
Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices


Abstract
:
1. Introduction
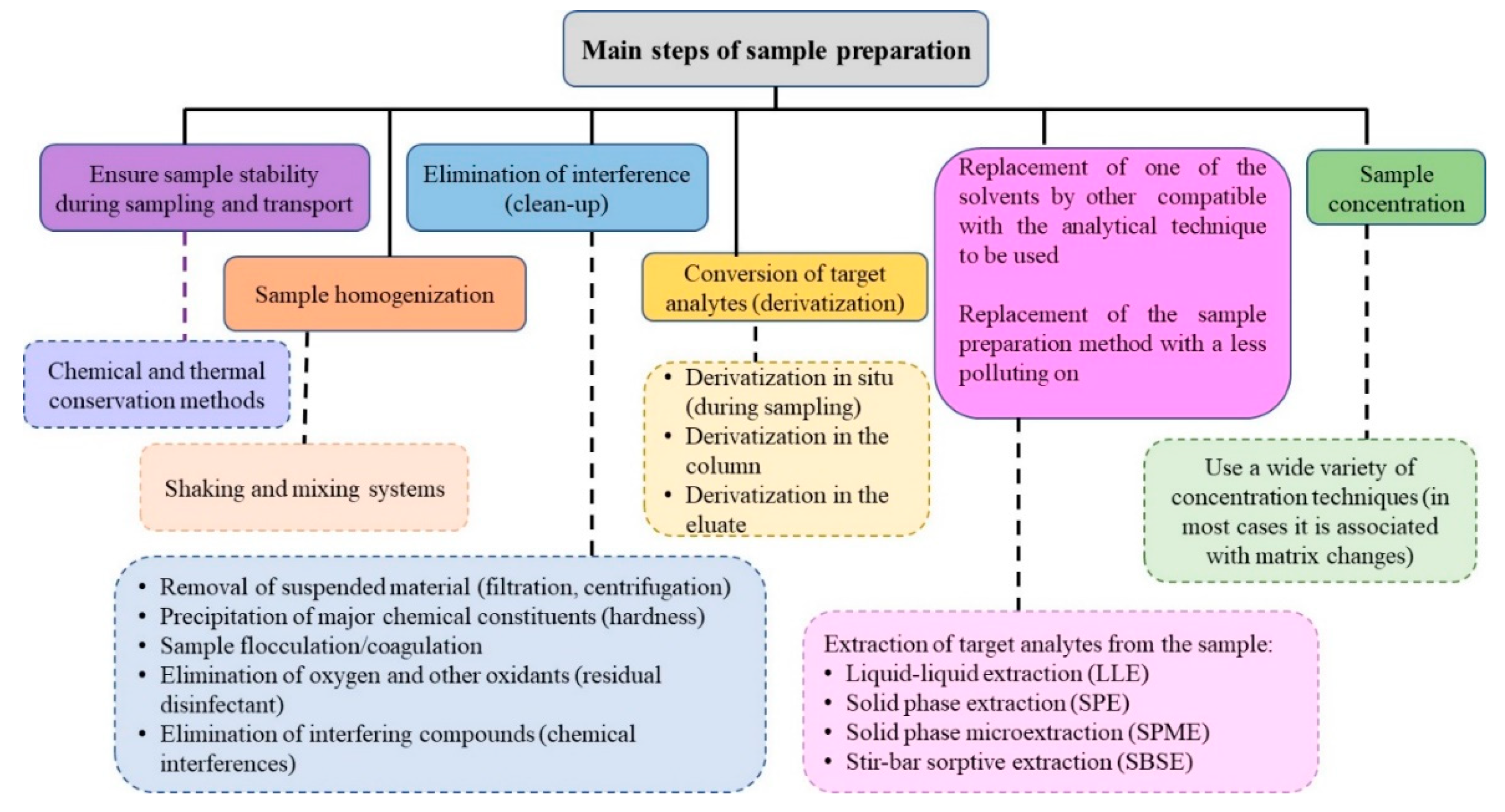
2. Sample Preparation Methods
2.1. Solid-Phase Extraction
2.1.1. Dispersive Solid-Phase Extraction
2.1.2. Magnetic Solid-Phase Extraction
2.1.3. Molecularly Imprinted Polymers
2.2. Sorbent-Based Microextraction
3. Analytical Instrumentation
3.1. Chromatography in Water Analysis
3.2. Mass Spectrometry: Interfaces and Analysers
3.3. High-Resolution Mass Spectrometry (HRMS)

{kind=link}
{kind=link}
| Matrix | Nº PhACs Therapeutical Classes | Extraction Configuration/Sample Volume | Phase System | Analytical Instrumentation | Stationary Phase/Column | Cal./R2 | Precision | Recovery | LOD/LOQ (ng/L) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Surface water | 1 PhACs (stimulant/caffeine) | LLE 1000 mL | --- | LC–APCI–MS | Luna HPLC (150 × 4.6 mm, 5 μm) | ISC 0.9997 | RSDr 8.3% RSDR 8.3% | 89% | LODs 4 | [188] |
| Surface waters (river and lakes) | 9 PhACs (lipid regulators, NSAIDs) | SPE | SDB-XC Empore disk | GC/MS after derivatization | DB5-MS | ESC | --- | Rel Rec 21–152% | --- | [21] |
| Wastewater and surface water | 10 PhACs (analgesic, lipid regulators, NSAIDs) | SPE WWE: 500 mL Surface water: 1000 mL | C18 Bondelut, (200 mg) Oasis HLB (60 mg) | GC-MS, after derivatization with MSTFA | HP5/MS (30 m × 0.2 5 mm × 0.25 μm) | ESC 0.9916–0.9999 | RSDr 5–13% | 53–94% (Oasis HLB) | --- | [215] |
| Wastewaters, surface waters, marine water and drinking waters | 18PhACs (antidepressant, antiepileptic, β-blockers, lipid regulators, NSAIDs, stimulants) | SPE | Oasis MCX (60 mg) | GC-MS, after derivatization with MSTFA | HP5/MS (30 m × 0.2 5 mm × 0.25 μm) | ISC | RSDr <20% | Rel Recoveries 54–120% | LODs WWE. 3.2–28.6 MW: 1.2–2.6 SW: 0.3–2.5 DW:0.1–1.5 | [216] |
| Tap water | 6 PhACs (NSAIDs) | On-line SPE 100 mL Off-line SPE 500 mL | LiChrospher RP-18e (500 mg) LiChrolut RP-18 (500 mg) | HPLC-DAD-MS | LiChrospher RP-18 (250 × 4.6 mm, 5 μm) | ESC 0.9992–0.9995 | RSDr On line: 0.2–6.4% Off-line: 3.3–10.4% | On line 96–104% Off-line 96.5–101.7 | On line LODS: 3.5–94 Off-line LODS: 20–950 | [217] |
| Drinking and Aquaculture Water | 6 PhACs Antibiotics | On-line SPE 25 mL | MIP | HPLC-DAD-FLD | Mediterranea Sea C18 (250 × 4.6 mm, 3 μm) | MMC >0.998 | RSDr 2–5% RSDR 2–6% | 62–102% | LODs DW: 1–11 AW: 1–12 | [110] |
| Surface water | 10 PhACs (analgesic, β-Blockers, Corticosteroids, NSAIDs) | SPE 1000 mL | Oasis HLB (500 mg) | HPLC-DAD-FLD | Purospher STAR C18e (125 × 3 mm, 5 μm) | ESC 0.9987–0.9999 | RSDr SW: 1.2–11.1% | Rec SW: 62–105% | LOQs SW: 6.5–3145 | [60] |
| Surface water and wastewater | 12 PhACs (antiepileptic, lipid regulators, NSAIDs, steroid hormones) | SPE 1000 mL | Strata X polymeric | HPLC-UV-FLD | Alltima C18 (250 × 4.6 mm, 5 μm) | ESC 0.9781–0.9980 | RSDr SW: 4–13% WWI: 7–15% WWE: 5–16% | SW: 86–104% WWI: 62–92% WWE: 65–100% | LOQs SW: 10–800 WWI: 30–1100 WWE: 10–850 | [177] |
| Wastewater | 65 PhACs (analgesic, antiepileptic, antibiotics, β-blockers, lipid regulators, natural hormones, NSAIDs, stimulants) | SPE 200 mL (acidic PhACs) 500 mL (neutral PhACs) | OASIS HLB (60 mg) (acidic PhACs) RP-C18ec cartridges (500 mg) (neutral PhACs) | HPLC-DAD-MS(ESI+) | Purospher® STAR RP-18 endcapped (5 µm) LiChroCART® 250-4 | ESC | RSDr, RSDR 1%–12% | Rel Rec WWI 64–104% | LOQs 4–95 | [23] |
| Surface water | 23 PhACs (analgesic/antipyretic, antibiotics, antiepileptic, antipsychotic, β-blockers, glucocorticoids, hormones, H2 receptor antagonist, lipid regulators, NSAIDs, stimulant) | SPE 1000 mL | SDB-disks | LC-UV/VIS-(ESI)-MS | Restek C18, (150 × 4.6 mm, 5 μm) | MMC >0.99 | RSDr 2.1–11.3% RSDR 5.9–21.5% | 46.8–92.1% | LOQs 11.1–354 | [176] |
| Surface water | 4 PhACs (estrogenic hormones) | SPE 2 L | C18 C18 + Florisil C18 + Florisil + NH2 | LC–ESI-MS | CAPCELL PAK C18 UG 120 (250 × 2.0 mm, 5 μm) | ---- | --- | 72–81% | LOQs 0.1–0.2 | [106] |
| Surface and wastewater water | 15 PhACs (analgetics, angiotensin converting enzyme inhibitors, angiotensin receptor antagonists, calcium antagonists, β-blockers, antidepressants, anticonvulsants, platelet antiaggregants, and cholesterol lowering agentes) | SPE 50 mL | Oasis Max | HPLC-MS/MS | Kinetex C18 (100 × 2.1 mm, 2.6 µm) | SAC >0.99 | <15% | 40–110% | LOQs 0.5–25 | [218] |
| Wastewater | 6 PhACs (analgesic, antiepileptic, antidepressants, stimulant) | SPE 100 mL | Oasis HLB (500 mg) | LC-APCI-MS/MS | Genesis C18 (150 × 3 mm, 4 μm) | ISC | RSDr WWI: 1–8% WWE:2–11% | Abs Rec WWI 79–161% WWE 75–178% Rel Rec WWI 84– 109% WWE 76–136% | --- | [183] |
| Surface, groundwater and drinking water | 8 PhACs (natural hormones, contraceptive hormones) | SPE 500 mL | Oasis HLB (200 mg) | HPLC-MS/MS | --- | ESC 0.9963–0.9998 | RSDr GW: 3.8–10% SW: 2.3–14% DW: 4.1–9.3% | GW: 41–77% SW: 40–95% DW: 54–82% | MDLs 0.69–11 | [59] |
| Surface and wastewater | 39 PhACs (corticosteroids) | SPE SW, WWE: 1000 mL WWI: 500 mL | In-house SPE Env + (150 mg), Strata-X-CW (100 mg), Strata-X-AW (100 mg) and Oasis HLB (200 mg) | HPLC-MS/MS | Poroshell 120 EC18 (100 ×2.1 mm, 2.7 μm) | --- | --- | 36–100% | LODs 0.5– 8 | [108] |
| Surface water and wastewater | 90 PhACs (analgesics, antidiabetic, antipropulsive, psychiatric drugs, antihistamine, anti-parkinson, antibiotics, anti-ulcer, antihypertensives, diuretics, antidiabetic, contraceptives, hormon therapy, anti-cancer) | SPE 100 mL | Oasis HLB (500 mg) | HPLC-MS/MS | Fully endcapped C18 Hypersil GOLD aQ (50 × 2.1 mm, 5 μm) GOLD Phenyl (50 × 2.1 mm, 3 μm) Porous grafite Hypercarb (50 × 2.1 mm, 5 μm) | ISC >0.980 | RSDr SW: 1–56% WWE: 2.7–50% | Rel Rec SW: 5–132% WWE: 35–246% | LOQs SW: 0.06–196 WWE: 0.07–78 | [46] |
| Surface and wastewater | 50 PhACs (antibiotics) | SPE SW: 1000 mL WWE: 500 mL WWI: 200 mL | Oasis HLB (500 mg) | HPLC-(ESI)-MS/MS | Eclipse Plus-C18 (100 mm × 2.1 mm, 1.8 μm) | ISC >0.995 | RSDr 0.63–9.67% RSDR 2.74–21.3% | SW: 49–292% WWE: 61–188% WWI: 32–446% | MQLs SW: 0.63–4.43 WWE: 1.42–9.52 WWI: 2.35–20 | [219] |
| Groundwater and surface water | 11 PhACs (antibiotics) | SPE 1000 mL | Oasis HLB (60 mg) | LC-(ESI)-MS/MS | Luna C8 (100 × 4.6-mm, 3 μm) | --- | --- | 84 - 130% | --- | [90] |
| Wastewater | 11 PhACs (analgesics, antibiotics, anxiolytics and hipnotics, lipid regulators, NSAIDs) | SPE WWI: 50 mL WWE: 100 mL | Oasis MAX (500 mg) | LC-(ESI)-MS/MS | Pursuit UPS C18 (2.1 × 50 mm, 2.4 µm) | ESC 0.9926–0.9992 | RSDr WWI: 7.2%–23% WWE: 5.9%–13% | Rel Rec WWI: 65.2–75.2% WWE: 64.1–88.2% | MQLs WWE: 1.4–200 WWI: 1.4–204 | [26] |
| Wastewater | 23 PhACs (antibiotics, antidepressant, antiepileptic, antiulcer, antihypertensives, lipid regulators, NSAIDs, stimulant) | SPE 250 mL | Oasis HLB (500 mg) | LC-MS/MS | SunFireTM C18 (100 × 3.0 mm, 3.5 μm) | --- | --- | --- | --- | [32] |
| Wastewater | 23 PhACs (analgesic, antibiotics, Antidepressives, diuretics, antipyretic, antiulcer, anxiolytics, cardiotonics, lipid regulators, NSAIDs) | SPE WWI: 50 mL WWE: 100 mL | LC sílica (500 mg) LC Florisil (1 g) GCB (1 g) LC SAX (500 mg) LC-NH2, WAX (500 mg) Oasis MAX (500 mg) | HPLC–(ESI)–MS/MS | Pursuit UPS C18 (50 × 2.1 mm, 2.4 µm) | ISC 0.9823–0.9988 | MAX cartridges RDSr WWI: 5%-17% WWE: 3%-18% RDSR WWI: 4%–15% WWE: 6%–26% | MAX cartridges LSL WWI:34–72% WWE: 3–90% MSL WWI:12–95% WWE: 12–129% HSL WWI:10–95% WWE: 9–176% | MAX cartridges MQLS WWI:3–19 WWE: 3–208 | [27] |
| Wastewater | 17 PhACs (analgesics, β-blockers, lipid regulators, NSAIDs, H2 histamine receptor antagonista, illicit drugs) | SPE WWE: 50 mL WWI: 25 mL | SCX (200 mg) Novel in-house SPE sorbents 2 Different sorbents: A and B | LC–(ESI)-MS/MS | Ascentis®Express C18 with Fused-Core technology (100 × 4.6 mm, 2.4 µm) | ESC ≥0.996 | RSDr and RSDR 16–23% | Sorbent A WWE: 19–106% WWI: 31–107% Sorbent B WWE: 43–98% WWI: 24–97% | Sorbent B LOQs WWE: 5–10 WWI: 10–20 | [220] |
| Wastewater, groundwater, and surface water | 8 PhACs (antibiotics, antiepileptic, β-blockers) | SPE WWI, WWE, SW, and GW: 100, 250, 500 and 1000 mL | Oasis HLB (60 mg) | HPLC-(ESI)-MS/MS | Zorbax XDB-C18 (50 × 2.1 mm, 5 μm) | ESC >0.99 | --- | Abs Rec GW: 40–97% SW:24–111% WWI: 18–121% WWE:52–105% Rel Rec GW: 48–121% SW: 19–118% WWI: 18–152% WWE: 59–127% | LOQs GW: 1–10 SW: 1–24 WWE:1.4–29 WWI: 3.5–163 | [109] |
| Drinking water | 27 PhACs (antibiotics, cardiotonic, neuroleptics, hormones, NSAIDs) | SPE 200 mL | Oasis HLB | UPLC-MS/MS | Acquity C18 (BEH) (100 × 2.1 mm, 1.7μm) | SAC | --- | - -- | --- | [80] |
| Wastewater and surface water | 5 PhACs (β-blockers) | SPE 250 mL | CNW MCX (60 mg) | HPLC-(ESI)-MS/MS | Chirobiotic T (150 mm × 2.1 mm, 5 μm) | ESC, ≥0.998 | RSDr <6.9% | Rel Rec SW 70.3%–99.4% WWI 59.5–97.9% | LOQs WWI 0.066–0.227 SW 0.059–0.227 | [82] |
| Wastewater | 15 PhACs (analgesics, NSAIDs, β-blockers, antiepileptics, antidepressants, lipid regulators) | SPE 250 mL MISEP 250 mL | Oasis HLB (150 mg) Oasis WAX (150 mg) Oasis MAX (150 mg) Affinilute MIP—NSAIDs (150 mg) | LC–(ESI)-MS/MS | Fused-CoreTM Ascentis Express C18 (100 × 4.6 mm, 2.7 μm) | ESC ≥0.996 | RSDr <15% (Rec > 25%) | WWE Oasis HLB 71–103% Oasis MAX 60–100% Oasis WAX 14–105% MIP 45–102% | MISEP LODs 0.5–2.0 | [107] |
| Groundwater, surface water, and wastewater | 14 PhACs (antibiotics, stimulant) | SPE 500 mL | Oasis HLB (500 mg) | Ion trap HPLC-MS/MS | Beta Basic C18 (100 × 2.1 mm, 3 µm) | ISC | RSDr 1.0–16% | Rel Rec (0.25 µg/L) GW:51–120% SW: 74–127% 82–126% | LOQs 100–650 | [92] |
| Wastewater | 6 PhACs (lipid regulators, NSAIDs) | SPE (MISEP) WWE: 50 mL WWI: 10 mL | Affinilute MIP—NSAIDs (150 mg) | LC–(ESI)-MS/MS | Fused-CoreTM Ascentis Express C18 (100 mm × 4.6 mm, 2.7 μm) | MMC ≥0.987 | RSDr <19% | WWE: 62–102% WWI: 69%–103% | LODSs 50–100 | [81] |
| Drinking water, surface water and reclaimed waters | 31 PhACs (antibiotics) | On-line SPE 1 mL, 5 mL and 10 mL | HyperSep retain PEP (porous polystyrene divinylbenzene, (20 × 3.0 mm, 12 μm) Hypersil gold aQ (polar endcapped C18, (20 ×2.1 mm, 12 μm) Hypercarb (porous graphitic carbon, 20 ×2.1 mm, 7 μm) | HPLC-(ESI)-MS/MS | Hypersil Gold C18 (50 × 2.1 mm, 1.9 μm) | ISC >0.99 | RSDr, RSDR <20% | 50–150% | LODSs 1.2–63 | [103] |
| Surface, groundwater and drinking water | 31 PhACs (analgesics, antibiotics, anticonvulsant, anti-depressants, β-blockers, corticosteroid, lipid regulators, nsaids, psychostimulant, sexual hormones) | SPE 500 mL | Oasis HLB (200 mg) | UPLC-(ESI)-MS/MS | Acquity BEH C18 (50 x 2.1 mm, 1.7 μm) | MMC 0.9951–0.9987 | RSDr <15% RSDR ≤20% | S: 43–111% GW: 31–120% DW: 49–88% | LOQs SW: 0.030–5.2 GW: 0.040–5.0 DW: 0.03–1.7 | [10] |
| Surface water and wastewater | 7 PhACs (antidepressants) | SPE (MISEP) 100 mL | MIP | UPLC-(ESI)-MS/MS | BEH C18 (50 × 2.1 mm, 1.7 μm) | ISC ≥0.992 | RSDr <15% | SW: 6–94% WWI: 6–90% WWE: 14–93% | MDLs SW: 0.3–12 WWI: 0.3–14 WWE: 0.3–11 | [132] |
| Wastewater | 16 PhACs (analgesic, antibiotics, antidepressant, antiepileptic, β-blockers, lipid regulators, NSAIDs, stimulant) | SPE WWE: 200 mL WWI: 100 mL | Oasis HLB (200 mg) | UPLC-MS/MS | UPLC BEH C18 (50 × 2.1 mm, 1.7 μm) | ISC >0.99 | RSDr 7.9% RSDR 10.4% | WWI: 70% WWE: 81% | LOQs WWE: 0.3–5.4 WWI: 1.0–20.2 | [179] |
| Surface and wastewater | 50 PhACs (antibiotics, antiulcer, cardiovascular, lipid regulators, NSAIDs, psychiatric drugs) | SPE | Oasis HLB (60 mg) | UHPLC-MS/MS | Acquity HSS T3 (100 × 2.1 mm, 1.8 µm) | ISC | <15% | Abs Rec 24–53% Rel Rec 48–147% | LOQs 2–170 | [175] |
| Surface water and wastewater | 20 PhACs (analgesic, anti-anxiety, antiarrhythmic, antiepileptic, antihistamine, contraceptives, corticosteroids, lipid regulators, stimulant, NSAIDs, steroid hormones) | SPE 1000 mL | Oasis HLB (200 mg) | UHPLC-MS/MS | ZORBAX Eclipse Plus C18 (50 × 2.1 mm, 1.8 μm) | ISC >0.99 | RSDr SW: 1.4–5.4% WWE: 3.2–21.6% | SW: 39–121% WW: 38–141% | MRLs 0.1–15 | [221] |
| Surface water and wastewater | 5 PhACs (antibiotics, steroid hormones) | 0n-line SPE 10 mL | Hypersil GOLD aQ (20 × 2.1 mm, 12 μm) | UHPLC-MS/MS | Kinetex Biphenyl (100 × 2.1 mm, 1.7 μm) Kinetex EVO C18 (100 × 2.1 mm, 1.7 μm) | ISC >0.99 | RSDr 2.3–8% RSDR 3.6–15% | 98.8–102% | LOQs WWE: 0.42–1.9 WWI: 0.49–1.9 | [222] |
| Surface water and wastewater | 20 PhACs (analgesics, anti-inflamatories, ansiolitics, Antidepressants, anti-ulcer, cardiovasculars, lipid regulators, psychiatric drugs) | SPE 100 mL | Oasis HLB (200 mg) | UHPLC-MS/MS | Acquity UPLC BEH C18 (50 × 2.1 mm, 1.7 μm) | ISC >0.99 | RSDr SW: 2–22% WWE: 2–18% WWI: 2–24% | Rel Rec SW: 27–117% WWE: 66–120% WWI: 55–124% | LOQs SW: 0.2–44 WWE: 3.6–85 WWI: 13–974 | [45] |
| Surface, groundwater, and wastewater | 73 PhACs (analgesic, antibiotics, antidepressants, anti-diabetic, antiepileptic, antihypertensive, β-blockers, diuretic, histamine H2 receptor antagonists, lipid regulators, NSAIDs, anti-cancer) | SPE SW, GW: 500 mL WWE: 200 mL WWI: 100 mL | Oasis HLB (60 mg) | UHPLC-MS/MS | BEH C18 (100 × 2.1 mm, 1.7 µm) | ISC >0.99 | RSDr 0.2–5% RSDR 0.1–10% | 50–150% | MDLs 0.01–8.98 | [94] |
| Surface and wastewater | 8 PhACs (antiepileptic, cardiovascular, lipid regulators, NSAIDs) | SPE | Oasis HLB (60 mg) | UHPLC-MS/MS | UPLC BEH C18 (50 × 2.1 mm, 1.7 µm) | ISC >0.98 | --- | 20–130% | LOQs 0.2–280 | [223] |
| Wastewater (efluente) | 33 PhACs (amphetamines, antidepressants, β-blockers) | SPE 50 mL | Oasis HLB (60 mg) | UPLC-MS/MS | Chirobiotic V (250 × 2.1 mm, 5 µm) (chiral separations) | ESC ≥0.997 | RSDr 2.8–28.2% RSDR 5–27.3% | Rel Rec 25.9–240.6% Abs Rec 46.9–142% | MQLs 0.09–109 | [64] |
| Surface water | 28 PhACs (analgesics, antibiotics, antidepressants, antiepileptics, β-blockers, bronchodilators, lipid regulators, histamine-2-blockers, calcium channel blockers, angiotensin-II antagonists, NSAIDs) | SPE | Oasis MCX | UPLC–MS/MS | ACQUITY UPLC BEH C18 (100 × 1 mm, 1.7 μm) | ISC | RSDr 3.1–23.9% (10 ng/L) RSDR 7.1–24.8% (10 ng/L) | Rel Rec 7.3–121% Abs Rec 5.2–131% | MQLs 0.3–50 | [98] |
| Surface and wastewater | 3 PhACs (antibiotics) | SPE 120 mL | Oasis HLB (60 mg) | LC–(ESI)-IT-MS/MS | Xterra MS C18 (50 × 2.1 mm, 2.5 μm) | --- | RSDr SW: 3.2–11.7% WWI: 8–18% | SW: 78.6–104.9% WWI: 70.1–99.5% | MDLs 30–70 | [197] |
| Surface and wastewater | 48 PhACs (analgesic, antibiotics, Anticoagulants, antidepressants, antidiabetic, antiepileptic Antihypertensive, β-blockers, diuretic, corticosteroides, histamine H2 receptor antagonists, hormones, lipid regulators, NSAIDs) | SPE 500 mL | Oasis MCX (150 mg) | UPLC-(ESI)-MS/MS | ACQUITY UPLC BEH C18 (100 × 1 mm, 1.7 μm) | ISC >0.99 | RSDr SW:3–25% WWE: 2–27% | SW: 48–123% WWE: 43–157% | LOQs SW: 3.4–85 WWE: 1.6–39 | [101] |
| Wastewater and surface water | 53 PhACs (antibiotics) | SPE 50 mL | Oasis HLB (60 mg) | UHPLC–(ESI)-QqLIT | Acquity HSS T3 (50 × 2.1 mm, 1.8 μm) | ISC 0.9878–1.0000 | RSDr 1–16% RSDR 4–30% | HWW: 30–176% SW: 20–121% WWI: 720–163% WWE: 20–180% | MQLs HWW: 3.97–164 SW: 1.44–44.6 WWI: 9.81–272 WWE: 4.93–183 | [174] |
| Surface and wastewater | 73 PhACs (analgesic, antibiotics, anti- cancer, antidiabetic, antidepressants, antiepileptic, antihypertensive, β-blockers, diuretic, histamine H2 receptor antagonists, lipid regulators, NSAIDs) | SPE SW: 500 mL WWI: 100 mL WWE: 200 mL | Oasis HLB (60 mg) Oasis MCX (150 mg) | LC-ESI-(QqLIT) MS/MS | Purospher Star RP-18 endcapped (125 × 2.0 mm, 5 μm) | ISC 0.9870–1.0000 | RSDr SW: 1–25% WWI: 2–21% WWE: 1–15% | Rel Rec SW: 10–194% WWI: 21–148% WWE: 30–121% | LOQs SW: 0.2–13 WWE: 0.6–28 WWI: 1–62 | [96] |
| Wastewater, groudwater and surface water | 19 PhACs (antibiotic/sulfonamides) | On-line SPE WWI: 5 mL WWE: 15 mL GW: 40 mL SW: 15 mL | Oasis HLB Hysphere C18 EC PRLPs cartridges Oasis HLB was selected | HPLC-QqLIT-MS/MS | Atlantis C18 (150 × 2.1 mm, 3 μm) | MMC WWI: 0.9948–0.9999 WWI: 0.9948–0.9999 SW: 0.9991–0.9999 GW: 0.9962–0.9999 | RSDr WWI: 1.5–10.3% WWE: 1.2–12.9% GW: 1.8–14.8% SW:1.0–25.6% RSDR <10% | Osais HLB 5–125% | MDLs WWI: 0.05–7.84 WWE: 0.01–6.90 SW: 0.02–4.52 GW: 0.02–5.13 | [62] |
| Wastewater, surface water, sea water and drinking water | 90 PhACs (analgesics, antibiotics, NSAIDs, anticoagulant, antidiabetic, β-Blockers, antihelmintics, antihypertensives, antiplatelet, diuretics, histamine H1 and H2 receptor antagonists, lipid regulators, prostatic hyperplasia, psychiatric drugs, sedation and muscle relaxation statin drugs, glucocorticoids, anti-asthma, tranquilizer, X-ray contrast agentes) | SPE MW: 200 mL WWI: 25 mL WWE: 50 mL DW: 500 mL SW: 100 mL | Sea waters Oasis HLB (200 mg) Other waters Oasis MCX (60 mg) | UPLC-QqLIT | PI Acquity HSS T3 (50 × 2.1 mm, 1.8 μm) NI Acquity BEH C18 (50 × 2.1 mm, 1.7 μm) | ISC | RSDr MW: 1.5–20% WWI: 2.0–33.7% WWE: 1.0–20% DW: 0.3–23% SW: 1.0–27.8% | MW: 30–147% WWI: 50–150% WWE: 40–146% DW: 12–157% SW: 30–158% | MQLs MW: 0.04–20 WWI: 0.7–140 WWE: 0.6–51 DW: 0.1–20 SW: 0.2–50.7 | [37] |
| Surface waters | 17 PhACs (antidepressant, antiepileptic, β-blockers, lipid regulators, NSAIDs Stimulants) | On-line SPE 0.5 mL | Oasis HLB, (2.1 × 20 mm, 25 μm) | UPLC-MS/MS | Acquity HSS T3 (150 × 2.1 mm, 1.7 μm) | ISC | --- | --- | LOQs 20–70 | [83] |
| Surface and drinking water | 28 PhACs (antibiotics, anticholinergic, antidepressant, antidiabetic, antifungal, anti-inflammatories, anti-ulcer agente, β-blockers, corticosteroids, histamine H2 receptor antagonists, lipid regulatorss, stimulant) | SPE 1000 mL | C18 (500 mg) | HPLC-ESI-microOTOF-QII | Shim-pack XR-ODS C18 (50 x 2.0 mm, 2.0 μm) | ESC 0.9907–0.9986 | RSDr 0.07–14.6% RSDR 0.85–22% | S: 9–293% DW: 4–286% | MQLs SW: 2.9–460 DW: 1.5–470 | [224,225] |
| Drinking water, groundwate, surface water, and wastewater | 100 PhACs (analgesic, antibiotics, antidepressants, antiepileptic, β-blockers, lipid regulators, NSAIDs, stimulant) | SPE 100 mL | Oasis HLB (200 mg) | HPLC-QTOF-MS | Zorbax Eclipse XDB-C8 (150 × 4.6 mm, 3.5 μm) | MMC >0.98 | RSDr 2–7% RSDR 5–12% | 65–105% | LODs 5–500 | [104] |
| Surface water and wastewater | 7 PhACs (antimycotic drugs) | SPE 500 mL | Oasis MCX | HPLC-Q-TOF-MS | ZORBAX Eclipse XBD C18 (100 × 2 mm, 3.5 μm) | ISC 0.9980–0.9999 | --- | SW: 84–104% WWE:71–109% WWI: 80–90% | LOQs 2–15 | [226] |
| Surface, groundwater, and sewage water | 74 PhACs (analgesics, anti-inflammatories, antibiotics, antidiabetic, antihypertensives, barbiturates, β-agonists, diuretics, histamine H2 receptor antagonists, lipid regulators and cholesterol lowering stain drugs, psychiatric drugs, anti-cancer drugs) | 0n-line SPE 2.5 mL | HySphere Resin GP PRLP-s Oasis HLB (1, 2.5 and 5 mL) | LC-ESI-(QqLIT) MS/MS | Purospher Star RP-18 endcapped (125 × 2.0 mm, 5 μm) | ISC 0.9637–1.0000 | RSDr 0.34–51% RSDR 0.51–89% | Abs Rec GW: 8.9–179% SW: 6.3–165% WWE: 3.3–174% WWI: 2.2–134% Rel Rec GW: 39–328% SW: 42–287% WWE: 12–535% WWI: 7.6–600% | MQLs GW: 0.11–260 SW: 0.02–78 WWE: 0.02–805 WWI: 0.01–1278 | [44] |
| Surface and wastewater | 105 PhACs (analgesics, antiepileptic anti-inflammatories, antibiotics, bronchodilatadors, β-blockers, diuretics, hormones, lipid regulators, psychiatrics, ulcer healings) | SPE 100 mL | Oasis HLB (200 mg) | HPLC-Q-TOF-MS | ZORBAX Eclipse Plus C18 (50 × 4.6 mm, 1.8 μm) | MMC >0.998 | <10% | 50–130% | LOQs 0.7–592.4 | [99] |
| Wastewater | 67 PhACs (illicit drugs, prescription drugs with potential for abuse and metabolites) | SPE 50 mL | Oasis MCX | UHPLC-QTOF-MS | UPLC BEH C18 (50 × 2.1 mm, 1.7 µm) | --- | --- | --- | IDLs 10–3500 | [227] |
| Surface and Wastewater | 29 PhACs (analgesics/anti-inflammatories, antibiotics, anti-ulcer, β-blockers, lipid regulators, histamine H2 receptor antagonist, psychiatric) | SPE SW, WWE, and WWI: 500, 200, and 100 mL | Oasis HLB (60 mg) | UPLC-QTOF-MS | Acquity C18 (50 ×2.1 mm, 1.7 μm) | RSDr 0.5–5.3% RSDR 2.1–9.1% | --- | MDLs 10–500 | [173] | |
| Surface, drinking, and groundwater. | 13 PhACs (analgesic, antibiotics, antiepileptic, lipid regulators, NSAIDs) | SPE 100 mL | Oasis-MCX (150 mg) | LC-(ESI)-QTOF-MS | RP-18 (100 × 2.1 mm, 3.5 μm) | MMC 0.995–0.999 | RSDr 2.0–12% (100 ng/L) RSDR 5–44% (25 ng/L) 13–34% (100 ng/L) | 63–195% | LOQs 5–25 | [91] |
| Wastewater | 160 PhACs (metabolites) | SPE 100 mL | Oasis HLB (60 mg) | UHPLC-QTOF-MS | UPLC BEH C18 (100 ×2.1 mm, 1.7 μm) | --- | --- | --- | --- | [105] |
| Surface and wastewaters | 87 PhACs (analgesics, antibiotics, antiepileptic, antipyretics, β-blockers, chemotherapy drugs, corticosteroids, diuretics, lipid regulators NSAIDs, psychiatric drugs) | SPE WWE: 200 mL SW: 400 mL | Oasis HLB (200 mg) | LC–(ESI)-QTOF-MS/MS | RP XDB-C18 (50 × 4.6 mm, 1.8 μm) | MMC 0.9000–0.9987 | RSDr 2–23% | 22–127% | MDLs <5–50 | [51] |
| Surface water, and influent and effluent wastewater | 22 PhACs (drugs of abuse stimulant, tranquilising drugs) | Direct injection | --- | Screening by HPLC- (ESI) QqQLIT-MS/MS Confirmation by LC–MS/MS | ZorbaxEclipse XDB, RP C8 (150 × 4.6 mm, 5 μm) | MMC R > 0.99 | RSDr, RSDR 4%–16% | --- | IDLs (LOQs) SW: 0.5–600 WWI: 10–700 WWE: 10–600 | [3] |
| Wastewater | 13 PhACs (antibiotic, antidepressant, anti-diabetic, lipid regulators, stimulant) | SPE 30 mL | Oasis HLB (60 mg) | UHPLC- Q-Orbitrap-MS | Agilent Extend-C18 (50 × 2.1 mm, 1.8 μm) | ESC 0.9875–0.9993 | --- | 95–101% | LOQs 40–2500 | [207] |
| Wastewater, surface water and drinking water | 27 PhACs (analgesic, antibiotics, antidepressant, antiepileptic, β-blockers, corticosteroids, lipid regulators, NSAIDs, steroid hormones, stimulant) | SPE 250 mL | Oasis MC X | UPLC-Q ExactiveTM Orbitrap-MS | Accucore RP-MS (100 x 2.1 μm) | ISC 0.9619–0.9990 | RSDr 2.0–7.7% RSDR 3.9–7.6% | 76–104% | MQLs 2.7–83.8 | [206] |
| Milli Water | 17 PhACs (antibiotic/sulfonamides) | --- | --- | UPLC- LTQ/Orbitrap MS/MS | --- | --- | --- | --- | --- | [55] |
| Marine waters | 11 PhACs (antibiotics) | SPE disks 1500 mL | Bakerbond Speedisk (H2O-Philic DVB) | HPLC-(ESI)-QqQIT HPLC-(ESI)-MS/MS | Eclipse XDB C18 (150 × 4.6 mm, 5 μm) | ESC 0.9984–1.0000 | RSDr 0.1–6.0 | Abs Rec 37.5–117% | LOQs 0.5–50 | [87] |
| Wastewater | 8 PhACs (steroid hormones) | SPE | RP Strata-X (surface-modified styrene divinylbenzene polymer) (200 mg) | LDTD-APCI-MS/MS | --- | MMC | RSDr 5–14% RSDR 5–17% | 77–121% | MDLs 13–42 | [190] |
| Wastewater | 9 PhACs (antibiotic, antiepileptic, NSAIDs, steroid hormones, stimulant) | SPE 250 mL | Strata ABW cartridge (500 mg) | LDTD-APCI-MS/MS | --- | MMC ≥0.991 | RSDr, RSDR <15 % | Rel Rec 76–106% | LODs 30–122 | [191] |
| Wastewater | 1 PhACs (NSAIDs/diclofenac) | --- | --- | LDTD-APCI-MS/MS | --- | ESC >0.999 | RSDr 7.1% RSDR 9.2 | 98.2–104.6% | MDLs 300 | [182] |
| Wastewater | 1 PhACs (antiepileptic/ carbamazepine) | --- | --- | LDTD-APCI-MS/MS | --- | ESC >0.999 | RSDr 8% RSDR 11% | 98–113% | LODs 12 | [184] |
| Wastewater | 3 PhACs (antibiotics) | DSPE | Activated carbon-decorated PAN nanofibers | HPLC-DAD | RP C-18 | ESC 0.9998 | RSDr <3.6% | 90–99%. | LODs 530–2170 | [111] |
| Mineral waters | 11 PhACs (antibiotic/sulfonamides) | DSPE 250 mL | MWCNTs m- MWCNTs) | UPLC-DAD | Hypersil Gold C18 (100 × 2.1 mm, 1.9 μm) | MMC 0.995–0.997 | RSDr 0.9–3.8% RSDR 1.7–2.9 | MWCNTs 40–100% m- MWCNTs 32 and 53% | LOQs 27–89 | [113] |
| Surface water | 4 PhACs (natural hormones) | DSPE 250 mL | mag-MFMIP Fe3O4/Poly(Stc- MPS)/SiO2 | HPLC-FLD | XDB-C18 | ESC 0.996–0.9999 | RSDr <7.0% | 72–102% | LODs 2.5–5.8 | [118] |
| Wastewater | 4 PhACs (NSAIDs) | MMSPD 1 mL | SiO2, C8, Magnetite particles Fe3O4 | HPLC-UV | Microsorb 100-C18 (150 × 4.6 mm, 5 μm) | --- | RSDr 1.0–2.1% RSDR 1.4–2.0% | >90% | LOQs 3000–5000 | [127] |
| Tap water, groundwater, and river water | 6 PhACs (NSAIDs) | MSPE 10 mL | Fe3O4@PEI-RGO (10 mg) | HPLC-DAD | BDS Hypersil C18 (250 × 4.6 mm, 5 μm) | ESC 0.9972–0.9986 | RSDr 0.39–6.67% RSDR 0.70%–8.75%, | 91.2–101.1% | LOQs 1000 | [114] |
| Surface water | 7 PhACs (antibiotics) | MSPE 3 mL | EMMIPs (10 mg) | HPLC-DAD | Ultimate XB-C18 (250 × 2.5 mm, 5 μm) | ESC 0.999–0.9999 | RSDr 0.9%–7.3%. | 79.3–92.4% | LOQs 100–310 | [134] |
| Surface water and wastewater | 6 PhACs (antibiotics/ sulfonamides) | MSPE | Fe3O4@SiO2/G (0.3 mg) | HPLC-UV | Inertsil® ODS-4 (150 × 4.6 mm, 5 μm) | --- | RSDr 3.3%–10.7% RSDR 4.6–9.8% | SW 76.4–104% WWI 74.2–89.3% | LOQs 320–530 | [123] |
| Wastewater | 1 PhACs (lipid regulator/gemfibrozil) | MSPE 200 mL | β-CD/ Fe3O4/GO NP (150 mg) | HPLC-FLD | --- | ESC 0.9989 | RSDr 1.09% RSDR 2.67% | 96.0–104.0% | LOQs 3 | [124] |
| Marine water, wastewater and drinking water | 5 PhACs (lipid regulators) | MSPE 10 mL | Fe3O4@Fe-BTC MMOF (5 mg) | HPLC-UV-Vis | Microsorb-MV 100-8 C18 (250 × 4.6 mm, 2.5 μm) | ESC 0.9992–0.996 | RSDr MW: 2.6–5.4% WW: 1.8–7.2% DW: 2.2–3.2% | MW 80.1–99.4% WWI 59.7–90% DW 83–100.5% | LOQs 460×103–725×103 | [125] |
| Surface water | 3 PhACs (antibiotics/macrolids) | MSPE 50 mL | Ol-coated Fe3O4MNP (50 mg) | LC-(ESI)-MS/MS | Kinetex XB-C18 (100 × 3 mm, 2.6 μm) | MMC | RSDr <6.0% RSDR <13% | 54–117% | LOQs 34–77 | [122] |
| Surface water | 24 PhACs (steroid hormones) | MSPE 100 mL | EDA@Mag-CNTs (50 mg) | UFLC-MS/MS | Shimpack XR-ODS II (150 × 2.0 mm, 2.2 μm) | ISC 0.9980–1.0000 | RSDr 1.6–6.4% RSDR 2.6–8.1% | 82.1–113%. | LOQs 0.02–1.0 | [126] |
| Mineral, tap and wastewater using | 12 PhACs (estrogenic hormones) | MSPE 25 mL | Fe3O4@poly(dopamine) (60 mg) | UFLC-(ESI)-MS/MS | X-Bridge C18 (100 mm × 4.6 mm, 3.5 μm) | ISC 0.9943–0.9991 | RSDr 3–15% | 70–119% | LOQs 10–1100 | [128] |
| Surface waters | 58 PhACs (antibiotics, steroid hormones) | MSPE 100 mL | 3D-Mag-CMGO (100 mg) | UFLC-(ESI)-MS/MS | Shimpack XR-ODS II (150 mm × 2.0 mm, 2.2 μm) | MMC 0.9981–0.9991 | RSDr 1.7–2.2% RSDR 7.3–9.0% | 78–109% | LOQs 0.10–2.09 | [130] |
| Surface water | 2 PhACs (antibiotics) | SBSE 25 mL | MIP MWCNTs | HPLC-DAD | ZORBAX C18 (4.6 × 150 mm, 5 μm) | ESC 0.9929–0.9945 | RSDr 5.1–5.5% RSDR 6.4–6.5% | 86.5–98.6% | LOQs 10×103–12×103 | [161] |
| Surface water | 4 PhACs (estrogen hormones) | SBSE 15 mL | dual-templates MIP | HPLC-DAD | C18 (250 × 4.6 mm, 5 μm) | ESC 0.9978–0.9992 | RSDr 2.0–5.5% | 62.8–98.0% | LOQs 1000–5000 | [162] |
| Surface and wastewater | 7 PhACs (analgesic, antiepileptic, β-blockers, NSAIDs, stimulants) | SBSE 100 mL | poly(MAA-co-DVB | LC-(ESI)-MS/MS | Kinetex C18 (100 × 4.6 mm, 2.6 μm) | ESC >0.999 | SW RSDr 1–7% WWE RSDr 3–19% | SW: 11–90% WWE: 10–85% | LODs 10--50 | [163] |
| Wastewater | 12 PhACs (analgesics, antibiotics, antipyretics, β-blockers, lipid regulators) | SBSE 50 mL | 32 µL EG Silicone Twister® 25 µL Acrylate Twister® | LC-(ESI)MS/MS | Kromasil 100 C18 (150 × 4.6 mm, 5 μm) | ESC, >0.997 | RSDr <15% (Rec > 16%) | PA: 1 - 43% EG silicone: 1–80% | LODSs 150–5000 | [61] |
| Tap water, groundwater, surface, and wastewater | 3 PhACs (NSAIDs) | DI-SPME 20 mL | PEG-g-MWCNTs) | GC-FID | CP-Sil 24CB - WCOT Fused silica (30 m × 0.32 mm × 0.25 μm) | ESC 0.9985–0.9992 | RSDr 5.9–8.1% RSDR 7.2–9.1% | 84–107%. | LOQs 50–70 | [143] |
| Surface water | 8 PhACs (analgesics, lipid regulators, NSAIDs) | On-line HS-SPME Derivatization with DMS 10 mL | PDMS–DVB, 65 μm | GC-MS | HP5MS (30 m × 0.25 mm × 0.25 μm) | ESC 0.994–0.999 | RSDr 0.6–12.3% | 92.5–110.8% | LOQs 0.34–4.13 | [145] |
| Tap water and wastewater | 6 PhACs (NSAIDs) | HS_SPME Derivatization with DMS 6 mL | PDMS 100 μm | GC-MS | HP5MS (30 m × 0.25 mm × 0.25 μm) | ISC 0.9920–0.9980 | RSDr 7.9–17.2% | 76–133% | LODs 0.3–2.9 | [146] |
| Surface water | 8 PhACs (antiepileptic, NSAIDs) | On-line SPME | PDMS–DVB, 65 μm | HPLC-DAD | Discovery RP-Amide C16 (150 × 4.6 mm, 5 μm) | ESC 0.994–0.999 | RSDr 4.4–8.2% RSDr 5.1–9.3% | 71.6–122.8% | LOQs 1000–4000 | [150] |
| Wastewater | 17 PhACs (analgesics, ansiolitics, Antibiotics, antidepressants, antiepileptic, antipsychotic, β-blockers, NSAIDs) | On-line dual-SPME 2 ×2 mL (pH=3, pH=11) | CW/TPR (50 µm) | LC–(ESI)-ITMS | Mediterranea Sea18) C18 (100 × 2.1 mm, 3 μm) | ISC 0.988–0.998 | RSDr 6.2–10.2% RSDR 7.4–13.6% | 89.2–109.7% | LOQs 0.005–0.01 | [93] |
| Wastewater | 9 PhACs (antibiotics) | SPME 1.5 mL | CW/TPR (50 µm) | HPLC-MS/MS | ZORBAX Eclipse XDB C18 (250 × 4.6 mm, 5 μm) | SAC WWI 0.9080–0.9992 WWE 0.9336–0.9987 | RSDr 7–50% | --- | LOQs WWI:9.2–1380 WWE: 14–260 | [152] |
| Tap water and surface water | 6 PhACs (hormones) | SPME 20 mL | Home made MMF/AMED | HPLC-DAD | Hypersil BDS C18 (250 × 4.6 mm, 5 μm) | ESC 0.9910–0.9980 | RSDr 3.5–5.0% RSDR 5.1–6.0% | 75.6–118% | LODs 130–340 | [148] |
| Wastewater | 16 PhACs (analgesic, antibiotic, antidepressant, lipid regulators, NSAIDs, stimulant) | SPME 10 mL | FPSE (FG)@PEG300 | UHPLC–LTQ Orbitrap MS | Hypersil Gold C18 analytical (100 × 2.1 mm, 1.9 μm) | ESC 0.9928–0.9989 | RSDr 8% RSDR 11% | 83–114% | LOQs 9.3–447.7 | [210] |
4. Methods Validation
5. Conclusions
Funding
Conflicts of Interest
References
- Sauve, S.; Desrosiers, M. A review of what is an emerging contaminant. Chem. Cent. J. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, M.O.; Moreira, N.F.F.; Ribeiro, A.R.; Pereira, M.F.R.; Silva, A.M.T. Occurrence and removal of organic micropollutants: An overview of the watch list of EU Decision 2015/495. Water Res. 2016, 94, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Ucles, S.; Hernando, M.; Fernandez-Alba, A. Development of a solvent-free method for the simultaneous identification/quantification of drugs of abuse and their metabolites in environmental water by LC-MS/MS. Talanta 2011, 85, 157–166. [Google Scholar] [CrossRef]
- Deblonde, T.; Hartemann, P. Environmental impact of medical prescriptions: Assessing the risks and hazards of persistence, bioaccumulation and toxicity of pharmaceuticals. Public Health 2013, 127, 312–317. [Google Scholar] [CrossRef]
- Grizzetti, B.; Lanzanova, D.; Liquete, C.; Reynaud, A.; Cardoso, A. Assessing water ecosystem services for water resource management. Environ. Sci. Policy 2016, 61, 194–203. [Google Scholar] [CrossRef]
- Grizzetti, B.; Liquete, C.; Antunes, P.; Carvalho, L.; Geamana, N.; Giuca, R.; Leone, M.; McConnell, S.; Preda, E.; Santos, R.; et al. Ecosystem services for water policy: Insights across Europe. Environ. Sci. Policy 2016, 66, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Daughton, C.; Ternes, T. Pharmaceuticals and personal care products in the environment: Agents of subtle change? Environ. Health Perspect. 1999, 107, 907–938. [Google Scholar] [CrossRef]
- Daughton, C. Pharmaceuticals as environmental pollutants: The ramifications for human exposure. In International Encyclopedia of Public Health; Academic Press: Oxford, UK, 2008. [Google Scholar] [CrossRef]
- Kummerer, K. Pharmaceuticals in the environment. Ann. Rev. Environ. Resour. 2010, 35, 57–75. [Google Scholar] [CrossRef] [Green Version]
- Gaffney, V.D.J.; Cardoso, V.V.; Rodrigues, A.; Ferreira, E.; Benoliel, M.J.; Almeida, C.M.M. Analysis of pharmaceutical compounds in waters by SPE-UPLC-ESI-MS/MS. Química Nova 2014, 37, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Kummerer, K. The presence of pharmaceuticals in the environment due to human use—Present knowledge and future challenges. J. Environ. Manag. 2009, 90, 2354–2366. [Google Scholar] [CrossRef]
- Fong, P.; Molnar, N. Norfluoxetine induces spawning and parturition in estuarine and freshwater bivalves. Bull. Environ. Contam. Toxicol. 2008, 81, 535–538. [Google Scholar] [CrossRef]
- Franzellitti, S.; Buratti, S.; Valbonesi, P.; Fabbri, E. The mode of action (MOA) approach reveals interactive effects of environmental pharmaceuticals on Mytilus galloprovincialis. Aquat. Toxicol. 2013, 140, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Franzellitti, S.; Buratti, S.; Capolupo, M.; Du, B.; Haddad, S.P.; Chambliss, C.K.; Brooks, B.W.; Fabbri, E. An exploratory investigation of various modes of action and potential adverse outcomes of fluoxetine in marine mussels. Aquat. Toxicol. 2014, 151, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Koutsogiannaki, S.; Franzellitti, S.; Fabbri, E.; Kaloyianni, M. Oxidative stress parameters induced by exposure to either cadmium or 17β-estradiol on Mytilus galloprovincialis hemocytes. The role of signaling molecules. Aquat. Toxicol. 2014, 146, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, V.; Mota-Filipe, H.; Pinto, R.; Thiemermann, C.; Loureiro, M.; Cardoso, V.; Benoliel, M.; Almeida, C. Chemical and biochemical characterization and in vivo safety evaluation of pharmaceuticals in drinking water. Environ. Toxicol. Chem. 2016, 35, 2674–2682. [Google Scholar] [CrossRef]
- WHO. Pharmaceuticals in Drinking-Water; WHO Press: Geneva, Switzerland, 2011. [Google Scholar]
- Bu, Q.; Wang, B.; Huang, J.; Deng, S.; Yu, G. Pharmaceuticals and personal care products in the aquatic environment in China: A review. J. Hazard. Mater. 2013, 262, 189–211. [Google Scholar] [CrossRef]
- Grenni, P.; Ancona, V.; Caracciolo, A. Ecological effects of antibiotics on natural ecosystems: A review. Microchem. J. 2018, 136, 25–39. [Google Scholar] [CrossRef]
- Patrolecco, L.; Rauseo, J.; Ademollo, N.; Grenni, P.; Cardoni, M.; Levantesi, C.; Luprano, M.; Caracciolo, A. Persistence of the antibiotic sulfamethoxazole in river water alone or in the co-presence of ciprofloxacin. Sci. Total Environ. 2018, 640, 1438–1446. [Google Scholar] [CrossRef]
- Boyd, G.; Palmeri, J.; Zhang, S.; Grimm, D. Pharmaceuticals and personal care products (PPCPs) and endocrine disrupting chemicals (EDCs) in stormwater canals and Bayou St. John in New Orleans, Louisiana, USA. Sci. Total Environ. 2004, 333, 137–148. [Google Scholar] [CrossRef]
- Wen, Z.H.; Chen, L.; Meng, X.Z.; Duan, Y.P.; Zhang, Z.S.; Zeng, E.Y. Occurrence and human health risk of wastewater-derived pharmaceuticals in a drinking water source for Shanghai, East China. Sci. Total Environ. 2014, 490, 987–993. [Google Scholar] [CrossRef]
- Salgado, R.; Noronha, J.; Oehmen, A.; Carvalho, G.; Reis, M. Analysis of 65 pharmaceuticals and personal care products in 5 wastewater treatment plants in Portugal using a simplified analytical methodology. Water Sci. Technol. 2010, 62, 2862–2871. [Google Scholar] [CrossRef]
- Salgado, R.; Pereira, V.J.; Carvalho, G.; Soeiro, R.; Gaffney, V.; Almeida, C.; Cardoso, V.V.; Ferreira, E.; Benoliel, M.J.; Ternes, T.A.; et al. Photodegradation kinetics and transformation products of ketoprofen, diclofenac and atenolol in pure water and treated wastewater. J. Hazard. Mater. 2013, 244, 516–527. [Google Scholar] [CrossRef] [PubMed]
- De Jesus Gaffney, V.; Almeida, C.M.M.; Rodrigues, A.; Ferreira, E.; Benoliel, M.J.; Cardoso, V.V. Occurrence of pharmaceuticals in a water supply system and related human health risk assessment. Water Res. 2015, 72, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Silva, L.; Meisel, L.; Lino, C.; Pena, A. Environmental impact of pharmaceuticals from Portuguese wastewaters: Geographical and seasonal occurrence, removal and risk assessment. Environ. Res. 2015, 136, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Sousa, M.; Goncalves, C.; Cunha, E.; Hajslova, J.; Alpendurada, M. Cleanup strategies and advantages in the determination of several therapeutic classes of pharmaceuticals in wastewater samples by SPE-LC-MS/MS. Anal. Bioanal. Chem. 2011, 399, 807–822. [Google Scholar] [CrossRef] [PubMed]
- European Union. Directive 2013/39/EU of the European Parliament and of the Council of 12 august 2013. Off. J. Eur. Union 2013, 226, 13–15. [Google Scholar]
- European Union. Commission implementing decision (EU) 2015/495 of 20 march 2015 establishing a watch list of substances for Union-wide monitoring in the field of water policy pursuant to Directive 2008/105/EC of the European Parliament and of the Council. Off. J. Eur. Union 2015, 78, 40–42. [Google Scholar]
- European Union. Commission implementing decision (EU) 2018/840 of 5 june 2018 establishing a watch list of substances for Union-wide monitoring in the field of water policy pursuant to Directive 2008/105/EC of the European Parliament and of the Council and repealing Commission Implementing Decision (EU) 2015/495. Off. J. Eur. Union 2018, 141, 9–12. [Google Scholar]
- European Community. Communication from the Commission of the Council and the European Parliament. In A European One Heath Action Plan against Antimicrobial Resistance (AMR); European Community: Brussels, Belgium, 2017. [Google Scholar]
- Afonso-Olivares, C.; Sosa-Ferrera, Z.; Santana-Rodríguez, J.J. Occurrence and environmental impact of pharmaceutical residues from conventional and natural wastewater treatment plants in Gran Canaria (Spain). Sci. Total Environ. 2017, 599, 934–943. [Google Scholar] [CrossRef]
- Agüera, A.; Martínez Bueno, M.J.; Fernández-Alba, A.R. New trends in the analytical determination of emerging contaminants and their transformation products in environmental waters. Environ. Sci. Pollut. Res. Int. 2013, 20, 3496–3515. [Google Scholar] [CrossRef]
- Merone, G.M.T.; Locatelli, A.; D’Ovidio, M.; Rosato, C.; de Grazia, E.; Santavenere, U.; Rossi, F.; Savini, S.F. Analytical chemistry in the 21st century: Chalenges, solutions, and future perspectives of complex matrices quantitative analysis in biological/clinical field. Analytica 2020, 1, 44–59. [Google Scholar] [CrossRef]
- Patnaik, P. Handbook of Environmental Analysis: Chemical Pollutants in Air, Water, Soil, and Solid Wastes; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Loos, R.; Carvalho, R.; Antonio, D.; Cornero, S.; Locoro, G.; Tavazzi, S.; Paracchini, B.; Ghiani, M.; Lettieri, T.; Blaha, L.; et al. EU-wide monitoring survey on emerging polar organic contaminants in wastewater treatment plant effluents. Water Res. 2013, 47, 6475–6487. [Google Scholar] [CrossRef]
- Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. Fast and comprehensive multi-residue analysis of a broad range of human and veterinary pharmaceuticals and some of their metabolites in surface and treated waters by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. J. Chromatogr. A 2012, 1248, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Jelic, A.; Petrovic, M.; Barceló, D. Multi-residue method for trace level determination of pharmaceuticals in solid samples using pressurized liquid extraction followed by liquid chromatography/quadropole-linear ion trap mass spectrometry. Talanta 2009, 80, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Chiaia-Hernandez, A.; Krauss, M.; Hollender, J. Screening of lake sediments for emerging contaminants by liquid chromatography atmospheric pressure photoionization and electrospray ionization coupled to high resolution mass spectrometry. Environ. Sci. Technol. 2013, 47, 976–986. [Google Scholar] [CrossRef]
- Barron, L.; Tobin, J.; Paull, B. Multi-residue determination of pharmaceuticals in sludge and sludge enriched soils using pressurized liquid extraction, solid phase extraction and liquid chromatography with tandem mass spectrometry. J. Environ. Monit. 2008, 10, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Sancho, J.V.; Ibáñez, M.; Abad, E.; Portolés, T.; Mattioli, L. Current use of high-resolution mass spectrometry in the environmental sciences. Anal. Bioanal. Chem. 2012, 403, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Farré, M.; Kantiani, L.; Petrovic, M.; Pérez, S.; Barceló, D. Achievements and future trends in the analysis of emerging organic contaminants in environmental samples by mass spectrometry and bioanalytical techniques. J. Chromatogr. A 2012, 1259, 86–99. [Google Scholar] [CrossRef]
- Petrovic, M.; Farré, M.; de Alda, M.L.; Perez, S.; Postigo, C.; Köck, M.; Radjenovic, J.; Gros, M.; Barcelo, D. Recent trends in the liquid chromatography-mass spectrometry analysis of organic contaminants in environmental samples. J. Chromatogr. A 2010, 1217, 4004–4017. [Google Scholar] [CrossRef] [PubMed]
- López-Serna, R.; Pérez, S.; Ginebreda, A.; Petrović, M.; Barceló, D. Fully automated determination of 74 pharmaceuticals in enctrvironmental and waste waters by online solid phase extraction-liquid chromatography-electrospray-tandem mass speometry. Talanta 2010, 83, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Lor, E.; Sancho, J.V.; Hernández, F. Simultaneous determination of acidic, neutral and basic pharmaceuticals in urban wastewater by ultra high-pressure liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 622–632. [Google Scholar] [CrossRef]
- Grabic, R.; Fick, J.; Lindberg, R.H.; Fedorova, G.; Tysklind, M. Multi-residue method for trace level determination of pharmaceuticals in environmental samples using liquid chromatography coupled to triple quadrupole mass spectrometry. Talanta 2012, 100, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Fontela, M.; Galceran, M.T.; Ventura, F. Fast liquid chromatography-quadrupole-linear ion trap mass spectrometry for the analysis of pharmaceuticals and hormones in water resources. J. Chromatogr. A 2010, 1217, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, V.; Cardoso, V.; Cardoso, E.; Teixeira, A.; Martins, J.; Benoliel, M.; Almeida, C. Occurrence and behaviour of pharmaceutical compounds in a Portuguese wastewater treatment plant: Removal efficiency through conventional treatment processes. Environ. Sci. Pollut. Res. 2017, 24, 14717–14734. [Google Scholar] [CrossRef]
- Richardson, S. Environmental mass spectrometry: Emerging contaminants and current issues. Anal. Chem. 2012, 84, 747–778. [Google Scholar] [CrossRef] [PubMed]
- Petrović, M.; Barceló, D. LC-MS for identifying photodegradation products of pharmaceuticals in the environment. Trends Anal. Chem. 2007, 26, 486–493. [Google Scholar] [CrossRef]
- Gómez, M.J.; Gómez-Ramos, M.M.; Malato, O.; Mezcua, M.; Férnandez-Alba, A.R. Rapid automated screening, identification and quantification of organic micro-contaminants and their main transformation products in wastewater and river waters using liquid chromatography-quadrupole-time-of-flight mass spectrometry with an accurate-mass database. J. Chromatogr. A 2010, 1217, 7038–7054. [Google Scholar] [CrossRef]
- Gaffney, V.D.; Cardoso, V.V.; Benoliel, M.J.; Almeida, C.M.M. Chlorination and oxidation of sulfonamides by free chlorine: Identification and behaviour of reaction products by UPLC-MS/MS. J. Environ. Manag. 2016, 166, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Escher, B.I.; Fenner, K. Recent advances in environmental risk assessment of transformation products. Environ. Sci. Technol. 2011, 45, 3835–3847. [Google Scholar] [CrossRef]
- Küster, A.; Alder, A.C.; Escher, B.I.; Duis, K.; Fenner, K.; Garric, J.; Hutchinson, T.H.; Lapen, D.R.; Péry, A.; Römbke, J.; et al. Environmental risk assessment of human pharmaceuticals in the European Union: A case study with the β-blocker atenolol. Integr. Environ. Assess. Manag. 2010, 6, 514–523. [Google Scholar] [CrossRef]
- Xia, B.; Liu, X.; Gu, Y.; Zhang, Z.; Wang, H.; Ding, L.; Zhou, Y. Non-target screening of veterinary drugs using tandem mass spectrometry on smart mass. J. Am. Soc. Mass Spectrom. 2013, 24, 789–793. [Google Scholar] [CrossRef]
- Beccaria, M.; Cabooter, D. Current developments in LC-MS for pharmaceutical analysis. Analyst 2020, 145. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gong, Z.; Kelly, B.C. Rapid analysis of pharmaceuticals and personal care products in fish plasma micro-aliquots using liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2015, 1383, 104–111. [Google Scholar] [CrossRef]
- Ferhi, S.; Bourdat-Deschamps, M.; Daudin, J.; Houot, S.; Nelieu, S. Factors influencing the extraction of pharmaceuticals from sewage sludge and soil: An experimental design approach. Anal. Bioanal. Chem. 2016, 408, 6153–6168. [Google Scholar] [CrossRef]
- Henriques, M.; Cardoso, V.; Rodrigues, A.; Ferreira, E.; Benoliel, M.; Almeida, C. Experimental and statistical validation of several endocrine disrupters by solid-phase extraction, liquid chromatography tandem mass spectrometry. Water Resour. Prot. 2010, 2, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Baranowska, I.; Kowalski, B. An analytical procedure for the determination of different therapeutic drugs in surface waters. Water Sci. Technol. 2009, 60, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Gilart, N.; Miralles, N.; Marce, R.; Borrull, F.; Fontanals, N. Novel coatings for stir bar sorptive extraction to determine pharmaceuticals and personal care products in environmental waters by liquid chromatography and tandem mass spectrometry. Anal. Chim. Acta 2013, 774, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Galan, M.; Diaz-Cruz, M.; Barcelo, D. Determination of 19 sulfonamides in environmental water samples by automated on-line solid-phase extraction-liquid chromatography-tandem mass spectrometry (SPE-LC-MS/MS). Talanta 2010, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, I.; Javad, S.; Yousaf, Z.; Iqbal, S.; Jabeen, K. Review: Microwave assisted extraction of phytochemicals an efficient and modern approach for botanicals and pharmaceuticals. Pak. J. Pharm. Sci. 2019, 32, 223–230. [Google Scholar]
- Evans, S.E.; Davies, P.; Lubben, A.; Kasprzyk-Hordern, B. Determination of chiral pharmaceuticals and illicit drugs in wastewater and sludge using microwave assisted extraction, solid-phase extraction and chiral liquid chromatography coupled with tandem mass spectrometry. Anal. Chim. Acta 2015, 882, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez, M.; Borrull, F.; Pocurull, E.; Fontanals, N. Pressurized liquid extraction followed by liquid chromatography with tandem mass spectrometry to determine pharmaceuticals in mussels. J. Sep. Sci. 2016, 39, 741–747. [Google Scholar] [CrossRef]
- Löffler, D.; Ternes, T.A. Determination of acidic pharmaceuticals, antibiotics and ivermectin in river sediment using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2003, 1021, 133–144. [Google Scholar] [CrossRef]
- Sun, H.; Ge, X.; Lv, Y.; Wang, A. Application of accelerated solvent extraction in the analysis of organic contaminants, bioactive and nutritional compounds in food and feed. J. Chromatogr. A 2012, 1237, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.; Albino, S.; Silva, S.; Cravo, A.; Cardoso, V.; Benoliel, M.; Almeida, C. Development of a multiresidue method for the determination of 24 pharmaceuticals in clams by QuEChERS and liquid chromatography-triple quadrupole tandem mass spectrometry. Food Anal. Methods 2019, 12, 838–851. [Google Scholar] [CrossRef]
- Núñez, M.; Borrull, F.; Fontanals, N.; Pocurull, E. Determination of pharmaceuticals in bivalves using QuEChERS extraction and liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 3841–3849. [Google Scholar] [CrossRef] [PubMed]
- González, A.G.; Herrador, M.Á. A practical guide to analytical method validation, including measurement uncertainty and accuracy profiles. Trends Anal. Chem. 2007, 26, 227–238. [Google Scholar] [CrossRef]
- González, A.G.; Herrador, M.A.; Asuero, A.G. Intra-laboratory assessment of method accuracy (trueness and precision) by using validation standards. Talanta 2010, 82, 1995–1998. [Google Scholar] [CrossRef]
- Wasik, A.; Kot-Wasik, A.; Namiesnik, J. New trends in sample preparation techniques for the analysis of the residues of pharmaceuticals in environmental samples. Curr. Anal. Chem. 2016, 12, 280–302. [Google Scholar] [CrossRef]
- Ali, I.; Suhail, M.; Alharbi, O.; Hussain, I. Advances in sample preparation in chromatography for organic environmental pollutants analyses. J. Liq. Chromatogr. Relat. Technol. 2019, 42, 137–160. [Google Scholar] [CrossRef]
- Moldoveanu, S. Solutions and challenges in sample preparation for chromatography. J. Chromatogr. Sci. 2004, 42, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Maciel, E.; de Toffoli, A.; Lancas, F. Recent trends in sorption-based sample preparation and liquid chromatography techniques for food analysis. Electrophoresis 2018, 39, 1582–1596. [Google Scholar] [CrossRef]
- Hyötyläinen, T.; Riekkola, M.L. Sorbent- and liquid-phase microextraction techniques and membrane-assisted extraction in combination with gas chromatographic analysis: A review. Anal. Chim. Acta 2008, 614, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Iparraguirre, A.; Navarro, P.; Rodil, R.; Prieto, A.; Olivares, M.; Etxebarria, N.; Zuloaga, O. Matrix effect during the membrane-assisted solvent extraction coupled to liquid chromatography tandem mass spectrometry for the determination of a variety of endocrine disrupting compounds in wastewater. J. Chromatogr. A 2014, 1356, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Perez-Fernandez, V.; Rocca, L.; Tomai, P.; Fanali, S.; Gentili, A. Recent advancements and future trends in environmental analysis: Sample preparation, liquid chromatography and mass spectrometry. Anal. Chim. Acta 2017, 983, 9–41. [Google Scholar] [CrossRef]
- Tranchida, P.; Maimone, M.; Purcaro, G.; Dugo, P.; Mondello, L. The penetration of green sample-preparation techniques in comprehensive two-dimensional gas chromatography. Trends Anal. Chem. 2015, 71, 74–84. [Google Scholar] [CrossRef]
- Cimetiere, N.; Soutrel, I.; Lemasle, M.; Laplanche, A.; Crocq, A. Standard addition method for the determination of pharmaceutical residues in drinking water by SPE-LC-MS/MS. Environ. Technol. 2013, 34, 3031–3041. [Google Scholar] [CrossRef] [Green Version]
- Gilart, N.; Marcé, R.M.; Fontanals, N.; Borrull, F. A rapid determination of acidic pharmaceuticals in environmental waters by molecularly imprinted solid-phase extraction coupled to tandem mass spectrometry without chromatography. Talanta 2013, 110, 196–201. [Google Scholar] [CrossRef]
- Xu, J.; Sun, H.; Zhang, Y.; Alder, A.C. Occurrence and enantiomer profiles of β-blockers in wastewater and a receiving water body and adjacent soil in Tianjin, China. Sci. Total Environ. 2019, 650, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Togola, A.; Baran, N.; Coureau, C. Advantages of online SPE coupled with UPLC/MS/MS for determining the fate of pesticides and pharmaceutical compounds. Anal. Bioanal. Chem. 2014, 406, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Huntscha, S.; Singer, H.P.; McArdell, C.S.; Frank, C.E.; Hollender, J. Multiresidue analysis of 88 polar organic micropollutants in ground, surface and wastewater using online mixed-bed multilayer solid-phase extraction coupled to high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2012, 1268, 74–83. [Google Scholar] [CrossRef]
- Diaz-Cruz, M.; de Alda, M.; Barcelo, D. Environmental behavior and analysis of veterinary and human drugs in soils, sediments and sludge. Trends Anal. Chem. 2003, 22, 340–351. [Google Scholar] [CrossRef]
- Richardson, S.; Ternes, T. Water analysis: Emerging contaminants and current issues. Anal. Chem. 2011, 83, 4614–4648. [Google Scholar] [CrossRef]
- Borecka, M.; Białk-Bielińska, A.; Siedlewicz, G.; Kornowska, K.; Kumirska, J.; Stepnowski, P.; Pazdro, K. A new approach for the estimation of expanded uncertainty of results of an analytical method developed for determining antibiotics in seawater using solid-phase extraction disks and liquid chromatography coupled with tandem mass spectrometry technique. J. Chromatogr. A 2013, 1304, 138–146. [Google Scholar] [CrossRef]
- Daniels, K.; Park, M.; Huang, Z.; Jia, A.; Flores, G.; Lee, H.; Snyder, S. A review of extraction methods for the analysis of pharmaceuticals in environmental waters. Crit. Rev. Environ. Sci. Technol. 2020, 50, 2271–2299. [Google Scholar] [CrossRef]
- Kim, S.; Carlson, K. Quantification of human and veterinary antibiotics in water and sediment using SPE/LC/MS/MS. Anal. Bioanal. Chem. 2007, 387, 1301–1315. [Google Scholar] [CrossRef]
- Lindsey, M.E.; Meyer, T.M.; Thurman, E.M. Analysis of trace levels of sulfonamide and tetracycline antimicrobials in groundwater and surface water using solid-phase extraction and liquid chromatography/mass spectrometry. Anal. Chem. 2001, 73, 4640–4646. [Google Scholar] [CrossRef]
- Stolker, A.A.M.; Nielsing, W.; Hogendoorn, E.A.; Versteegh, J.F.M.; Fuchs, R.; Brinkman, U.A.T. Liquid chromatography with triple-quadrupole or quadrupole-time of flight mass spectrometry for screening and confirmation of residues of pharmaceuticals in water. Anal. Bioanal. Chem. 2004, 378, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Batt, A.L.; Aga, D.S. Simultaneous analysis of multiple classes of antibiotics by ion trap LC/MS/MS for assessing surface water and groundwater contamination. Anal. Chem. 2005, 77. [Google Scholar] [CrossRef] [PubMed]
- Unceta, N.; Sampedro, M.C.; Abu Bakar, N.K.; Gómez-Caballero, A.; Goicolea, M.A.; Barrio, R.J. Multi-residue analysis of pharmaceutical compounds in wastewaters by dual solid-phase microextraction coupled to liquid chromatography electrospray ionization ion trap mass spectrometry. J. Chromatogr. A 2010, 1217, 3392–3399. [Google Scholar] [CrossRef]
- López-Serna, R.; Petrović, M.; Barceló, D. Development of a fast instrumental method for the analysis of pharmaceuticals in environmental and wastewaters based on ultra high performance liquid chromatography (UHPLC)-tandem mass spectrometry (MS/MS). Chemosphere 2011, 85, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Seifrtová, M.; Nováková, L.; Lino, C.; Pena, A.; Solich, P. An overview of analytical methodologies for the determination of antibiotics in environmental waters. Anal. Chim. Acta 2009, 649, 158–179. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Petrovic, M.; Barcelo, D. Tracing pharmaceutical residues of different therapeutic classes in environmental Waters by using liquid chromatography/quadrupole-linear ion trap mass spectrometry and automated library searching. Anal. Chem. 2009, 81, 898–912. [Google Scholar] [CrossRef]
- Wong, C.S.; MacLeod, S.L. JEM spotlight: Recent advances in analysis of pharmaceuticals in the aquatic environment. J. Environ. Monit. 2009, 11, 923–936. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Dinsdale, R.M.; Guwy, A.J. Multi-residue method for the determination of basic/neutral pharmaceuticals and illicit drugs in surface water by solid-phase extraction and ultra performance liquid chromatography-positive electrospray ionisation tandem mass spectrometry. J. Chromatogr. A 2007, 1161, 132–145. [Google Scholar] [CrossRef]
- Robles-Molina, J.; Lara-Ortega, F.J.; Gilbert-López, B.; García-Reyes, J.F.; Molina-Díaz, A. Multi-residue method for the determination of over 400 priority and emerging pollutants in water and wastewater by solid-phase extraction and liquid chromatography-time-of-flight mass spectrometry. J. Chromatogr. A 2014, 1350, 30–43. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Dinsdale, R.; Guwy, A. The removal of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs during wastewater treatment and its impact on the quality of receiving waters. Water Res. 2009, 43, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Batt, A.L.; Kostich, M.S.; Lazorchak, J.M. Analysis of ecologically relevant pharmaceuticals in wastewater and surface water using selective solid-phase extraction and UPLC-MS/MS. Anal. Chem. 2008, 80, 5021–5030. [Google Scholar] [CrossRef] [PubMed]
- Ido, A.; Hiromori, Y.; Meng, L.; Usuda, H.; Nagase, H.; Yang, M.; Hu, J.; Nakanishi, T. Occurrence of fibrates and their metabolites in source and drinking water in Shanghai and Zhejiang, China. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panditi, V.R.; Batchu, S.R.; Gardinali, P.R. Online solid-phase extraction-liquid chromatography-electrospray-tandem mass spectrometry determination of multiple classes of antibiotics in environmental and treated waters. Anal. Bioanal. Chem. 2013, 405, 5953–5964. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Thurman, E.M. Analysis of 100 pharmaceuticals and their degradates in water samples by liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2012, 1259, 148–157. [Google Scholar] [CrossRef]
- Hernández, F.; Ibáñez, M.; Gracia-Lor, E.; Sancho, J.V. Retrospective LC-QTOF-MS analysis searching for pharmaceutical metabolites in urban wastewater. J. Sep. Sci. 2011, 34, 3517–3526. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, H.; Chang, H. Improved method for analyzing estrogens in water by liquid chromatography-electrospray mass spectrometry. J. Chromatogr. A 2005, 1070, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Gilart, N.; Marcé, R.M.; Borrull, F.; Fontanals, N. Determination of pharmaceuticals in wastewaters using solid-phase extraction-liquid chromatography-tandem mass spectrometry. J. Sep. Sci. 2012, 35, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Ammann, A.A.; Macikova, P.; Groh, K.J.; Schirmer, K.; Suter, M.J. LC-MS/MS determination of potential endocrine disruptors of cortico signalling in rivers and wastewaters. Anal. Bioanal. Chem. 2014, 406, 7653–7665. [Google Scholar] [CrossRef] [PubMed]
- Vieno, N.; Tuhkanen, T.; Kronberg, L. Analysis of neutral and basic pharmaceuticals in sewage treatment plants and in recipient rivers using solid phase extraction and liquid chromatography-tandem mass spectrometry detection. J. Chromatogr. A 2006, 1134, 101–111. [Google Scholar] [CrossRef]
- Rodriguez, E.; Navarro-Villoslada, F.; Benito-Pena, E.; Marazuela, M.; Moreno-Bondi, M. Multiresidue determination of ultratrace levels of fluoroquinolone antimicrobials in drinking and aquaculture water samples by automated online molecularly imprinted solid phase extraction and liquid chromatography. Anal. Chem. 2011, 83, 2046–2055. [Google Scholar] [CrossRef]
- Mogolodi Dimpe, K.; Nomngongo, P.N. Application of activated carbon-decorated polyacrylonitrile nanofibers as an adsorbent in dispersive solid-phase extraction of fluoroquinolones from wastewater. J. Pharm. Anal. 2019, 9, 117–126. [Google Scholar] [CrossRef]
- Wu, A.; Zhao, X.; Wang, J.; Tang, Z.; Zhao, T.; Niu, L.; Yu, W.; Yang, C.; Fang, M.; Lv, H.; et al. Application of solid-phase extraction based on magnetic nanoparticle adsorbents for the analysis of selected persistent organic pollutants in environmental water: A review of recent advances. Crit. Rev. Environ. Sci. Technol. 2020. [Google Scholar] [CrossRef]
- Herrera-Herrera, A.V.; Hernández-Borges, J.; Afonso, M.M.; Palenzuela, J.A.; Rodríguez-Delgado, M. Comparison between magnetic and non magnetic multi-walled carbon nanotubes-dispersive solid-phase extraction combined with ultra-high performance liquid chromatography for the determination of sulfonamide antibiotics in water samples. Talanta 2013, 116, 695–703. [Google Scholar] [CrossRef]
- Li, N.; Chen, J.; Shi, Y. Magnetic polyethyleneimine functionalized reduced graphene oxide as a novel magnetic sorbent for the separation of polar non-steroidal anti-inflammatory drugs in waters. Talanta 2019, 191, 526–534. [Google Scholar] [CrossRef]
- Capriotti, A.; Cavaliere, C.; La Barbera, G.; Montone, C.; Piovesana, S.; Lagana, A. Recent applications of magnetic solid-phase extraction for sample preparation. Chromatographia 2019, 82, 1251–1274. [Google Scholar] [CrossRef]
- Liu, D.; Huang, Z.; Li, M.; Li, X.; Sun, P.; Zhou, L. Construction of magnetic bifunctional beta-cyclodextrin nanocomposites for adsorption and degradation of persistent organic pollutants. Carbohydr. Polym. 2020, 230. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Huang, Z.; Li, M.; Sun, P.; Yu, T.; Zhou, L. Novel porous magnetic nanospheres functionalized by beta-cyclodextrin polymer and its application in organic pollutants from aqueous solution. Environ. Pollut. 2019, 250, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; He, Q.; Wang, L.; Wang, X.; Dong, Q.; Huang, C. Preparation of magnetic multi-functional molecularly imprinted polymer beads for determining environmental estrogens in water samples. J. Hazard. Mater. 2013, 252, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Soto, J.M.; Cárdenas, S.; Valcárcel, M. Evaluation of carbon nanocones/disks as sorbent material for solid-phase extraction. J. Chromatogr. A 2009, 1216, 5626–5633. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, L.; Li, J.; Liu, D.; Chen, L. Recent advances in solid-phase sorbents for sample preparation prior to chromatographic analysis. Trends Anal. Chem. 2014, 59, 26–41. [Google Scholar] [CrossRef]
- Li, X.; Zhu, G.; Luo, Y.; Yuan, B.; Feng, Y. Synthesis and applications of functionalized magnetic materials in sample preparation. Trends Anal. Chem. 2013, 45, 233–247. [Google Scholar] [CrossRef]
- Pérez, R.A.; Albero, B.; Férriz, M.; Tadeo, J.L. Analysis of macrolide antibiotics in water by magnetic solid-phase extraction and liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2017, 146, 79–85. [Google Scholar] [CrossRef]
- Luo, Y.; Shi, Z.; Gao, Q.; Feng, Y. Magnetic retrieval of graphene: Extraction of sulfonamide antibiotics from environmental water samples. J. Chromatogr. A 2011, 1218, 1353–1358. [Google Scholar] [CrossRef]
- Abdolmohammad-Zadeh, H.; Talleb, Z. Magnetic solid phase extraction of gemfibrozil from human serum and pharmaceutical wastewater samples utilizing a beta-cyclodextrin grafted graphene oxide-magnetite nano-hybrid. Talanta 2015, 134, 387–393. [Google Scholar] [CrossRef]
- Pena-Mendez, E.; Mawale, R.; Conde-Gonzalez, J.; Socas-Rodriguez, B.; Havel, J.; Ruiz-Perez, C. Metal organic framework composite, nano-Fe3O4@Fe-(benzene-1,3,5-tricarboxylic acid), for solid phase extraction of blood lipid regulators from water. Talanta 2020, 207. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Zhan, P.; Chen, X.; Pan, S.; Jin, M. Fast determination of 24 steroid hormones in river water using magnetic dispersive solid phase extraction followed by liquid chromatography-tandem mass spectrometry. Environ. Sci. Pollut. Res. 2016, 23, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Arteaga, K.; Rodriguez, J.; Miranda, J.; Medina, J.; Barrado, E. Determination of non-steroidal anti-inflammatory drugs in wastewaters by magnetic matrix solid phase dispersion-HPLC. Talanta 2010, 80, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Socas-Rodríguez, B.; Hernández-Borges, J.; Salazar, P.; Martín, M.; Rodríguez-Delgado, M. Core-shell polydopamine magnetic nanoparticles as sorbent in micro-dispersive solid-phase extraction for the determination of estrogenic compounds in water samples prior to high-performance liquid chromatography-mass spectrometry analysis. J. Chromatogr. A 2015, 1397, 1–10. [Google Scholar] [CrossRef]
- Sajid, M.; Nazal, M.K.; Ihsanullah, I. Novel materials for dispersive (micro) solid-phase extraction of polycyclic aromatic hydrocarbons in environmental water samples: A review. Anal. Chim. Acta 2021, 1141, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, Y.; Qiu, Q.; Zhu, Y.; Min, J.; Jin, M. A fast and high throughput LC-MS/MS method for the determination of 58 human and veterinary drugs in river water. Anal. Methods 2017, 9, 4228–4233. [Google Scholar] [CrossRef]
- Wackerlig, J.; Schirhagl, R. Applications of molecularly imprinted polymer nanoparticles and their advances toward industrial use: A review. Anal. Chem. 2016, 88, 250–261. [Google Scholar] [CrossRef]
- Demeestere, K.; Petrovic, M.; Gros, M.; Dewulf, J.; Van Langenhove, H.; Barcelo, D. Trace analysis of antidepressants in environmental waters by molecularly imprinted polymer-based solid-phase extraction followed by ultra-performance liquid chromatography coupled to triple quadrupole mass spectrometry. Anal. Bioanal. Chem. 2010, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, X.; Lu, W.; Wu, X.; Li, J. Molecular imprinting: Perspectives and applications. Chem. Soc. Rev. 2016, 45, 2137–2211. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, C.; Li, X.; Liu, L. Preparation and application of epitope magnetic molecularly imprinted polymers for enrichment of sulfonamide antibiotics in water. Electrophoresis 2017, 38, 2462–2467. [Google Scholar] [CrossRef]
- Rebelo, P.; Costa-Rama, E.; Seguro, I.; Pacheco, J.G.; Nouws, H.P.A.; Cordeiro, M.N.D.S.; Delerue-Matos, C. Molecularly imprinted polymer-based electrochemical sensors for environmental analysis. Biosens. Bioelectron. 2021, 172, 112719. [Google Scholar] [CrossRef] [PubMed]
- Wackerlig, J.; Lieberzeit, P. Molecularly imprinted polymer nanoparticles in chemical sensing—Synthesis, characterisation and application. Sens. Actuators B Chem. 2015, 207, 144–157. [Google Scholar] [CrossRef]
- Ouyang, G. Handbook of Solid Phase Microextraction; Pawliszyn, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 251–290. [Google Scholar]
- Montesdeoca-Esponda, S.; Torres-Padrón, M.E.; Sosa-Ferrera, Z.; Santana-Rodríguez, J.J. Analytical Separation Science; Jared, L., Anderson, A.B., Pino, V., Stalcup, A., Eds.; Wiley: Hoboken, NJ, USA, 2015; pp. 1897–1927. [Google Scholar]
- Pawliszyn, J. Solid Phase Microextraction, Theory and Practice; Wiley-VCH: New York, NY, USA, 1997. [Google Scholar]
- Almeida, C.; Boas, L. Analysis of BTEX and other substituted benzenes in water using headspace SPME-GC-FID: Method validation. J. Environ. Monit. 2004, 6, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Simoes, N.G.; Cardoso, V.V.; Ferreira, E.; Benoliel, M.J.; Almeida, C.M.M. Experimental and statistical validation of SPME-GC-MS analysis of phenol and chlorophenols in raw and treated water. Chemosphere 2007, 68, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Castells, P.; Santos, F.; Galceran, M. Solid-phase extraction versus solid-phase microextraction for the determination of chlorinated paraffins in water using gas chromatography—Negative chemical ionisation mass spectrometry. J. Chromatogr. A 2004, 157–162. [Google Scholar] [CrossRef]
- Sarafraz-Yazdi, A.; Amiri, A.; Rounaghi, G.; Eshtiagh-Hosseini, H. Determination of non-steroidal anti-inflammatory drugs in water samples by solid-phase microextraction based sol-gel technique using poly(ethylene glycol) grafted multi-walled carbon nanotubes coated fiber. Anal. Chim. Acta 2012, 720, 134–141. [Google Scholar] [CrossRef]
- López-Serna, R.; Marín-de-Jesús, D.; Irusta-Mata, R.; García-Encina, P.A.; Lebrero, R.; Fdez-Polanco, M.; Muñoz, R. Multiresidue analytical method for pharmaceuticals and personal care products in sewage and sewage sludge by online direct immersion SPME on-fiber derivatization—GCMS. Talanta 2018, 186, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Zhu, F.; Jiang, R.; Zhou, S.; Zhu, D.; Liu, H.; Ouyang, G. Determination of eight pharmaceuticals in an aqueous sample using automated derivatization solid-phase microextraction combined with gas chromatography-mass spectrometry. Talanta 2015, 136, 198–203. [Google Scholar] [CrossRef]
- Araujo, L.; Wild, J.; Villa, N.; Camargo, N.; Cubillan, D.; Prieto, A. Determination of anti-inflammatory drugs in water samples, by in situ derivatization, solid phase microextraction and gas chromatography-mass spectrometry. Talanta 2008, 75, 111–115. [Google Scholar] [CrossRef]
- Yu, H.; Merib, J.; Anderson, J.L. Crosslinked polymeric ionic liquids as solid-phase microextraction sorbent coatings for high performance liquid chromatography. J. Chromatogr. A 2016, 1438, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, M.; Yu, J.; Huang, X.; Li, H.; Lin, L.; Yuan, D. Monitoring of selected estrogen mimics in complicated samples usingpolymeric ionic liquid-based multiple monolithic fiber solid-phasemicroextraction combined with high-performance liquidchromatography. J. Chromatogr. A 2015, 1385, 12–19. [Google Scholar] [CrossRef]
- Gil García, M.D.; Cañada Cañada, F.; Culzoni, M.J.; Vera-Candioti, L.; Siano, G.G.; Goicoechea, H.C.; Martínez Galera, M. Chemometric tools improving the determination of anti-inflammatory and antiepileptic drugs in river and wastewater by solid-phase microextraction and liquid chromatography diode array detection. J. Chromatogr. A 2009, 1216, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Vera-Candioti, L.; Gil García, M.D.; Martínez Galera, M.; Goicoechea, H.C. Chemometric assisted solid-phase microextraction for the determination of anti-inflammatory and antiepileptic drugs in river water by liquid chromatography-diode array detection. J. Chromatogr. A 2008, 1211, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.M.; Chen, L.; Volmer, D.A. Rapid analysis of tetracycline antibiotics by combined solid phase microextraction/high performance liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 1999, 13, 1744–1754. [Google Scholar] [CrossRef]
- McClure, E.L.; Wong, C.S. Solid phase microextraction of macrolide, trimethoprim, and sulfonamide antibiotics in wastewaters. J. Chromatogr. A 2007, 1169, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, V.K.; Terry, K.A.; Toito, J. Determination of sulfonamide antibiotics in wastewater: A comparison of solid phase microextraction and solid phase extraction methods. J. Chromatogr. A 2006, 1131, 1–10. [Google Scholar] [CrossRef]
- Sánchez-Rojas, F.; Bosch-Ojeda, C.; Cano-Pavó, J.M. A review of stir bar sorptive extraction. Chromatographia 2009, 69, S69–S84. [Google Scholar] [CrossRef]
- He, M.; Chen, B.; Hu, B. Recent developments in stir bar sorptive extraction. Anal. Bioanal. Chem. 2014, 406, 2001–2026. [Google Scholar] [CrossRef]
- Camino-Sánchez, F.J.; Rodríguez-Gómez, R.; Zafra-Gómez, A.; Santos-Fandila, A.; Vílchez, J.L. Stir bar sorptive extraction: Recent applications, limitations and future trends. Talanta 2014, 130, 388–399. [Google Scholar] [CrossRef]
- Hashemi, S.H.; Kaykhaii, M. Nanoparticle coatings for stir bar sorptive extraction, synthesis, characterization and application. Talanta 2021, 221, 121568. [Google Scholar] [CrossRef]
- Fan, W.; Mao, X.; He, M.; Chen, B.; Hu, B. Development of novel sol-gel coatings by chemically bonded ionic liquids for stir bar sorptive extraction--application for the determination of NSAIDS in real samples. Anal. Bioanal. Chem. 2014, 406, 7261–7273. [Google Scholar] [CrossRef] [PubMed]
- Gilart, N.; Marcé, R.M.; Cormack, P.A.; Fontanals, N.; Borrull, F. Development of new polar monolithic coatings for stir bar sorptive extraction. J. Sep. Sci. 2014, 37, 2225–2232. [Google Scholar] [CrossRef]
- Mao, X.; He, M.; Chen, B.; Hu, B. Membrane protected C. J. Chromatogr. A 2016, 1472, 27–34. [Google Scholar] [CrossRef]
- Peng, J.; Liu, D.; Shi, T.; Tian, H.; Hui, X.; He, H. Molecularly imprinted polymers based stir bar sorptive extraction for determination of cefaclor and cefalexin in environmental water. Anal. Bioanal. Chem. 2017, 409, 4157–4166. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yang, Z.; Liu, Z. Development of dual-templates molecularly imprinted stir bar sorptive extraction and its application for the analysis of environmental estrogens in water and plastic samples. J. Chromatogr. A 2014, 1358, 52–59. [Google Scholar] [CrossRef]
- Bratkowska, D.; Fontanals, N.; Cormack, P.A.; Borrull, F.; Marcé, R.M. Preparation of a polar monolithic stir bar based on methacrylic acid and divinylbenzene for the sorptive extraction of polar pharmaceuticals from complex water samples. J. Chromatogr. A 2012, 1225, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ministério do Ambiente. Decreto-Lei n.º 236/1998, de 1 de Agosto de 1998, I Série—A; Ministério do Ambiente, do Ordenamento do Território e do Desenvolvimento Regional, Qualidade da Água: Lisbon, Portugal, 1998.
- Ministério do Ambiente. Decreto-Lei n.º 306/2007, de 27 de Agosto, Diário da República, 1.ª série, n.º 164, 27 de Agosto de 2007; Ministério do Ambiente, do Ordenamento do Território e do Desenvolvimento Regional, Qualidade da Água: Lisbon, Portugal, 2007.
- Petrovic, M.; Hernando, M.; Diaz-Cruz, M.; Barcelo, D. Liquid chromatography-tandem mass spectrometry for the analysis of pharmaceutical residues in environmental samples: A review. J. Chromatogr. A 2005, 1067, 1–14. [Google Scholar] [CrossRef]
- Gros, M.; Petrovic, M.; Barcelo, D. Multi-residue analytical methods using LC-tandem MS for the determination of pharmaceuticals in environmental and wastewater samples: A review. Anal. Bioanal. Chem. 2006, 386, 941–952. [Google Scholar] [CrossRef]
- Ternes, T. Analytical methods for the determination of pharmaceuticals in aqueous environmental samples. Trends Anal. Chem. 2001, 20, 419–434. [Google Scholar] [CrossRef]
- Ternes, T.A.; Bonerz, M.; Herrmann, N.; Loffler, D.; Keller, E.; Lacida, B.B.; Adler, A.C. Determination of pharmaceuticals, iodinated contrast media and musk fragrances in sludge by LC/tandem MS and GC/MS. J. Chromatogr. A 2005, 1067, 213–223. [Google Scholar] [CrossRef]
- O’Mahony, J.; Clarke, L.; Whelan, M.; O’Kennedy, R.; Lehotay, S.J.; Danaher, M. The use of ultra-high pressure liquid chromatography with tandem mass spectrometric detection in the analysis of agrochemical residues and mycotoxins in food—Challenges and applications. J. Chromatogr. A 2013, 1292, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Van de Steene, J.C.; Lambert, W.E. Comparison of matrix effects in HPLC-MS/MS and UPLC-MS/MS analysis of nine basic pharmaceuticals in surface waters. J. Am. Soc. Mass Spectrom. 2008, 19, 713–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchwell, M.I.; Twaddle, N.C.; Meeker, L.R.; Doerge, D.R. Improving LC-MS sensitivity through increase in chromatographic performance: Comparisons of UPLC-ES/MS/MS to HPLC-ES/MS/MS. J. Chromatogr. B 2005, 825, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Petrovic, M.; Gros, M.; Barcelo, D. Multi-residue analysis of pharmaceuticals in wastewater by ultra-performance liquid chromatography-quadrupole-time-of-flight mass spectrometry. J. Chromatogr. A 2006, 1124, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Rodríguez-Mozaz, S.; Barceló, D. Rapid analysis of multiclass antibiotic residues and some of their metabolites in hospital, urban wastewater and river water by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. J. Chromatogr. A 2013, 1292, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Gracia-Lor, E.; Sancho, J.V.; Hernández, F. Multi-class determination of around 50 pharmaceuticals, including 26 antibiotics, in environmental and wastewater samples by ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 2264–2275. [Google Scholar] [CrossRef]
- Nannou, C.; Kosma, C.; Albanis, T. Occurrence of pharmaceuticals in surface waters: Analytical method development and environmental risk assessment. Int. J. Environ. Anal. Chem. 2015, 95, 1242–1262. [Google Scholar] [CrossRef]
- Patrolecco, L.; Ademollo, N.; Grenni, P.; Tolomei, A.; Caracciolo, A.B.; Capti, S. Simultaneous determination of human pharmaceuticals in water samples by solid phase extraction and HPLC with UV- fluorescence detection. Microchem. J. 2013, 107, 165–171. [Google Scholar] [CrossRef]
- Gracia-Lor, E.; Sancho, J.; Serrano, R.; Hernandez, F. Occurrence and removal of pharmaceuticals in wastewater treatment plants at the Spanish Mediterranean area of Valencia. Chemosphere 2012, 87, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Sui, Q.; Huang, J.; Deng, S.; Yu, G. Rapid determination of pharmaceuticals from multiple therapeutic classes in wastewater by solid-phase extraction and ultra-performence liquid chromatography tandem mass spectrometry. Chin. Sci. Bull. 2009, 54, 4633–4643. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Aller, M.; Gurny, R.; Veuthey, J.L.; Guillarme, D. Coupling ultra high-pressure liquid chromatography with mass spectrometry: Constraints and possible applications. J. Chromatogr. A 2013, 1292, 2–18. [Google Scholar] [CrossRef] [PubMed]
- De Hoffman, E.; Stroobant, V. Mass Spectrometry—Principles and Applications; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Lonappan, L.; Pulicharla, R.; Rouissi, T.; Brar, S.K.; Verma, M.; Surampalli, R.Y.; Valero, J.R. Diclofenac in municipal wastewater treatment plant: Quantification using laser diode thermal desorption—Atmospheric pressure chemical ionization—Tandem mass spectrometry approach in comparison with an established liquid chromatography-electrospray ionization-tandem mass spectrometry method. J. Chromatogr. A 2016, 1433, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Metcalfe, C.D. Characterizing and compensating for matrix effects using atmospheric pressure chemical ionization liquid chromatography-tandem mass spectrometry: Analysis of neutral pharmaceuticals in municipal wastewater. Anal. Chem. 2008, 80, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, D.P.; Brar, S.K.; Tyagi, R.D.; Picard, P.; Surampalli, R.Y. Carbamazepine in municipal wastewater and wastewater sludge: Ultrafast quantification by laser diode thermal desorption-atmospheric pressure chemical ionization coupled with tandem mass spectrometry. Talanta 2012, 99, 247–255. [Google Scholar] [CrossRef]
- Horimoto, S.; Mayumi, T.; Aoe, K.; Nishimura, N.; Sato, T. Analysis of beta-lactam antibiotics by high performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry using bromoform. J. Pharm. Biomed. Anal. 2002, 30, 1093–1102. [Google Scholar] [CrossRef]
- Pfeifer, T.; Tuerk, J.; Bester, K.; Spiteller, M. Determination of selected sulfonamide antibiotics and trimethoprim in manure by electrospray and atmospheric pressure chemical ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2002, 16, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Doerge, D.R.; Bajic, S. Multiresidue determination of quinolone antibiotics using liquid chromatography coupled to atmospheric-pressure chemical ionization mass spectrometry and tandem mass spectrometry. Rapid Commun. Mass Spectrom. 1995, 9, 1012–1016. [Google Scholar] [CrossRef]
- Gardinali, P.R.; Zhao, X. Trace determination of caffeine in surface water samples by liquid chromatography—Atmospheric pressure chemical ionization—Mass spectrometry (LC-APCI-MS). Environ. Int. 2002, 28, 521–528. [Google Scholar] [CrossRef]
- Matić, I.; Grujić, S.; Jauković, Z.; Laušević, M. Trace analysis of selected hormones and sterols in river sediments by liquid chromatography-atmospheric pressure chemical ionization-tandem mass spectrometry. J. Chromatogr. A 2014, 1364, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Fayad, P.B.; Prévost, M.; Sauvé, S. Laser diode thermal desorption/atmospheric pressure chemical ionization tandem mass spectrometry analysis of selected steroid hormones in wastewater: Method optimization and application. Anal. Chem. 2010, 82, 639–645. [Google Scholar] [CrossRef]
- Boisvert, M.; Fayad, P.B.; Sauvé, S. Development of a new multi-residue laser diode thermal desorption atmospheric pressure chemical ionization tandem mass spectrometry method for the detection and quantification of pesticides and pharmaceuticals in wastewater samples. Anal. Chim. Acta 2012, 754, 75–82. [Google Scholar] [CrossRef]
- Wu, J.; Hughes, C.S.; Picard, P.; Letarte, S.; Gaudreault, M.; Lévesque, J.F.; Nicoll-Griffith, D.A.; Bateman, K.P. High-throughput cytochrome P450 inhibition assays using laser diode thermal desorption-atmospheric pressure chemical ionization-tandem mass spectrometry. Anal. Chem. 2007, 79, 4657–4665. [Google Scholar] [CrossRef]
- Crotti, S.; Isak, I.; Traldi, P. Liquid Chromatgraphy; Salvatore Fanali, P.R.H., Colin, F., Poole, P., Schoenmakers, D.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 307–331. [Google Scholar]
- Alonso, S.G.; Catalá Maroto, R.R.; Gil, J.L.R.; Miguel, A.G.; Valcárcel, Y. Pollution by psychoactive pharmaceuticals in the Rivers of Madrid metropolitan area (Spain). Environ. Int. 2010, 36, 195–201. [Google Scholar] [CrossRef]
- Pozo, O.; Guerrero, C.; Sancho, J.; Ibanez, M.; Pitarch, E.; Hogendoorn, E.; Hernandez, F. Efficient approach for the reliable quantification and confirmation of antibiotics in water using on-line solid-phase extraction liquid chromatography/tandem mass spectrometry. J. Chromatogr. A 2006, 1103, 83–93. [Google Scholar] [CrossRef]
- Boix, C.; Ibanez, M.; Sancho, J.; Parsons, J.; de Voogt, P.; Hernandez, F. Biotransformation of pharmaceuticals in surface water and during waste water treatment: Identification and occurrence of transformation products. J. Hazard. Mater. 2016, 302, 175–187. [Google Scholar] [CrossRef]
- Yang, S.; Carlson, K.H. Solid-phase extraction-high-performance liquid chromatography-ion trap mass spectrometry for analysis of trace concentrations of macrolide antibiotics in natural and wastewater matrices. J. Chromatogr. A 2004, 1038, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Parada, A.; Agüera, A.; Gómez-Ramos, M.D.M.; García-Reyes, J.F.; Heinzen, H.; Fernández-Alba, A.R. Behavior of amoxicillin in wastewater and river water: Identification of its main transformation products by liquid chromatography/electrospray quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 731–742. [Google Scholar] [CrossRef]
- Bueno, M.J.; Agüera, A.; Hernando, M.D.; Gómez, M.J.; Fernández-Alba, A.R. Evaluation of various liquid chromatography-quadrupole-linear ion trap-mass spectrometry operation modes applied to the analysis of organic pollutants in wastewaters. J. Chromatogr. A 2009, 1216, 5995–6002. [Google Scholar] [CrossRef]
- Calza, P.; Medana, C.; Padovano, E.; Giancotti, V.; Baiocchi, C. Identification of the unknown transformation products derived from clarithromycin and carbamazepine using liquid chromatography/high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 1687–1704. [Google Scholar] [CrossRef] [PubMed]
- Calza, P.; Medana, C.; Padovano, E.; Dal Bello, F.; Baiocchi, C. Identification of the unknown transformation products derived from lincomycin using LC-HRMS technique. J. Mass Spectrom. 2012, 47, 751–759. [Google Scholar] [CrossRef]
- Makarov, A.; Scigelova, M. Coupling liquid chromatography to Orbitrap mass spectrometry. J. Chromatogr. A 2010, 1217, 3938–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bade, R.; Causanilles, A.; Emke, E.; Bijlsma, L.; Sancho, J.V.; Hernandez, F.; de Voogt, P. Facilitating high resolution mass spectrometry data processing for screening of environmental water samples: An evaluation of two deconvolution tools. Sci. Total Environ. 2016, 569, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Hogenboom, A.C.; van Leerdam, J.A.; de Voogt, P. Accurate mass screening and identification of emerging contaminants in environmental samples by liquid chromatography-hybrid linear ion trap Orbitrap mass spectrometry. J. Chromatogr. A 2009, 1216, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Bijlsma, L.; Emke, E.; Hernández, F.; de Voogt, P. Performance of the linear ion trap Orbitrap mass analyzer for qualitative and quantitative analysis of drugs of abuse and relevant metabolites in sewage water. Anal. Chim. Acta 2013, 768, 102–110. [Google Scholar] [CrossRef]
- Abou-Elwafa Abdallah, M.; Nguyen, K.H.; Ebele, A.J.; Atia, N.N.; Ali, H.R.H.; Harrad, S. A single run, rapid polarity switching method for determination of 30 pharmaceuticals and personal care products in waste water using Q-Exactive Orbitrap high resolution accurate mass spectrometry. J. Chromatogr. A 2019, 1588, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Althakafy, J.T.; Kulsing, C.; Grace, M.R.; Marriott, P.J. Liquid chromatography—Quadrupole Orbitrap mass spectrometry method for selected pharmaceuticals in water samples. J. Chromatogr. A 2017, 1515, 164–171. [Google Scholar] [CrossRef]
- Bade, R.; Rousis, N.I.; Bijlsma, L.; Gracia-Lor, E.; Castiglioni, S.; Sancho, J.V.; Hernandez, F. Screening of pharmaceuticals and illicit drugs in wastewater and surface waters of Spain and Italy by high resolution mass spectrometry using UHPLC-QTOF MS and LC-LTQ-Orbitrap MS. Anal. Bioanal. Chem. 2015, 407, 8979–8988. [Google Scholar] [CrossRef] [Green Version]
- Chitescu, C.; Oosterink, E.; de Jong, J.; Stolker, A. Ultrasonic or accelerated solvent extraction followed by U-HPLC-high mass accuracy MS for screening of pharmaceuticals and fungicides in soil and plant samples. Talanta 2012, 88, 653–662. [Google Scholar] [CrossRef]
- Kalaboka, M.; Chrimatopoulos, C.; Jiménez-Holgado, C.; Boti, V.; Sakkas, V.; Albanis, T. Exploring the efficiency of UHPLC-orbitrap MS for the determination of 20 pharmaceuticals and acesulfame K in hospital and urban wastewaters with the aid of FPSE. Separations 2020, 7, 46. [Google Scholar] [CrossRef]
- Verlicchi, P.; Al Aukidy, M.; Galletti, A.; Petrovic, M.; Barceló, D. Hospital effluent: Investigation of the concentrations and distribution of pharmaceuticals and environmental risk assessment. Sci. Total Environ. 2012, 430, 109–118. [Google Scholar] [CrossRef]
- Al Aukidy, M.; Verlicchi, P.; Jelic, A.; Petrovic, M.; Barcelo, D. Monitoring release of pharmaceutical compounds: Occurrence and environmental risk assessment of two WWTP effluents and their receiving bodies in the Po Valley, Italy. Sci. Total Environ. 2012, 438, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Petrovic, M.; Ginebreda, A.; Barcelo, D. Removal of pharmaceuticals during wastewater treatment and environmental risk assessment using hazard indexes. Environ. Int. 2010, 36, 15–26. [Google Scholar] [CrossRef]
- Petrovic, M.; De Alda, M.; Diaz-Cruz, S.; Postigo, C.; Radjenovic, J.; Gros, M.; Barcelo, D. Fate and removal of pharmaceuticals and illicit drugs in conventional and membrane bioreactor wastewater treatment plants and by riverbank filtration. Philos. Trans. A Math. Phys. Eng. Sci. 2009, 367, 3979–4003. [Google Scholar] [CrossRef] [PubMed]
- Togola, A.; Budzinski, H. Analytical development for analysis of pharmaceuticals in water samples by SPE and GC-MS. Analytical and Bio. Anal. Chem. 2007, 388, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Togola, A.; Budzinski, H. Multi-residue analysis of pharmaceutical compounds in aqueous samples. J. Chromatogr. A 2008, 1177, 150–158. [Google Scholar] [CrossRef]
- Kot-Wasik, A.; Dębska, J.; Wasil, A.; Namiesnik, J. Determination of non-steroidal anti-inflammatory drugs in natural waters using off-line and on-line SPE followed by LC coupled with DAD-MS. Chromatographia 2006, 64, 13–21. [Google Scholar] [CrossRef]
- Tarcomnicu, I.; van Nuijs, A.L.; Simons, W.; Bervoets, L.; Blust, R.; Jorens, P.G.; Neels, H.; Covaci, A. Simultaneous determination of 15 top-prescribed pharmaceuticals and their metabolites in influent wastewater by reversed-phase liquid chromatography coupled to tandem mass spectrometry. Talanta 2011, 83, 795–803. [Google Scholar] [CrossRef]
- Zhou, L.J.; Ying, G.G.; Liu, S.; Zhao, J.L.; Chen, F.; Zhang, R.Q.; Peng, F.Q.; Zhang, Q.Q. Simultaneous determination of human and veterinary antibiotics in various environmental matrices by rapid resolution liquid chromatography-electrospray ionization tandem mass spectrometry. J. Chromatogr. A 2012, 1244, 123–138. [Google Scholar] [CrossRef]
- Gilart, N.; Cormack, P.A.; Marcé, R.M.; Fontanals, N.; Borrull, F. Selective determination of pharmaceuticals and illicit drugs in wastewaters using a novel strong cation-exchange solid-phase extraction combined with liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2014, 1325, 137–146. [Google Scholar] [CrossRef]
- Anumol, T.; Merel, S.; Clarke, B.O.; Snyder, S.A. Ultra high performance liquid chromatography tandem mass spectrometry for rapid analysis of trace organic contaminants in water. Chem. Cent. J. 2013, 7, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusmaroli, L.; Insa, S.; Petrovic, M. Development of an online SPE-UHPLC-MS/MS method for the multiresidue analysis of the 17 compounds from the EU “Watch list”. Anal. Bioanal. Chem. 2018, 410, 4165–4176. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Lor, E.; Martínez, M.; Sancho, J.V.; Peñuela, G.; Hernández, F. Multi-class determination of personal care products and pharmaceuticals in environmental and wastewater samples by ultra-high performance liquid-chromatography-tandem mass spectrometry. Talanta 2012, 99, 1011–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.V.; Couto, C.F.; Lebron, Y.A.R.; Moreira, V.R.; Foureaux, A.F.S.; Reis, E.O.; Santos, L.V.S.; de Andrade, L.H.; Amaral, M.C.S.; Lange, L.C. Occurrence and risk assessment of pharmaceutically active compounds in water supply systems in Brazil. Sci. Total Environ. 2020, 746, 141011. [Google Scholar] [CrossRef]
- Reis, E.O.; Foureaux, A.F.S.; Rodrigues, J.S.; Moreira, V.R.; Lebron, Y.A.R.; Santos, L.V.S.; Amaral, M.C.S.; Lange, L.C. Occurrence, removal and seasonal variation of pharmaceuticals in Brasilian drinking water treatment plants. Environ. Pollut. 2019, 250, 773–781. [Google Scholar] [CrossRef]
- Casado, J.; Rodríguez, I.; Ramil, M.; Cela, R. Selective determination of antimycotic drugs in environmental water samples by mixed-mode solid-phase extraction and liquid chromatography quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2014, 1339, 42–49. [Google Scholar] [CrossRef]
- Hernández, F.; Bijlsma, L.; Sancho, J.V.; Díaz, R.; Ibáñez, M. Rapid wide-scope screening of drugs of abuse, prescription drugs with potential for abuse and their metabolites in influent and effluent urban wastewater by ultrahigh pressure liquid chromatography-quadrupole-time-of-flight-mass spectrometry. Anal. Chim. Acta 2011, 684, 87–97. [Google Scholar] [CrossRef]
- EURACHEM. The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics. 1998. Available online: http://www.eurachem.org/guides/mval.htm (accessed on 9 January 2020).
- ISO/IEC 17025: 2017 (E). General Requirements for the Competence of Testing and Calibration Laboratories; International Organization for Standardization (ISO): Geneva, Switzerland, 2017. [Google Scholar]
- Thompson, M.; Ellison, S.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis—(IUPAC technical report). Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- EURACHEM. Guide to Quality in Analytical Chemistry. 2002. Available online: http://www.eurachem.org/guides/accr.htm (accessed on 9 January 2020).
- ISO 8466-1. Part 1: Statistical evaluation of the linear calibration function. In Water Quality—Calibration and Evaluation of Analytical Methods and Estimation of Performance Characteristics; International Organization for Standardization (ISO): Geneva, Switzerland, 1990. [Google Scholar]
- ISO. Accuracy (Trueness and Precision) of Measurement Methods and Results—Part 1: General Principles and Definitions; ISO 5725-1; International Organization for Standardization (ISO): Geneva, Switzerland, 1994. [Google Scholar]
- ISO. Statistics—Vocabulary and Symbols—Part 2: Applied statistics; ISO 3534-2; International Organization for Standardization (ISO): Geneva, Switzerland, 2006. [Google Scholar]
- Stuber, M.; Reemtsma, T. Evaluation of three calibration methods to compensate matrix effects in environmental analysis with LC-ESI-MS. Anal. Bioanal. Chem. 2004, 378, 910–916. [Google Scholar] [CrossRef]
- Petrovic, M.; Elijarrat, E.; Lopez de Alda, M.J.; Barceló, D. Endocrine disrupting compounds and other emerging contaminants in the environment: A survey on new monitoring strategies and occurence data. Anal. Bioanal. Chem. 2004, 378, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Konieczka, P.; Namieśnik, J. Estimating uncertainty in analytical procedures based on chromatographic techniques. J. Chromatogr. A 2010, 1217, 882–891. [Google Scholar] [CrossRef]
- EA (European Co-Operation for Accreditation). EA Guidelines on the Expression of Uncertainty in Quantitative Testing; EA-4/16; EA: Amsterdam, The Netherlands, 2003. [Google Scholar]
- EURACHEM/CITAC. Quantifying Uncertainty in Analytical Measurement. Guide CG 4, 2nd ed. 2000. Available online: http://www.eurachem.org/guides/ptguide2000.pdf (accessed on 27 July 2019).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, C.M.M. Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices. Separations 2021, 8, 16. https://doi.org/10.3390/separations8020016
Almeida CMM. Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices. Separations. 2021; 8(2):16. https://doi.org/10.3390/separations8020016
Chicago/Turabian StyleAlmeida, Cristina M. M. 2021. "Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices" Separations 8, no. 2: 16. https://doi.org/10.3390/separations8020016
APA StyleAlmeida, C. M. M. (2021). Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices. Separations, 8(2), 16. https://doi.org/10.3390/separations8020016