
Design of a Bench-Scale Tubular Reactor Similar to Plug Flow Reactor for Gas-Phase Kinetic Data Generation-Illustration with the Pyrolysis of Octanoic Acid


 , ,
, ,
Abstract
:Highlights
- Small sized tubular reactor appropriately designed to produce gas phase kinetic data.
- Reason: axial dispersion coefficient much smaller in gas phase than in liquid phase.
- Helpful tool for research dedicated to thermal conversion of liquid biomass.
1. Introduction
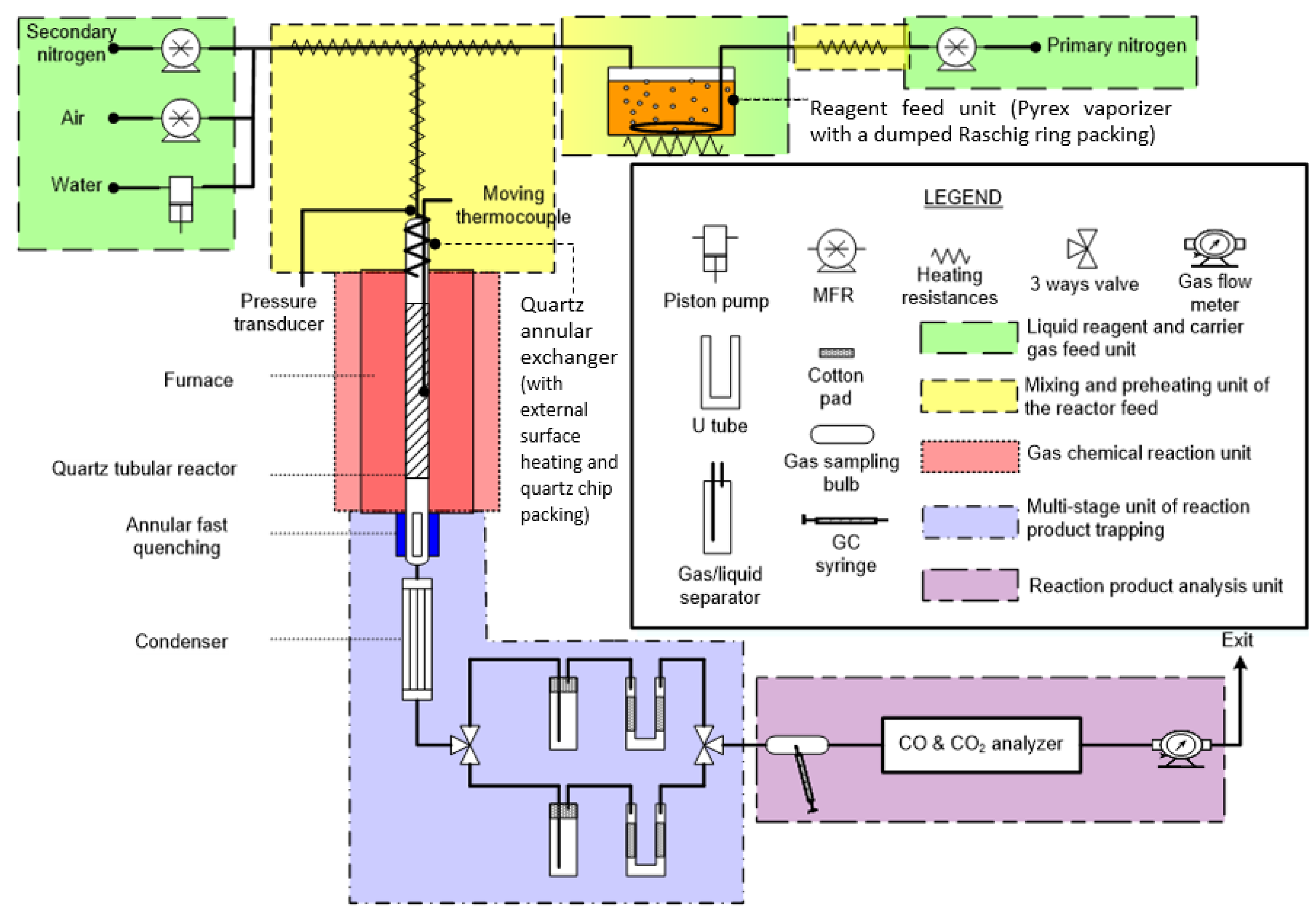
2. Experimental Setup Description
2.1. Liquid Reagent and Carrier Gas Feed Unit
2.2. Mixing and Preheating Unit of the Reactor Feed
2.3. Gas Phase Chemical Reaction Unit
2.4. Multi-Stage Unit of Reaction Product Trapping: Fast Quenching, Condensation, and Gas-Liquid Separation
2.5. Reaction Product Analysis Unit, Procedure, and Material Balances
3. Design of the Tubular Reactor and Fast Quenching System
3.1. Reactor Design
3.1.1. Equations Related to the Reactor Sizing
3.1.2. Calculation Procedure and Numerical Solving Related to the Reactor Sizing
- (1)
- Choice of an initial value for Pe. The choice was selected since that condition induces a real reactor flow sufficiently close to plug flow (as mentioned previously).
- (2)
- Selection of operating conditions typically used in pyrolysis process (key variables: , , and ) and first evaluation of the reactor length L. These conditions, together with the estimation of the property required for parameter-L calculation (Equation (10)), i.e., the molecular diffusion coefficient for the binary system [octanoic acid (reagent)–nitrogen (carrier gas)], are listed in Table 2. Details of the estimation method used for parameter- are given in Appendix C [16,17,18,19]. Solving Equation (9) led to the physical meaning solution that was rounded to because of the dimensions of the available furnaces.
- (3)
- Given the L rounded value obtained in step 2, calculation of the reactor internal diameter d (Equation (7)). Here, again, the value obtained for parameter-d () was rounded to the nearest inferior value corresponding to a standard diameter ().
- (4)
- Checking that the Pe criterion () is still satisfied in spite of the rounded values adopted for L and d parameters. Calculation of the mean real velocity for the gas reacting fluid and of the axial dispersion coefficient (u and , respectively, Table 2) led to , allowing to conclude that the flow inside the actual reactor would be close to plug flow under these process operating conditions.
- (5)
- Checking the applicability range of the correlation used (i.e., Equations (5b) and (5c)). Values calculated for Re and the terms involved in Equation (5c) (Table 2) helped to verify that this last step of L and d calculation procedure was achieved successfully.
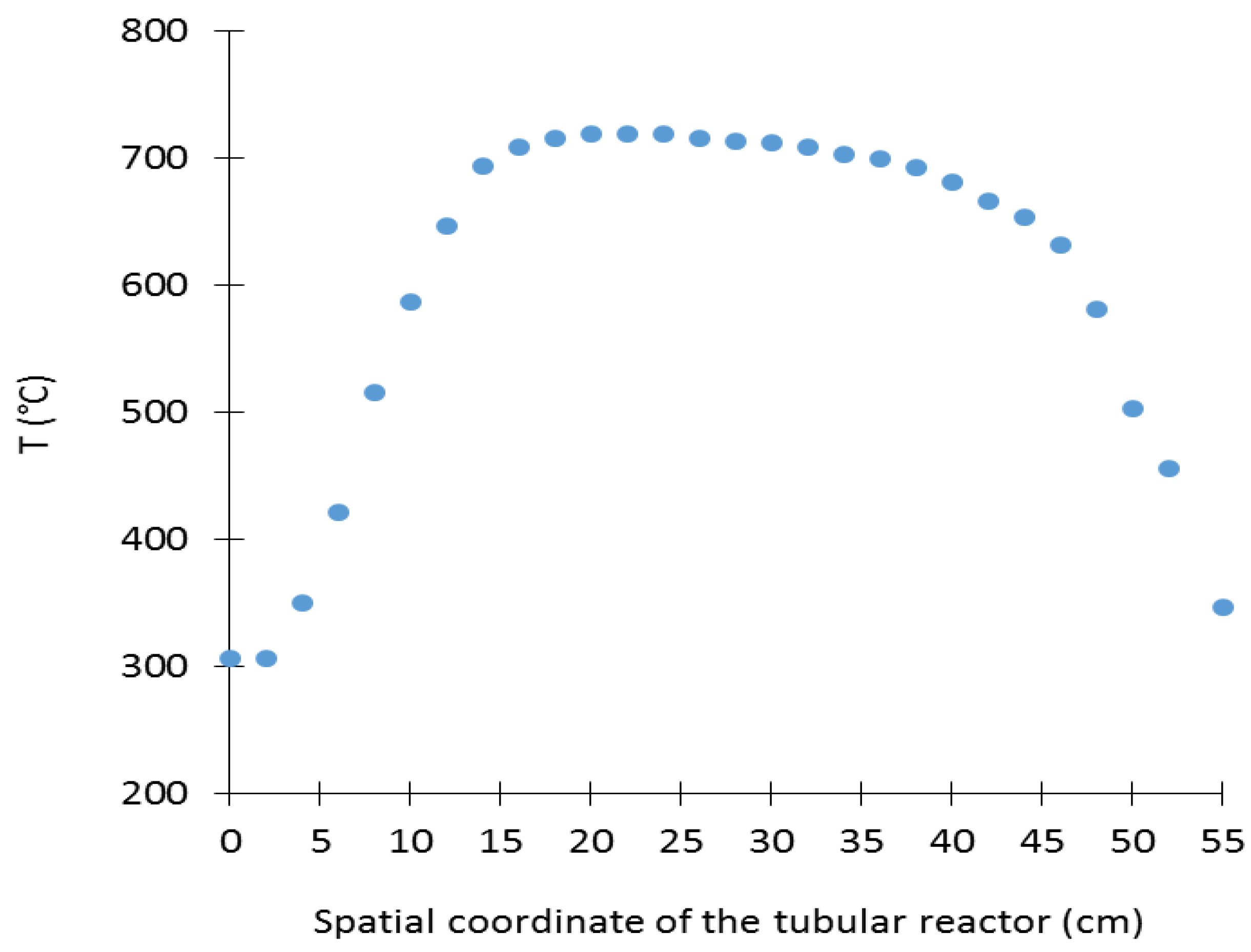
3.1.3. Checking the Validity of the Obtained Reactor Sizing for the Whole Experiments Planed in the Work
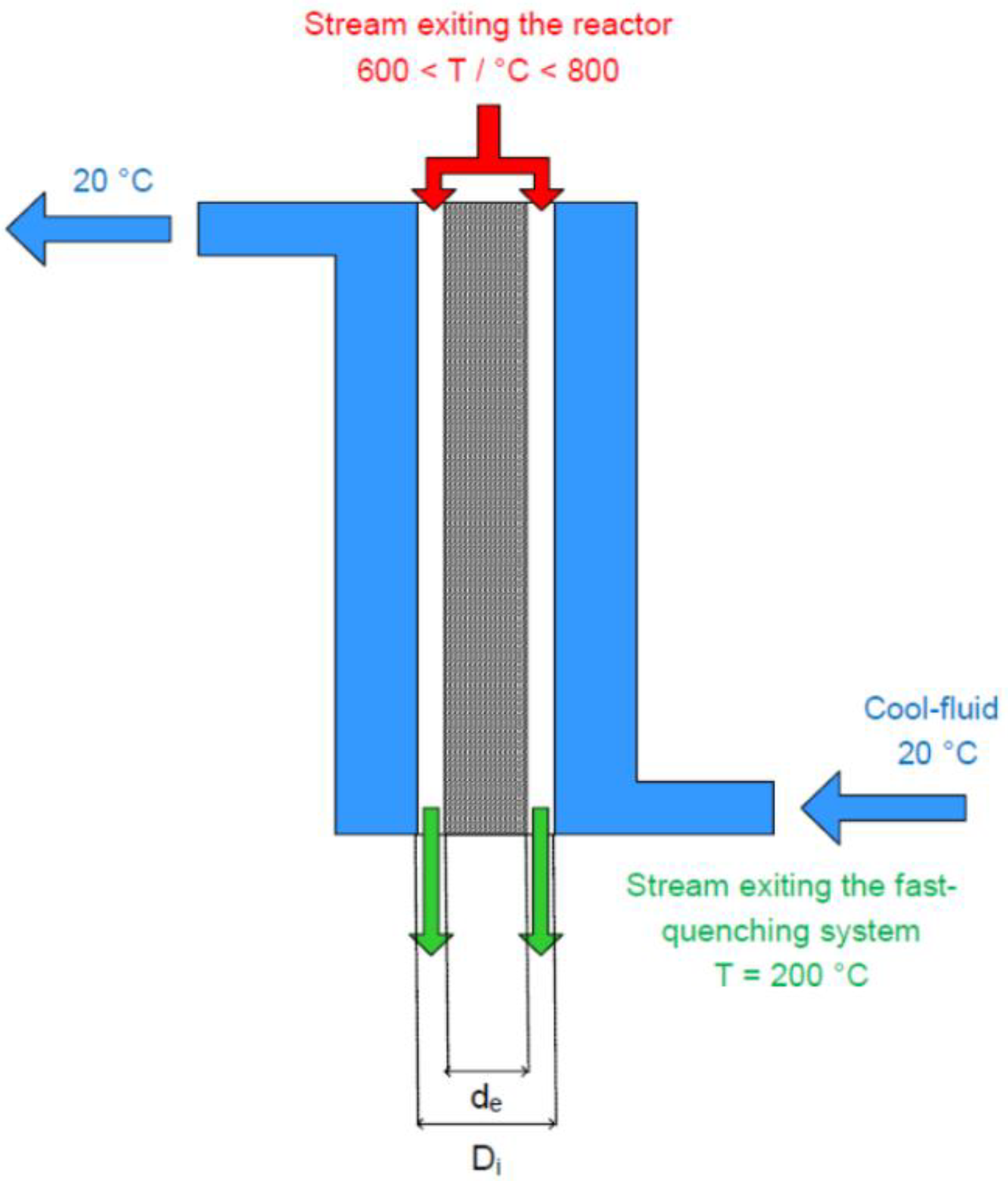
3.2. Fast Quenching System Design
3.2.1. Equations Related to the Fast-Quenching System Sizing
3.2.2. Calculation Procedure and Numerical Solving Related to the Fast-Quenching System Sizing
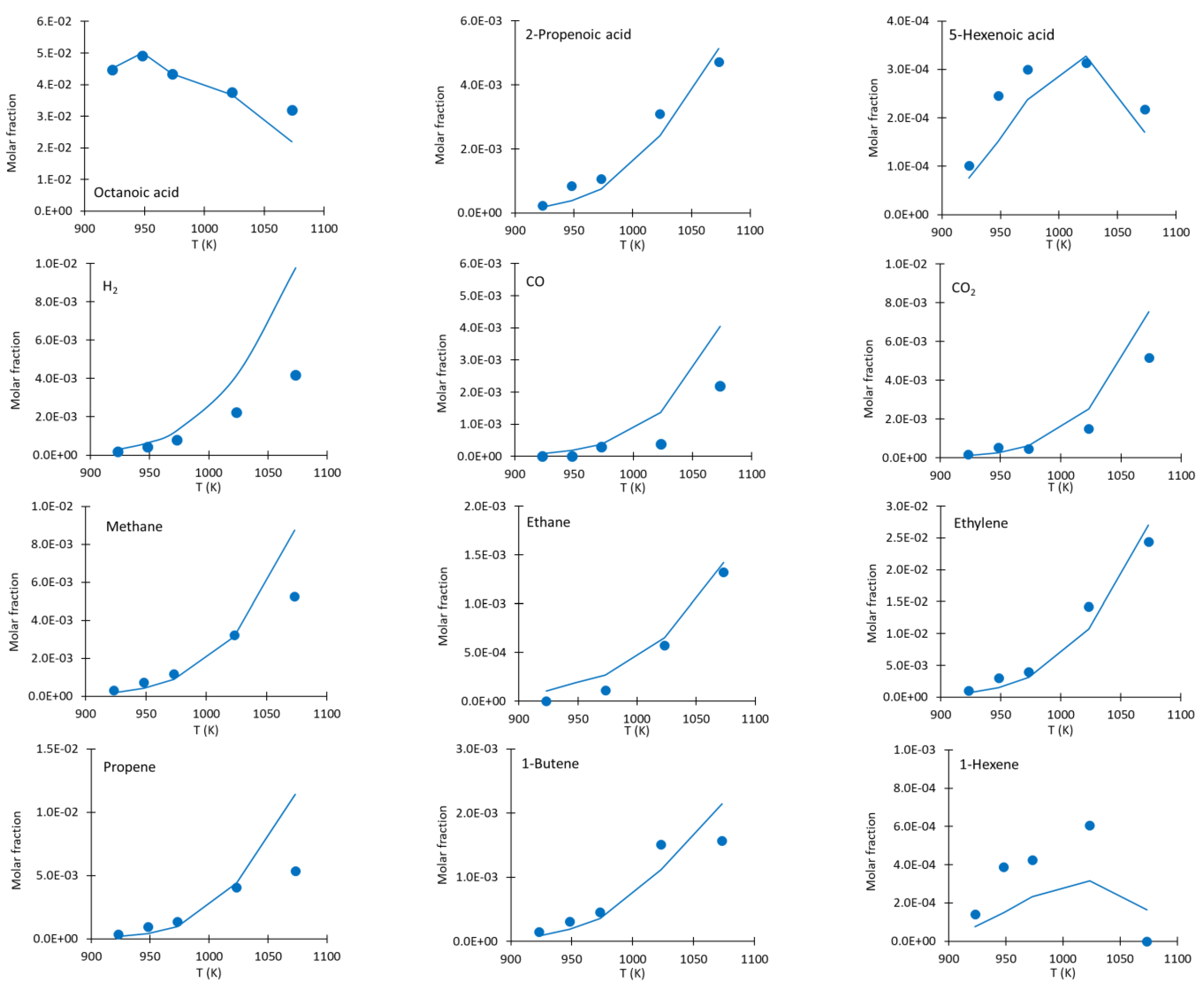
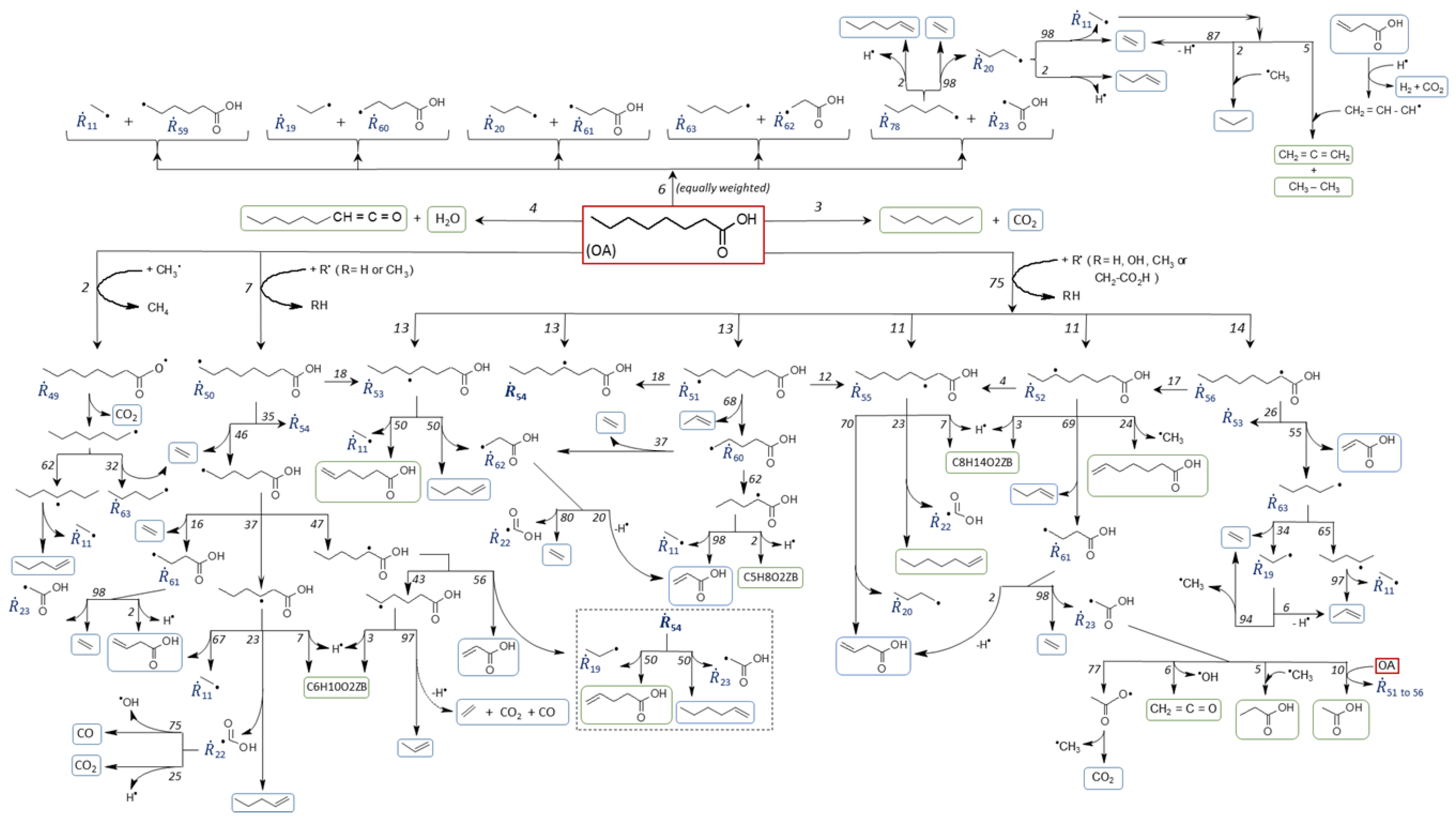
4. Sample of Kinetic Data Generated with the Designed Bench-Scale Setup
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Nomenclature
| Chromatographic area of internal standard E | |
| Chromatographic area of component i | |
| Inlet octanoic acid molar flowrate | |
| Inlet molar flowrate of the considered element | |
| Inlet nitrogen molar flowrate | |
| Inlet molar flowrate of the primary carrier gas feed (for partial vaporization of the liquid reagent) | |
| Inlet molar flowrate of the secondary carrier gas feed (for individual control of τ and reagent dilution) | |
| Inlet total molar flowrate | |
| Outlet molar flowrate of the considered element | |
| Molar flowrate of the outlet condensable component i | |
| Molar flowrate of product j encountered both in the gas phase and in the organic liquid phase | |
| Molar flowrate of the off-line detected incondensable product j in the outlet gas stream | |
| Molar flowrate of the on-line detected incondensable product j in the outlet gas stream | |
| Carbon response factor of component i compared with internal standard E for quantification by GC-FID analysis | |
| Inlet mass of octanoic acid collected at equally spaced time intervals | |
| Mass of coke deposited during each experiment | |
| Mass of internal standard E | |
| Mass of the outlet condensable component i | |
| Total mass of the detected incondensable products taken as a whole in the outlet gas stream | |
| Mass of the on-line detected incondensable product j in the outlet gas stream | |
| Mass of the off-line detected incondensable product j in the outlet gas stream | |
| Mass of the outlet organic liquid phase product | |
| Molecular weight of component j | |
| Molecular weight of the outlet condensable component i | |
| Molecular weight of the off-line detected incondensable product j present in the outlet gas stream | |
| Molecular weight of the on-line detected incondensable product j present in the outlet gas stream | |
| Pressure related to normal temperature and pressure conditions | |
| Pressure at the inlet of the reactor (assumed constant until the outlet) | |
| Inlet volume flowrate related to nitrogen at and | |
| Total inlet volume flowrate at and for a molar fraction in reactant A (octanoic acid here) | |
| R | Ideal gas constant ( ) |
| Temperature related to normal temperature and pressure conditions | |
| Average reactor temperature | |
| Reactor volume | |
| Molar fraction related to the outlet condensable component i | |
| Mass fraction related to the outlet condensable component i | |
| Molar fraction of component j in the gas phase | |
| Mass fraction of component j in the gas phase | |
| Molar fraction of the off-line detected incondensable product j in the outlet gas stream | |
| Mass fraction of the on-line detected incondensable product j present in the outlet gas stream | |
| Greek symbols | |
| Time intervals during which reactant A (octanoic acid) is collected in order to determine its molar flowrate () | |
| Duration of experiment | |
| Stoichiometric coefficient of “element” in the chemical formula of component k | |
| Stoichiometric coefficient of “element” in the chemical formula of octanoic acid | |
| Volume fraction of the on-line detected incondensable product j in the outlet gas stream | |
| Residence time of the feed | |
Appendix A. Reaction Product Analysis Description
Appendix A.1. Gaseous Products
Appendix A.2. Liquid Products
Appendix A.3. Coke

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | H2 | Non-Condensable Gas (Excluding H2, CO, and CO2) | Condensate a (Excluding Octanoic Acid) | Condensate a (Exclusively Octanoic Acid) |
|---|---|---|---|---|
| GC-type | Intersmat IGC 11-type | Schimadzu GC 17A | Schimadzu GC 17A | Stang ST 200 |
| Detector | TCD | FID | FID | FID |
| Detector temperature/°C | - | 310 | 310 | 280 |
| Injector temperature/°C | - | 300 | 300 | 270 |
| Carrier gas (N2) volume flowrate (NmL/min) or linear velocity (cm/s) | 30 NmL/min | 15 cm/s | 15 cm/s | 0.91 NmL/min |
| Split ratio | - | 1/100 | 1/100 | 1/160 |
| Column | Silica-gel filled column/molecular sieve (5Å thickness) 5 m length, 6 mm inner diameter | Bonded non-polar (methyl silicone) capillary column (P.O.N.A.-type, Hewlet Packard) 50 m, 0.21 mm, 0.5 μm film thickness | Bonded non-polar (methyl silicone) capillary column (P.O.N.A.-type, Hewlet Packard) 50 m, 0.21 mm, 0.5 μm film thickness | Polyethyleneglycol capillary column (CP-Wax 52CB, Chrompack) 50 m, 0.32 mm, 0.2 μm film thickness |
| Oven temperature program | 60 °C | 60 °C (4 min), 60–180 °C (10 °C/min, 10 min), 180–300 °C (10 °C/min) | 60 °C (20 min), 60–300 °C (2 °C/min, 40 min | 60 °C (10 min), 60–240 °C (10 °C/min, 32 min) |
| Column pressure program | - | 96 kPa (4 min), 96–153 kPa (2.4 kPa/min, 10 min), 153–182 kPa (2.4 kPa/min) | 124 kPa (20 min), 124–182 kPa (0.5 kPa/min, 44 min) | - |
Appendix B. Designed Bench-Scale Setup Procedure and Material Balances
Appendix B.1. Operating Procedure
Appendix B.2. Material Balances
| Equations Related to the Inlet of the Reactor | |
| • For the Reagent Liquid Feed (Octanoic Acid) | |
| (A6) | |
| • For the Carrier Gas Feed (Nitrogen) | |
| (A7) | |
| • Residence Time of the Feed | |
| (A8) | |
| (A9) | |
| (A10) | |
| Equations Related to the Outlet of the Reactor b | |
| • Partial Molar Flowrate for Organic Liquid Phase (OLP) Components | |
| -Weight and Molar Fractions | |
| (A11) | |
| (A12) | |
| • Intermediate Normalization of the Detected Incondensable Components (Gaseous Products) | |
| where j is either an on-line or an off-line quantified product. | (A13) |
| For an on-line quantified product: | |
| For an off-line quantified product: | (A14) |
| • Total Mass of All Incondensable Products | |
| (A15) | |
| • Partial Molar Flowrate for the On-line Detected Incondensable Products (H2, CO, and CO2) | |
| (A16) | |
| (A17) | |
| • Partial Molar Flowrate for the Off-line Detected Incondensable Products (Other Gases than H2, CO, and CO2) | |
| (A18) | |
| (A19) | |
| Partial Molar Flowrate for Components Encountered both in the Gas Phase and in the Organic Liquid Phase | |
| (A20) | |
| Final Normalization of all Detected Component Molar Fractions as a Whole | |
| (A21) | |
| for on-line detected gaseous products, | (A22) |
| for off-line detected gaseous products, | (A23) |
| for the organic liquid phase products, | (A24) |
| ), | (A25) |
| given by Equation (A20) for components encountered both in the organic liquid phase and in the gas phase. | |
| Carbon-Hydrogen-Oxygen Element Mole Balance Equations (%) | |
| (A26) | |
| , and element = C, H, or O. | |
Appendix C. Estimation Method for the Molecular Diffusion Coefficient –Application to Low Pressure Binary Gas-Phase Diffusion
References
- Ganley, J.C. A homogeneous chemical reactor analysis and design laboratory: The reaction kinetics of dye and bleach. Educ. Chem. Eng. 2015, 12, 20–26. [Google Scholar] [CrossRef]
- Kee, R.J.; Rupley, F.M.; Miller, J.A. Chemkin II. A Fortran Chemical Kinetics Package for the Analysis of a Gas-Phase Chemical Kinetics; Sandia Laboratories Report, SAND 89-8009B; Sandia National Lab.: Livermor, CA, USA, 1993. [Google Scholar]
- Warth, V.; Stef, N.; Glaude, P.A.; Battin-Leclerc, F.; Scacchi, G.; Côme, G.M. Computer-Aided Derivation of Gas-Phase Oxidation Mechanisms: Application to the Modeling of the Oxidation of n-Butane. Combust. Flame 1998, 114, 81. [Google Scholar] [CrossRef]
- Glaude, P.A.; Battin-Leclerc, F.; Judenherc, B.; Warth, V.; Fournet, R.; Côme, G.M.; Scacchi, G.; Dagaut, P.; Cathonnet, M. Experimental and Modelling Study of the Gas-Phase Oxidation of Methyl and Ethyl-tert-Butyl Ethers. Combust. Flame 2000, 121, 345–355. [Google Scholar] [CrossRef]
- Buda, F.; Bounaceur, R.; Warth, V.; Glaude, P.A.; Fournet, R.; Battin-Leclerc, F. Progress Toward a Unified Detailed Kinetic Model for the Autoignition of Alkanes from C4 to C10 Between 600 and 1200 K. Combust. Flame 2005, 142, 170–186. [Google Scholar] [CrossRef]
- Vanhove, G.; Minetti, R.; Touchard, S.; Fournet, R.; Glaude, P.A.; Battin-Leclerc, F. Experimental and Modelling Study of the Autoignition of 1-Hexene / Iso-octane Mixtures at Low Temperature. Combust. Flame 2006, 145, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.T.; Berkowitz, M.; Oehlschlaeger, M.A.; Biet, J.; Warth, V.; Glaude, P.A.; Battin-Leclerc, F. An Experimental and Kinetic Modelling Study of the Oxidation of the Four Isomers of Butanol. J. Phys. Chem. A 2008, 112, 10843–10855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gornay, J.; Coniglio, L.; Billaud, F.; Wild, G. Octanoic Acid Pyrolysis in a Stainless-Steel Tube: What is the Role of the Coke Formed on the Wall? J. Anal. Appl. Pyrolysis 2010, 87, 78–84. [Google Scholar] [CrossRef]
- Gornay, J.; Coniglio, L.; Billaud, F.; Wild, G. Steam Cracking and Steam Reforming of Waste Cooking Oil in a Tubular Stainless-Steel Reactor with Wall Effects. Energy Fuels 2009, 23, 5663–5676. [Google Scholar] [CrossRef]
- Coniglio, L.; Bennadji, H.; Glaude, P.A.; Herbinet, O.; Billaud, F. Combustion Chemical Kinetics of Biodiesel and Related Compounds (Methyl and Ethyl Esters): Experiments and Modeling Advances and Future Refinements. Prog. Energy Combust. Sci. 2013, 39, 340–382. [Google Scholar] [CrossRef] [Green Version]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Pyrolysis of Wood/Biomass for Bio-oil: A Critical Review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Ferrando, N.; Gedik, I.; Lachet, V.; Pigeon, L.; Lugo, R. Prediction of Phase Equilibrium and Hydration Free Energy of Carboxylic Acids by Monte Carlo Simulations. J. Phys. Chem. B 2013, 117, 7123–7132. [Google Scholar] [CrossRef] [PubMed]
- Gornay, J.; Glaude, P.A.; Billaud, F.; Coniglio, L. Experiments and Modeling of Octanoic Acid Pyrolysis in a Plug Flow Reactor. J. Anal. Appl. Pyrolysis 2020, 146, 104767. [Google Scholar] [CrossRef] [Green Version]
- Gollakota, A.R.K.; Kishore, N.; Gu, S. A Review on Hydrothermal Liquefaction of Biomass. Renew. Sustain. Energy Rev. 2018, 81, 138–1392. [Google Scholar] [CrossRef]
- Levenspiel, O. Chemical Reaction Engineering, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1999; ISBN 0-471-25424-X. [Google Scholar]
- ProII SimSci, 2018 Schneider Electric Software; Version 10.1.2; Schneider Electric: Paris, France, 2018.
- Wilke, C.R.; Lee, C.Y. Estimation of diffusion coefficients for gases and vapors. Ind. Eng. Chem. 1955, 47, 1253–1257. [Google Scholar] [CrossRef]
- Le Bas, G. The Molecular Volumes of Liquid Chemical Compounds; Longmans, Green: New York, NY, USA, 1915. [Google Scholar]
- Bruce, E.P.; Prausnitz, J.M.; O’Connell, J.P. The Properties of Gases and Liquids, 5th ed.; McGraw-Hill: New-York, NY, USA, 2001; ISBN 0-07-011682-2. [Google Scholar]






| Units | Features | Variable | Value/m |
|---|---|---|---|
| Carrier gas and reagent feed | Pyrex vaporizer with a dumped Raschig ring packing | ||
| di | 0.14 | ||
| hpacking | 0.1 | ||
| Mixing and preheating of the reactor feed | Quartz annular exchanger with external surface heating and quartz chip packing | ||
| L | 0.2 | ||
| Di | 0.03 | ||
| De | 0.01 | ||
| hpacking | 0.03 | ||
| Reactor | Quartz tube | L | 0.55 |
| di | 0.008 | ||
| Fast quenching | Quartz annular exchanger with external surface cooling | ||
| L | 0.1 | ||
| Di | 0.008 | ||
| de | 0.006 |
| Selected Pyrolysis Operating Conditions | |
|---|---|
| 1000 | |
| 1.067 | |
| 0.05 | |
| 0.36 | |
| Estimation of the Thermophysical Properties Related to the Reacting Fluid | |
| b | 5.644 |
| /(kg·m−3) c | 0.488 |
| c | |
| Regime Variables | |
| 1.657 | |
| Pe | 56 |
| Re | 179 |
| Reactor Sizing | |
| L/m | 0.49 rounded to 0.55 |
| d/m | rounded to |
| Correlation Used | |
| Re | 179 < 2300 (laminar flow) |
| L/d | 68.75 |
| 7.05 (Equation (5c) satisfied) | |
| Parameters | Numerical Value |
|---|---|
| Cooling Operating Conditions and Technical Parameters | |
| 600 | |
| 200 | |
| 400 | |
| 20 | |
| 341.86 | |
| 0.008 | |
| 0.006 | |
| 0.001 | |
| 0.002 | |
| Regime Variables | |
| 2.8028 | |
| Re | 89.320 |
| Pr | 0.70380 |
| Thermophysical Properties of the Warm Gas Fluid a | |
| 1.2500 | |
| 0.5070 | |
| 1091.3 | |
| 0.04937 | |
| Iteration Rank k | Gz | Nu | ||
|---|---|---|---|---|
| 1 | 5.0 | 25.145 | 7.0797 | 9.1 |
| 2 | 9.1 | 13.816 | 6.4627 | 10.0 |
| 3 | 10.0 | 12.573 | 6.3839 | 10.0 |
| Operating Conditions | R1 | R2 | Mean | SD | CV (%) |
|---|---|---|---|---|---|
| Furnace set point temperature (°C) | 700 | 700 | 700 | 0.0 | 0.0 |
| Average reactor temperature (°C) | 601 | 601 | 601 | 0.0 | 0.0 |
| Reactor pressure (Torr) | 860 | 862 | 861 | 1.4 | 0.2 |
| Residence time (ms) | 422 | 423 | 423 | 0.7 | 0.2 |
| Pe number | 46 | 46 | 46 | 0.0 | 0.0 |
| Conversion of reactant (%) a | 7.8 | 9.2 | 8.5 | 0.9 | 8.5 |
| Material balance in C atom a | −0.4 | −0.5 | −0.5 | 0.1 | 0.2 |
| Material balance in H atom a | −0.3 | −0.3 | −0.3 | 0.0 | 0.0 |
| Material balance in O atom a | 1.4 | 1.7 | 1.6 | 0.2 | 12.5 |
| Chemical species | Molar fractions (solvent-free) % | ||||
| Methane (CH4) | 2.13 | 2.40 | 2.27 | 0.14 | 6.1 |
| Ethylene (C2H4) | 7.21 | 8.23 | 7.72 | 0.51 | 6.6 |
| Propene (C3H6) | 2.48 | 2.43 | 2.46 | 0.02 | 1.0 |
| 1-Butene (C4H8) | 0.82 | 0.87 | 0.85 | 0.03 | 3.0 |
| 1-Pentene (C5H10) | 0.54 | 0.53 | 0.54 | 0.01 | 1.0 |
| 1-Hexene (C6H12) | 0.77 | 0.78 | 0.78 | 0.01 | 0.8 |
| 1-Heptene (C7H14) | 0.27 | 0.25 | 0.26 | 0.01 | 4.9 |
| Acetic acid (CH3COOH) | 0.74 | 0.80 | 0.77 | 0.03 | 3.9 |
| 2-Propenoic acid (C2H3COOH) | 1.92 | 2.32 | 2.12 | 0.20 | 9.7 |
| 4-Pentenoic acid (C4H7COOH) | 0.41 | 0.44 | 0.43 | 0.01 | 3.5 |
| 5-Hexenoic acid (C5H9COOH) | 0.54 | 0.59 | 0.57 | 0.02 | 3.7 |
| 6-Heptenoic acid (C6H11COOH) | 0.23 | 0.24 | 0.24 | 0.01 | 2.7 |
| Octanoic acid (C7H15COOH) | 78.42 | 75.14 | 76.78 | 1.64 | 2.1 |
| Hydrogen (H2) | 1.42 | 1.68 | 1.55 | 0.13 | 8.3 |
| Carbon monoxide (CO) | 0.53 | 0.00 | 0.27 | 0.26 | 100.0 |
| Carbon dioxide (CO2) | 0.84 | 1.03 | 0.94 | 0.09 | 10.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gornay, J.; Plasari, E.; Portha, J.-F.; Glaude, P.-A.; Billaud, F.; Coniglio, L. Design of a Bench-Scale Tubular Reactor Similar to Plug Flow Reactor for Gas-Phase Kinetic Data Generation-Illustration with the Pyrolysis of Octanoic Acid. Processes 2021, 9, 2270. https://doi.org/10.3390/pr9122270
Gornay J, Plasari E, Portha J-F, Glaude P-A, Billaud F, Coniglio L. Design of a Bench-Scale Tubular Reactor Similar to Plug Flow Reactor for Gas-Phase Kinetic Data Generation-Illustration with the Pyrolysis of Octanoic Acid. Processes. 2021; 9(12):2270. https://doi.org/10.3390/pr9122270
Chicago/Turabian StyleGornay, Julien, Edouard Plasari, Jean-François Portha, Pierre-Alexandre Glaude, Francis Billaud, and Lucie Coniglio. 2021. "Design of a Bench-Scale Tubular Reactor Similar to Plug Flow Reactor for Gas-Phase Kinetic Data Generation-Illustration with the Pyrolysis of Octanoic Acid" Processes 9, no. 12: 2270. https://doi.org/10.3390/pr9122270
APA StyleGornay, J., Plasari, E., Portha, J. -F., Glaude, P. -A., Billaud, F., & Coniglio, L. (2021). Design of a Bench-Scale Tubular Reactor Similar to Plug Flow Reactor for Gas-Phase Kinetic Data Generation-Illustration with the Pyrolysis of Octanoic Acid. Processes, 9(12), 2270. https://doi.org/10.3390/pr9122270