic Cooling Profiles and Oscillatory Flow. Processes, 9(12), 2268. https://doi.org/10.3390/pr9122268